蛋白质与小分子结合位点预测方法、预测装置与流程
本发明涉及蛋白质与小分子结合位点预测方法、预测装置,属于小分子结合蛋白结合位点预测技术领域。
背景技术:
蛋白质并不是独立存在的,它们必须和诸如dna、rna以及其它的蛋白质和小分子相互作用来发挥其生物学功能。其中蛋白质-小分子结合位点是蛋白质表面行使蛋白质生物功能的活性位点,知道蛋白质与其他小分子的相互作用位点,也能够帮助科学家更多的了解蛋白质的生物学功能,为蛋白质的药物设计提供技术支撑。
通常人们可以通过生物学实验方法来发现蛋白质表面小分子的结合位点,并且实验得到的结合亦非常可靠。但是受技术、时间和经济等因素的限制,使用实验来确定结合位点的方案往往效率较低且成本高昂。相比之下,使用生物信息学手段来预测蛋白质与小分子结合位点有着极大的生命力,能够节省可观的时间和经济成本。基于生物结构的药物设计是生物信息学极为重要的研究领域,通常基于结构化的药物设计的第一步是在蛋白质的表面准确的预测这种蛋白质与小分子结合位点的位置。
目前,已经开发了一些预测蛋白质-小分子结合位点的预测方法,如《蛋白质-小分子结合位点预测新算法研究开发,浙江大学硕士学位论文》中开发了一种新型的计算机预测算法,整合了ligsite、pass、q-sitefinder、surfnet等传统算法,达到了较高的预测效果。但是该算法在构建预测模型及计算时,往往仅关注单个氨基酸残基的性质,而没有从所在位置的整体去考虑,因此其预测的效果比较有限。
技术实现要素:
本发明的目的是提供蛋白质与小分子结合位点预测方法,该方法使用滑动采样窗口法提取小分子结合蛋白残基对应特征矩阵,能够更准确的预测蛋白质与小分子结合位点。
本发明还提供了蛋白质与小分子结合位点预测装置,能够更准确的预测蛋白质与小分子结合位点。
为了实现上述目的,本发明所采用的技术方案是:
蛋白质与小分子结合位点预测方法,包括如下步骤:
1)从蛋白质类数据库中提取能够与小分子相互作用的小分子结合蛋白数据集;使用滑动采样窗口法提取小分子结合蛋白残基对应特征矩阵,窗口中心位置的残基如果是结合位点,则采样窗口所提取的矩阵是正集;窗口中心位置的残基如果是非结合位点,则采样窗口所提取的矩阵是负集;
2)将小分子结合蛋白残基对应特征矩阵转化为一维向量,构建分类模型;
3)将待测蛋白质对应特征输入所述的分类模型中,对待测蛋白质的结合位点进行预测。
本发明中的蛋白质与小分子结合位点预测方法,使用滑动采样窗口法提取数据,蛋白质结合作用会受到结合残基周围环境影响,因此采用采样窗口法提取数据来表征中心位置残基的特征,具有更好的表征效果;在使用提取的特征构建分类模型后,分类模型具有更好的预测效果,能够更准确的预测蛋白与小分子的结合位点。
步骤1)和步骤3)中所述对应特征包括:氨基酸类别、pssm矩阵、亲水性、疏水性和静电电荷中的一种或多种。
本发明中通过分析发现残基的分布差异可能与结合位点与非结合位点的残基功能有关;蛋白质序列中不同位置残基进化的保守性可以通过蛋白质的位置特异性评分矩阵(pssm)反映出来;还发现小分子与蛋白质结合区域残基,表现出更强的亲水倾向,同时结合残基电荷也强于非结合残基;因此可以以上述五种特征中的一种或几种作为基础,构建分类模型。
步骤1)中所述滑动采样窗口法中窗口长度等于13-17。
蛋白质与小分子的结合作用,不仅与结合残基特征有关,还与该残基周围环境有关。当窗口长度较小时,采样提取的矩阵,不能有效地代表结合残基周围环境的特征;当采样长度太大时,采样所得矩阵中将包含大量无关信息,这会影响分类模型的分类效果。同时根据不同长度的重复性实验结果,最终选择采样窗口长度为13-17作为最终特征计算标准。
步骤1)中所述分类模型使用xgboost算法构建。
treeboosting是一种有效且广泛使用的机器学习方法。在本文中,使用可扩展的端到端treeboost系统:xgboost,科学家广泛使用这些算法在许多机器学习工作中取得了显著的结果。gradientboosting是在boosting的基础上的改进。该算法的思想是连续减少残差(residual),并进一步减少先前模型在梯度方向上的残差以获得新模型。
步骤1)中所述蛋白质类数据库为sc-pdb数据库。sc-pdb数据库中收集了较多的蛋白质和配体小分子数据。
步骤1)中对所有的小分子结合蛋白数据进行去除冗余的处理,得到小分子结合蛋白数据集;具体为:去除同源性≥30%的序列,去除序列长度≤40的序列,去除
进行去除冗余的处理后得到的数据集有效性更强。
步骤1)中筛选出aco、adp、anp、atp、coa、fad、fmn、gdp、gnp、nad、nap、ndp、sah和sam中的一个或多个种类的小分子结合蛋白作为小分子结合蛋白数据集。
这14类小分子结合蛋白数据量足够大,可用于分类,分类模型的训练和测试。
蛋白质与小分子结合位点预测装置,包括如下模块:
用于从蛋白质类数据库中提取出小分子结合蛋白数据集的模块;
用于获得小分子结合蛋白残基对应特征的模块;
用于采用滑动采样窗口法提取小分子结合蛋白残基对应特征矩阵的模块;窗口中心位置的残基如果是结合位点,则采样窗口所提取的矩阵是正集;窗口中心位置的残基如果是非结合位点,则采样窗口所提取的矩阵是负集;
用于将小分子结合蛋白残基对应特征矩阵转化为一维向量的模块;用于构建分类模型的模块;
用于将待测蛋白质对应特征输入所述的分类模型中,对待测蛋白质的结合位点进行预测的模块。
本发明中的蛋白质与小分子结合位点预测装置包括用于采用滑动采样窗口法提取小分子结合蛋白残基对应特征矩阵的模块,采用采样窗口法提取数据来表征中心位置的残基的特征,具有更好的表征效果;在使用提取的特征构建分类模型后,分类模型具有更好的预测效果,进而该预测装置能够更准确的预测蛋白与小分子的结合位点。
所述对应特征包括:氨基酸类别、pssm矩阵、亲水性、疏水性和静电电荷中的一种或多种。
本发明中通过分析发现残基的分布差异可能与结合位点与非结合位点的残基功能有关;蛋白质序列中不同位置残基进化的保守性可以通过蛋白质的位置特异性评分矩阵(pssm)反映出来;还发现小分子与蛋白质结合区域残基,表现出更强的亲水倾向,同时结合残基电荷也强于非结合残基;因此可以以上述五种特征中的一种或几种作为基础,构建分类模型。
所述滑动采样窗口法中窗口长度等于13-17。
蛋白质与小分子的结合作用,不仅与结合残基特征有关,还与该残基周围环境有关。当窗口长度较小时,采样提取的矩阵,不能有效地代表结合残基周围环境的特征;当采样长度太大时,采样所得矩阵中将包含大量无关信息,这会影响分类模型的分类效果。综上,最终选择采样窗口长度为13-17作为最终特征计算标准。
附图说明
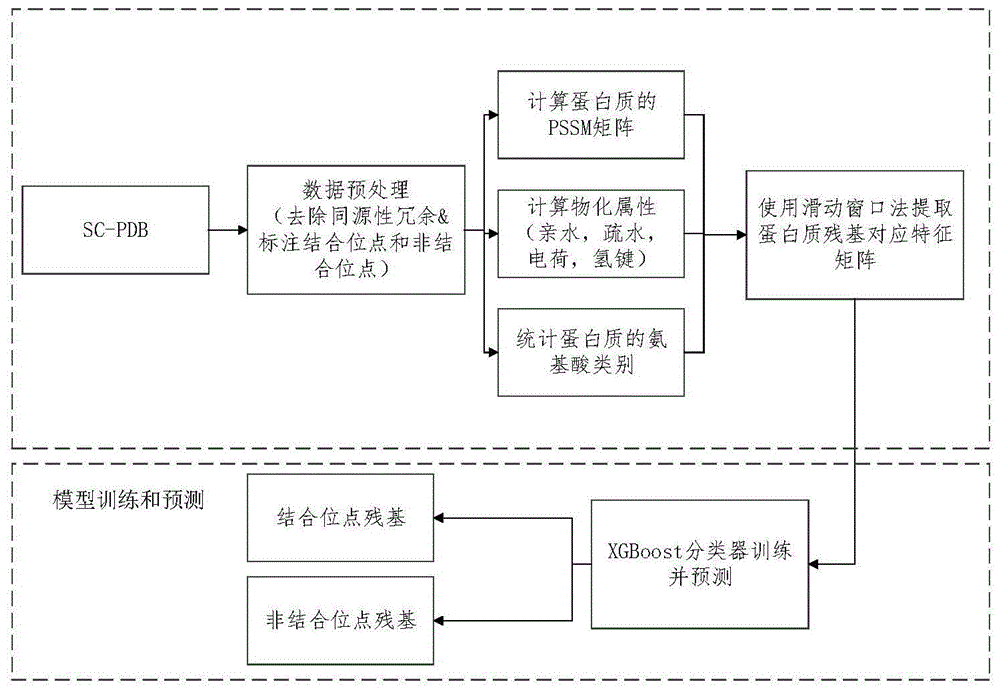
图1为本发明蛋白质与小分子结合位点预测方法的实施例1中蛋白质小分子结合位点的分析与预测流程图;
图2为本发明蛋白质与小分子结合位点预测方法的实施例1中pdb数据库中蛋白质编号为12gs对应的pssm矩阵文件展示图;
图3为本发明蛋白质与小分子结合位点预测方法的实施例1中rna结合蛋白的结合区域几何结构实例图;
图4为本发明蛋白质与小分子结合位点预测方法的实施例1中蛋白质序列滑动窗口采样示意图;
图5为本发明蛋白质与小分子结合位点预测方法的实施例1中蛋白质小分子序列中20类氨基酸的结合残基和非结合残基的分布情况图;
图6为本发明蛋白质与小分子结合位点预测方法的实施例1中蛋白质小分子结合位点和非结合位点残基亲水性属性值分布图;
图7为本发明蛋白质与小分子结合位点预测方法的实施例1中蛋白质小分子结合位点和非结合位点残基疏水性属性值分布图;
图8为本发明蛋白质与小分子结合位点预测方法的实施例1中蛋白质小分子结合位点和非结合位点残基静电电荷属性值分布图;
图9为本发明蛋白质与小分子结合位点预测方法的实施例1中蛋白质小分子结合位点和非结合位点残基氢键属性值分布图;
图10为本发明蛋白质与小分子结合位点预测方法的实施例1中采样窗口w与分类结果的auc之间的关系;
图11为本发明蛋白质与小分子结合位点预测方法的实施例1中采样窗口w与分类结果的准确率之间的关系;
图12为本发明蛋白质与小分子结合位点预测方法的实施例1中平均降低准确率法计算各种特征的重要性得分展示图。
具体实施方式
下面结合具体实施例对本发明做进一步的详细说明。
蛋白质与小分子结合位点预测方法的实施例1
本发明中,提出了一种基于蛋白质小分子pdb数据,预测小分子与蛋白质结合位点的新方法,本发明实验中构建的分类模型,可用于预测结合位点和非结合位点。通过提取蛋白质序列上残基的物理化学属性与进化信息等特征,生成训练和测试所需的数据集,训练并测试分类模型,最终将分类模型用于分类预测蛋白质的结合位点和非结合位点。蛋白质的pssm矩阵信息已被许多科学家应用于,如预测结合位点、二级结构和分析蛋白质的功能等工作中。由于蛋白质小分子序列中的残基是否突变,是由进化过程中的许多因素决定的,这些因素也影响蛋白质与小分子的结合作用。同样结合残基和非结合残基的物理化学性质也不同,为了研究这些区别,实验中分析了蛋白质序列上结合残基和非结合残基的疏水性、亲水性、静电电荷和氢键,与此同时还以独热码形式,表示氨基酸残基的类型信息。在上述工作的基础上,构建xgboost分类模型,对蛋白质小分子的结合位点和非结合位点,进行分类预测。用14个类别的蛋白质小分子的数据集,训练和测试构建的分类模型,最终获得了非常显着的分类结果,实验流程如图1所示。
1、构建蛋白质小分子数据集
蛋白质配体小分子数据集从sc-pdb数据库提取。截至2018年12月,sc-pdb数据库中收集了4782个蛋白质和6326个配体小分子数据。在实验过程中使用pisces程序(http://dunbrack.fccc.edu/guoli/pisces.php),对所有的小分子结合蛋白数据进行去除冗余的处理。软件中筛选蛋白质数据所设定的第一个条件是序列同源性不超过30%,同时为去除蛋白质序列过短数据,还设置了序列长度应大于40的标准为获取精度相对较高的蛋白质数据,实验中去掉了低分辨率结构,要求只留下了分辨率高于
表1蛋白质小分子数据集不同类别的序列与结合位点信息
2、特征计算
1)氨基酸类别的分布
实验共从sc-pdb数据库中共收集了与14个种类配体小分子结合的6582个蛋白质序列数据。通过计算蛋白质序列中结合残基和非结合残基的氨基酸类别,得到各类残基的分布情况。
2)pssm矩阵
蛋白质序列中不同位置残基进化的保守性可以通过蛋白质的位置权重矩阵反映出来。位置权重矩阵(pwm),也称为位置特定权重矩阵(pswm)或位置特异性评分矩阵(pssm),是生物序列中的基序(模式)的常用表示。位置权重矩阵(positionweightmatrix)由美国遗传学家garystormo及其同事在1982年引入。
psi-blast是位置特定的迭代基本局部对齐搜索工具。该程序用于查找蛋白质的远亲。首先,创建所有密切相关的蛋白质列表,这些蛋白质组合成一般的“概况”序列,其总结了这些序列中存在的重要特征。然后使用该概况运行针对蛋白质数据库的查询,并且发现更大的蛋白质组。这个较大的组用于构建另一个配置文件,并重复该过程。psi-blast在获取远程进化关系方面比标准的blast程序更敏感。实验中用psi-blast程序来扫描ncbi数据库,其中参数e值设为0.001,迭代次数设为3。如图2所示为psi-blast程序处理蛋白质数据后得到的pssm文件。
3)物理化学性质
aaindex是包含多种氨基酸属性指数的数据库。目前数据库共搜集了544种氨基酸指数,如图3所示,氨基酸指数代表氨基酸物理化学属性;h表示属性类型的索引号;d表示属性的类型描述,a为属性值发布的作者,t表示相关论文的标题,j表示文章发表的期刊信息,c为与本属性和其他属性的相关性系数。在蛋白质组研究中,蛋白质序列是由20种氨基酸无规律构成的符号序列。在实验中可以将符号序列转换成为氨基酸属性指数序列,以将蛋白质进行数字编码。
hopp为20种氨基酸提供了具有固定值的亲水性标度(aaindexid:hopt810101)。jones给出了20种氨基酸的疏水性标度(aaindexid:hopt810101)。通常认为具有疏水性的氨基酸倾向存在于蛋白质结构的内部,具有亲水性的氨基酸倾向分布于蛋白质分子的表面。因此,亲水性氨基酸与小分子相互作用的可能性更大,这种特性有助于构建分类模型以分类预测结合残基和非结合残基。
蛋白质与小分子结合区域的静电荷,是蛋白质与配体分子相互作用最具影响力的特性之一。静电互补有助于蛋白质与小分子的非特异性结合。在20种标准氨基酸中,通常认为arg,his和lys带正电荷,asp和glu带负电荷(aaindexid:fauj880111,aaindexid:fauj880112)。
氢键是一种相互作用力,其比分子间作用力略强,但略弱于共价键和离子键。氢键在确定配体分子结合的特异性中起关键作用。因此,氨基酸的氢键是需要研究的重要物理化学性质之一。氨基酸氢键特性值可以从aaindex数据库获得(aaindexid:fauj880109)。
3、滑动采样窗口
在预测蛋白质与小分子的结合位点工作中,从蛋白质序列中提取信息构建特征向量是关键的步骤。由于蛋白质数据结合残基与非结合残基是交替排列的,在蛋白质序列上使用滑动窗口来采样。如图4所示,蛋白质序列中任意氨基酸残基为中心点,以(w-1)/2为边界,提取该残基属性值矩阵;取样窗口的长度为(w-1)/2,窗口中包括了结合位点残基和两侧残基信息,使用八个值作为窗口长度。窗口中心位置的残基如果是结合位点,则采样窗口所提取的矩阵是正集。窗口中心位置的残基如果是非结合位点,则采样窗口所提取的矩阵是负集。以上步骤中,之所以用采样窗口法提取数据,是因为蛋白质结合作用会受到结合残基周围环境影响。
4、结果与讨论
1)氨基酸类别的分布
氨基酸类别的分布如图5所示,对于非结合残基,ala,leu,gly,val和glu分布较多。gly,glu,cys和lys这四种残基表现出统计学意义上的差异。上述残基的分布差异可能与结合位点与非结合位点的残基功能有关。
2)蛋白质结合位点理物理化学性质分析
在统计氨基酸类别分布情况外,实验中还计算了结合位点与非结合位点的亲水性、疏水性、静电电荷和氢键的分布。结果如图6和7中所示,蛋白质结合位点和非结合位点之间,亲水性值和疏水性值的差异非常显著。这是因为结合位点残基主要分布在蛋白质表面,因此有更大的概率与水分子接触。如图8中所示,非结合区域静电电荷分布小于结合区域,这因为静电互补有助于蛋白质与小分子的结合作用。先前研究发现氢键在配体和蛋白质的结合作用中发挥重要作用,但计算结果显示结合残基和非结合残基之间,氢键分布没有表现出统计学意义上的差异(如图9中所示)。综上所述,蛋白质小分子残基的亲水性、疏水性和静电电荷,是有助于构建预测结合位点和非结合位点的分类模型的。
3)确定采样窗口的长度
本实验基于14类蛋白质小分子数据集,训练并评估测试了构建的分类模型。14类小分子分别是aco、adp、anp、atp、coa、fad、fmn、gdp、gnp、nad、nap、ndp、sah和sam。特征提取过程中,采样窗口的长度会决定提取数据集的质量,也会影响分类实验效果。因此,实验针对14类小分子数据集,分别测试了3、5、7、9、11、13、15、17和19这几个窗口长度。由于数据集中,蛋白质结合位点与非结合位点个数相差较大,为了数据集的正负样例平衡,实验中使用下采样来处理数据集不平衡问题。针对每个种类蛋白质数据集,融合亲水性、疏水性、静电电荷、氨基酸类别和pssm矩阵组成训练和测试用的特征矩阵。
最后xgboost分类模型使用10折交叉验证方法进行训练和测试。为了结果对比,选择auc和准确率作为评判标准。auc值与窗口长度的关系如图10中所示,当窗口长度从3开始增加时,auc的值显示出逐渐上升趋势,窗口长度等于15时,大部分蛋白质小分子的auc达到峰值;随着窗口长度的继续增加,auc开始逐渐减小。如图11中所示,准确率与采样窗口长度的关系走势与auc有类似的表现,随采样窗口长度增加,准确率值逐渐增加,w=15时所有准确率值达到峰值,随后开始降低。之所以会有这种现象,其原因是由于蛋白质与小分子的结合作用,不仅与结合残基特征有关,还与该残基周围环境有关。当窗口长度较小时,采样提取的矩阵,不能有效地代表结合残基周围环境的特征,因此auc和准确率很小。但由于小分子的原子个数相对比蛋白质较少,所以序列中结合位点的个数有限。当采样长度太大时,采样所得矩阵中将包含大量无关信息,这会影响分类模型的分类效果,从而导致auc和准确率的值降低。综上最终选择采样窗口长度为15作为最终特征计算标准。
4)特征重要性测试
本发明之前共分析了6类特征:氨基酸种类、pssm矩阵、亲水性、疏水性、静电电荷和氢键。为衡量特征对构建分类模型的重要程度,实验中使用了平均降低准确率法(meandecrease准确率uracy),计算其中6类特征(氨基酸种类、pssm矩阵、亲水性、疏水性、静电电荷和氢键)对分类模型的影响。算法基本思想是将一种特征的值更改为随机数,再将特征矩阵输入分类模型进行分类预测,观察分类模型的准确率降低幅度。对于重要程度较低的特征,分类模型输出的准确值影响不大,如将较重要的特征值改为随机数,那么会大大影响分类模型的准确率。使用平均降低准确率法测试结果如图12所示,从图中可知pssm矩阵具有最高的重要性得分,其次是氨基酸类别、亲水性、疏水性和电荷,氢键对分类模型的贡献程度最小。该现象与前述对蛋白质结合位点的分析结果相符合。
5、构建分类模型
实验中构建的特征矩阵融合了氨基酸类别、pssm矩阵、亲水性、疏水性和静电电荷的信息。将小分子结合蛋白残基对应特征矩阵转化为一维向量,xgboost算法构建分类模型。特征计算过程中采样窗口长度为15。
随后,对14类蛋白质小分子数据集,使用xgboost分类模型执行10折交叉验证测试。测试结果如表2所示,分类模型在atp小分子数据集上训练和测试,auc和准确率的结果分别是0.935和0.927,这样的结果是所有小分子数据集中最高的。所有数据集的平均auc和准确率的结果分别为0.918和0.913,auc与准确率值越大表示训练后的分类模型分类效果越好,这也表明实验构建的分类模型的分类效果是非常显著的。表中的精确率、召回率、f1(精确率和召回率综合数值)数值作为参考。
表2xgboost模型蛋白质小分子数据集上取得的分类预测结果
6、未知蛋白的预测
将待测蛋白质对应特征输入所述的分类模型中,对待测蛋白质的结合位点进行预测。该处的对应特征是指上文中提到的氨基酸类别、pssm矩阵、亲水性、疏水性和静电电荷的特征在输入分类模型前,将蛋白残基对应特征矩阵转化为一维向量。
7、小结
本发明关注于蛋白质小分子结合位点与非结合位点的分类预测工作。针对蛋白质小分子的结合位点和非结合位点,首先,分析了结合位点和非结合位点氨基酸的组成,从结果可知,ala和leu等残基分布多于其他残基。gly,glu,cys和lys四类残基,在结合位点和非结合位点上的分布差别是明显的。与此同时,实验还分析残基的亲水性、疏水性、静电电荷和氢键这几种性质,从而对比两类残基之间物理化学性质的差异程度。分析结果说明小分子与蛋白质结合区域残基,表现出更强的亲水倾向,同时结合残基电荷也强于非结合残基,而两类残基的氢键值差别却不大,这印证了特征的重要性分析的结果。随后,基于上述特征,同时融合pssm矩阵构造特征向量。基于14种蛋白质小分子数据集,使用xgboost算法构建分类模型执行10折交叉验证测试。最终分类模型获得了显著的效果,平均auc和准确率分别为0.918和0.913。这说明本发明中提出的方法能够较好的预测蛋白质小分子的结合位点和非结合位点,实验过程中的分析同样有助于理解蛋白质和小分子之间的结合机制。
蛋白质与小分子结合位点预测装置的实施例1
本实施例中蛋白质与小分子结合位点预测装置,包括如下模块:
用于从蛋白质类数据库中提取出小分子结合蛋白数据集的模块;
用于获得小分子结合蛋白残基对应特征的模块;所述对应特征包括:氨基酸类别、pssm矩阵、亲水性、疏水性和静电电荷中的一种或多种;
用于采用滑动采样窗口法提取小分子结合蛋白残基对应特征矩阵的模块;窗口中心位置的残基如果是结合位点,则采样窗口所提取的矩阵是正集;窗口中心位置的残基如果是非结合位点,则采样窗口所提取的矩阵是负集。所述滑动采样窗口法中窗口长度等于13-17,优选为15;
用于将小分子结合蛋白残基对应特征矩阵转化为一维向量的模块;用于构建分类模型的模块;
用于将待测蛋白质对应特征输入所述的分类模型中,对待测蛋白质的结合位点进行预测的模块。所述对应特征包括:氨基酸类别、pssm矩阵、亲水性、疏水性和静电电荷中的一种或多种。
- 还没有人留言评论。精彩留言会获得点赞!