一种定量PCR检测转基因细胞中外源基因拷贝数的方法与流程
本发明属于分子生物学技术领域,涉及一种转基因动物细胞中抗体分子的轻重链基因拷贝数的定量检测方法。
背景技术:
动物细胞已广泛地应用于生产各种生物活性物质如疫苗、生长因子、抗原、单克隆抗体等重组蛋白药物,这些动物细胞包括293细胞、Vero细胞、BHK细胞、PER.C6细胞、SP2/0细胞、NS0细胞、CHO细胞等,其中应用最为广泛的当属CHO细胞。经美国FDA批准的仅重组表达的抗体药物就达二十几种,每年创造的市场价值高达几百亿美元。随着基因工程技术的发展,越来越多的外源蛋白如抗体通过转基因的方法来获得。
但是对于转基因细胞来说,外源基因是非内源性的物质,细胞在生长繁殖过程中,会产生外源基因丢失的情况,这就是基因的遗传稳定性。转基因细胞中普遍存在着基因不稳定的情况,对于工业化生产的细胞来说,特别是对于制药企业,细胞株的稳定性显得特别重要,其对于产品质量的稳定起到了非常重要的作用(Barnes LM等Biotechnol.Bioeng.,2003,81(6):631-9.)。所以非常有必要从分子水平、从遗传的角度来深入分析整合入细胞株的外源基因的稳定性。目前,研究外源基因的稳定性主要是通过检测基因拷贝数的变化来分析的(Chusainow J等,Biotechnol.Bioeng.,2009,102(4):1182-1196.)。
基因拷贝数的检测目前常用Southern Blotting(Kaneko Y等,Journal of Bioscience and Bioengineering,2010,109(3):274–280)、Real-time PCR(Lattenmayer C等,J Biotechnol.2007,128(4):716-725)等方法,而采用Real-time PCR的方法可以省却了Southern Blotting方法的繁琐的步骤,可以简单快速准确地检测基因组的拷贝数。Real time PCR(实时PCR)又叫Q-PCR(quantitative PCR,定量PCR,实时定量PCR),是指在PCR反应体系中加入荧光基团(如SYBR-Green、TaqMan荧光探针等),利用荧光信号的积累实时监测整个PCR进程,最后通过荧光强度的变化来对未知模板进 行定量分析的方法。定量PCR按照荧光材料的不同,主要分为SYBR-Green定量PCR、TaqMan定量PCR方法。
TaqMan定量PCR方法是在原有一对引物的基础之上,需要重新合成一条与正反向引物之间的靶序列相结合的特异性探针,荧光基团连接在探针的5'末端,而淬灭基团则在3'末端。当探针与靶序列配对时,荧光基团发射的荧光因与3'端的淬灭基团接近而被淬灭;PCR扩增时,Taq酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和猝灭荧光基团分离,从而荧光监测系统可接收到荧光信号。
在SYBR-Green为荧光染料的PCR反应体系中,加入过量SYBR-Green染料,SYBR-Green染料非特异性地掺入双链DNA而非单链DNA后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射荧光信号,从而保证荧光信号随着PCR产物的增加而同步增加。与TaqMan定量PCR方法相比较,SYBR-Green定量PCR方法具有使用简单,DNA染料较便宜,成本较低,不需要加入特异性探针,而且适用于任何序列,通用性好。
在常规PCR扩增过程中,产物的累积一般遵循指数扩增的规律,Nn=N0(1+e)n(Nn为扩增n轮后的产物量,N0为起始模板量,e为扩增效率,n为循环数)。但到PCR扩增后期,随着产物的不断增加,扩增产物已不再呈指数性增加,PCR的终产物量与起始模板量之间没有线性关系,根据最终PCR产物量无法计算出起始DNA拷贝数。而在定量PCR反应中,可以实时检测产物产生的荧光强度,在荧光信号呈指数扩增阶段,PCR产物量的对数值与起始模板量之间存在线性关系,因此可以进行定量分析。
在定量PCR过程中,每一个PCR样品都有一个Ct阈值(Cycle threshold value),Ct值的含义是:每个反应管内的荧光信号到达设定的阈值时所经历的循环数,一般而言,PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光阈值的缺省设置是3-15个循环的荧光信号的标准偏差的10倍,即:Ct=10×SDcycle(3-15)。大量数据研究表明,每个模板的Ct值与该模板的起始拷贝数的对数存在线性关系,起始拷贝数越多,Ct值越小。
利用已知起始拷贝数的标准品可作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代表Ct值。因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。随着近几年基因组计划的研究进展,大量的物种基因组测序完成,包括人、小鼠、大鼠、中国仓鼠、食蟹猴、果蝇、线虫、牛、斑马鱼、恒河猴、家蚕、鸡、猩猩、山羊、猕猴、猪、马、熊猫等生物,相继阐明了基因组DNA的精确大小,则可推 算单位质量的细胞基因组中所含的DNA分子数,依据样品的起始拷贝数,进而可以绝对定量细胞中的外源基因的拷贝数量。
技术实现要素:
本发明的第一个目的是提供一种检测转基因动物细胞中抗体分子重链基因拷贝数的定量检测方法,该方法中采用的重链基因正向引物为JRTH3和重链基因反向引物为JRTH3R,其中
JRTH3的序列为:5'-CAAGCTGACCGTGGATAA-3'(如SEQ ID No.1所示),
JRTH3R的序列为:5'-GACAGGCTCTTCTGAGTG-3'(如SEQ ID No.2所示)。
为了实现上述目的,本发明采用如下的步骤:
a)合成针对抗体分子重链基因的特异性引物JHc-1F(如SEQ ID No.7所示的DNA分子)和JHc-1R(如SEQ ID No.8所示的DNA分子),以完整抗体分子的重链DNA作为模板进行普通PCR扩增,制备出带有重链片段的质粒DNA,从而制备重链质粒DNA标准品。
b)以JRTH3和JRTH3R为正向引物和反向引物、以步骤a)中的质粒DNA作为模板进行定量PCR反应,计算质粒DNA的拷贝数。从而建立标准曲线。
c)提取转染抗体分子的动物细胞株的基因组DNA和未转染抗体分子的动物细胞株的基因组DNA作为模板,以JRTH3和JRTH3R为正向引物和反向引物,进行定量PCR反应,同时用无菌超纯水作为空白对照。定量PCR反应结束后进行溶解曲线的试验,获得Ct值,根据Ct值和步骤b)中的标准曲线计算动物细胞中抗体分子的重链基因拷贝数。
上述步骤b)和c)中,在进行定量PCR扩增时,PCR反应的总体积是30μl:SYBR Green PCR Mix(SYBR Green PCR混合液)15μl;正向引物(10μM)0.5μl;反向引物(10μM)0.5μl;DNA模板10μl;无菌超纯水4μl。反应条件为95℃10min;95℃30s、60℃30s、72℃30s,共40个循环。
上述的抗体如可以是人源化抗TNF-α单克隆抗体,还可以是人-鼠嵌合性抗TNF-α单克隆抗体,还可以是抗HER2人源化单克隆抗体,还可以是抗VEGF人源化单克隆抗体等等。
本发明的第二个目的是提供一种检测转基因动物细胞中抗体分子轻链基因拷贝数的定量检测方法,该方法中采用的轻链基因正向引物为JRTL1F和轻链基因反向引物 JRTL1R,其中
JRTL1F的序列为:5'-GCAGTGGAAGGTGGATAA-3'(如SEQ ID No.3所示),
JRTL1R的序列为:5'-TTTGCTCAGTGTCAGGGT-3'(如SEQ ID No.4所示)。
为了实现上述目的,本发明采用如下的步骤:
a)合成针对抗体分子轻链基因的特异性引物JLc-1F(如SEQ ID No.5所示的DNA分子)和JLc-1R(如SEQ ID No.6所示的DNA分子),以完整抗体分子的轻链DNA作为模板进行普通PCR扩增,制备出带有轻链片段的质粒DNA,从而制备轻链质粒DNA标准品。
b)以JRTL1F和JRTL1R为正向引物和反向引物、以步骤a)中的质粒DNA作为模板进行定量PCR反应,获取Ct值和计算质粒DNA的拷贝数,从而建立标准曲线。
c)提取转染抗体分子的动物细胞株的基因组DNA和未转染抗体分子的动物细胞株的基因组DNA作为模板,以JRTL1F和JRTL1R为正向引物和反向引物进行定量PCR反应,同时用无菌超纯水作为空白对照。定量PCR反应结束后进行溶解曲线的试验,获得Ct值,根据Ct值和步骤b)中的标准曲线计算动物细胞中抗体分子的轻链基因拷贝数。
上述步骤b)和c)中,在进行定量PCR扩增时,PCR反应的总体积是30μl:SYBR Green PCR Mix(SYBR Green PCR混合液)15μl;正向引物(10μM)0.5μl;反向引物(10μM)0.5μl;DNA模板10μl;无菌超纯水4μl。反应条件为95℃10min;95℃30s、60℃30s、72℃30s,共40个循环。
上述的抗体如可以是人源化抗TNF-α单克隆抗体,还可以是人-鼠嵌合性抗TNF-α单克隆抗体,还可以是抗HER2人源化单克隆抗体,还可以是抗VEGF人源化单克隆抗体等。
本发明采用SYBR-Green Real-time PCR方法,可针对动物细胞中抗体分子的轻链基因和/或重链基因的拷贝数进行单独或同时测定,且是绝对定量,使得不同实验、不同细胞间的检测结果可以相互比较;可用于研究抗体细胞株的稳定性,筛选出稳定的高表达的工程化细胞株。
本发明的方法检测灵敏度高且线性范围宽,最低可以检测到细胞中仅含有1个基因拷贝数的样品,最大可以达到1500以上个拷贝数;线性关系良好,R2均大于0.99,足以胜任样品中基因拷贝数检测的需要。
本发明的方法检测的结果可靠,样品的测定Ct值的标准偏差<0.3。
本发明的方法,PCR检测反应特异性高,定量PCR产物的熔解曲线仅有单一吸收峰,无非特异性扩增信号。
本发明方法通用性好,可通过改变设计特异性引物,针对不同抗体分子的轻链基因或重链基因进行基因拷贝数的检测;可同时测定多种细胞内的基因拷贝数,这些细胞可以包括293细胞、Vero细胞、BHK细胞、PER.C6细胞、SP2/0细胞、NS0细胞、CHO细胞、DG44细胞等。
本发明的方法可以高通量测定多个样品的基因拷贝数。
本发明的方法采用SYBR-Green为荧光染料,具有简单便宜的优点,在PCR反应过程中可完全代替昂贵的TaqMan荧光探针,同时达到准确测定基因拷贝数的目的。
附图说明
图1.轻链标准品的定量PCR扩增曲线,其中横坐标为循环数,纵坐标为荧光强度。图中从左至右6条曲线是不同浓度的轻链质粒DNA标准品的定量PCR扩增曲线。
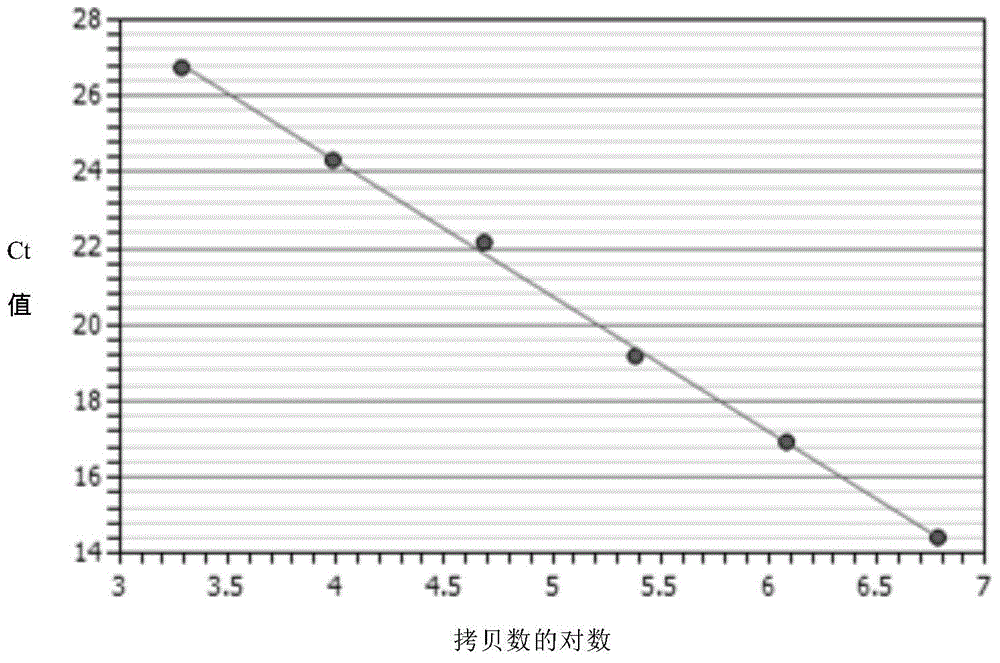
图2.轻链基因标准曲线,其中横坐标为轻链质粒DNA标准品质粒拷贝数的对数,纵坐标为定量PCR仪软件自动优化得到的Ct值。
图3.重链标准品的定量PCR扩增曲线,其中横坐标为循环数,纵坐标为荧光强度。图中从左至右6条曲线是不同浓度的重链质粒DNA标准品的定量PCR扩增曲线。
图4.重链基因标准曲线,其中横坐标为轻链质粒DNA标准品质粒拷贝数的对数,纵坐标为定量PCR仪软件自动优化得到的Ct值。
图5.轻链基因的定量PCR扩增曲线,其中横坐标为循环数,纵坐标为荧光强度。图中曲线为转染抗体分子的动物细胞株的基因组DNA和未转染抗体分子的动物细胞株的基因组DNA的定量PCR扩增曲线,基线部分的曲线为无菌超纯水的空白对照曲线。
图6.轻链基因的定量PCR熔解曲线,其中横坐标为温度;纵坐标为荧光相对时间变化率。图中曲线为转染抗体分子的动物细胞株的基因组DNA和未转染抗体分子的动物细胞株的基因组DNA的PCR熔解曲线,基线部分的曲线为无菌超纯水的空白对照曲线。
图7.重链基因的定量PCR扩增曲线,其中横坐标为循环数,纵坐标为荧光强度。图中曲线为转染抗体分子的动物细胞株的基因组DNA和未转染抗体分子的动物细胞株的基因组DNA的定量PCR扩增曲线,基线部分的曲线为无菌超纯水的空白对照曲线。
图8.重链基因的定量PCR熔解曲线,其中横坐标为温度,纵坐标为荧光相对时间变化率。图中曲线为转染抗体分子的动物细胞株的基因组DNA和未转染抗体分子的动物 细胞株的基因组DNA的PCR熔解曲线,基线部分的曲线为无菌超纯水的空白对照曲线。
具体实施方式
下面以具体的实施例,对本发明的技术方案做进一步的说明,其中动物细胞选最常用的CHO细胞作为示例,但本发明并不限于这些实施例。
本发明的技术方案包括标准质粒DNA和基因组DNA的制备、标准曲线的建立、轻重链基因的拷贝数的检测、基因拷贝数的计算等过程。本发明的技术方案包括以下实验流程:
实施例1轻链基因拷贝数的定量PCR检测方法
1.1、质粒DNA的获得:
a)设计针对轻链基因的特异性引物:特异性正向引物JLc1F的核苷酸序列为:5'-AGGACAGTGGCTGCACCA-3'(如SEQ ID No.5所示);特异性反向引物JLc1R的核苷酸序列为:5'-TCAACACTCTCCTCTGTT-3'(如SEQ ID No.6所示)。
b)获得轻链pLc质粒菌种:以完整抗体分子的轻链DNA为模板,进行普通PCR扩增,获得抗体分子轻链Lc片段,然后插入到p simple-19T载体上,转化DH5α感受态细胞,获得了含有抗体轻链基因的pLc质粒菌种。
c)经序列测定,获得了分子大小为3016bp的pLc质粒菌种。
1.2、轻链质粒DNA标准品的制备
将含有抗体轻链基因的pLc质粒菌种,接种于10ml的LB液体培养基中,37℃摇床培养过夜,取1ml培养液,10000rpm离心弃上清,菌液沉淀按Axygen公司“质粒DNA小量抽提试剂盒”说明书中的方法抽提质粒DNA,最终溶于60ml无菌超纯水中。获得的质粒用核酸蛋白分析仪(Bio-Rad公司的SmartSpecTM Plus Spectrophotometer)检测提取的DNA质量。用无菌超纯水将轻链质粒稀释至0.002ng/ml,然后采用5倍倍比稀释,制备得到轻链质粒DNA标准品。
1.3、标准曲线的建立
取10μl步骤1.2中制备的轻链质粒DNA标准品作为DNA模板、正向引物JRTL1F 0.5μl(浓度10μM)、正反引物JRTL1R 0.5μl(浓度10μM)、SYBR Green PCR Mix 15μl(购自Applied Biosystems公司)以及无菌超纯水4μl,进行定量PCR反应。其中采用的实时定量PCR仪为Bio-Rad公司的iQ5Real-Time PCR Detection System系统。定量PCR 反应条件为95℃10min;95℃30s、60℃30s、72℃30s,共40个循环。轻链的定量PCR扩增曲线见图1。
其中,正向引物JRTL1F的核苷酸序列为:5'-GCAGTGGAAGGTGGATAA-3'(如SEQ ID No.3所示);反向引物JRTL1R的核苷酸序列为:5'-TTTGCTCAGTGTCAGGGT-3'(如SEQ ID No.4所示)。
根据以下公式计算质粒DNA拷贝数:
计算得到:1ng轻链质粒含有的拷贝数为3.02×108个,则轻链质粒DNA标准品的拷贝数分别为:6.04×106,1.21×106,2.42×105,4.83×104,9.66×103,1.93×103。
以定量PCR仪软件(仪器自带的iQ5分析软件)自动优化得到的Ct值作为纵坐标,起始模板中标准质粒拷贝数的对数作为横坐标生成标准曲线见图2,PCR的扩增效率91.6%,二者的曲线相关系数R2=0.999,标准曲线的回归方程为:Y=-3.542X+38.459。
依据标准曲线,最低检测的拷贝数是1.93×103,即1.93×103/3723=0.5,也就是能检测到1个拷贝数,能够胜任转基因细胞中检测的需要;最大检测的拷贝数是6.04×106,即6.04×106/3723=1622,也就是最大能检测到1622个拷贝数。
1.4、细胞培养
复苏一支转染了抗体轻链基因的CHO细胞株,接种于无血清培养基中,37℃、5%CO2摇床中培养,待细胞活力达到98%以上时,分别接种于另外二瓶摇瓶中,一瓶加入MTX(氨甲喋呤,Methotrexate),另一瓶不另加入MTX,在相同条件下开始连续培养,分别在培养第10、20、30、35、40代时,取细胞并离心,沉淀保存于-80℃冰箱。
1.5、细胞基因组DNA的制备
按照QIAmp DNA Mini Kit说明书,分别提取未转染抗体分子的CHO细胞和转染抗体轻链基因的CHO细胞株的基因组DNA,溶于无菌超纯水中。获得的基因组DNA用核酸蛋白分析仪(Bio-Rad公司的SmartSpecTM Plus Spectrophotometer)检测提取的DNA质量,并分别用无菌超纯水将基因组DNA稀释至1ng/μl,作为定量PCR检测的阴性对照和样品,并用无菌超纯水作为空白对照。
1.6、轻链基因的拷贝数的检测:
各取基因组DNA 10μl作为PCR反应模板,JRTL1F/JRTL1R分别为正反引物,进行定量PCR反应,并进行熔解曲线的试验,结果见图5和6。
实时定量PCR反应体系如下,定量PCR反应的条件为95℃10min;95℃30s、60℃30s、72℃30s,共40个循环。
熔解曲线的试验设置:针对反应产物从55℃开始,以0.5℃为间隔进行升温,至95℃,每个温度保持30s并检测荧光。反应结束后,iQ5分析软件自动以荧光随温度变化率与温度的关系绘图,得到熔解曲线。不同浓度的标准品,样品基因组DNA,阴性对照基因组DNA和空白对照,同时进行扩增,供试样品设多个复孔进行平行检测(≥3)。
从图5、6中可以看出,样品PCR扩增反应良好,而水和未转染抗体分子的CHO细胞的基因组没有任何扩增,所有的样品都仅在81℃左右出现一个单一吸收峰,说明PCR扩增的特异性好。同时,所有的样品的Ct值均落在标准曲线的范围以内。反应结束后,分析软件会自动优化得到Ct值,根据Ct值与轻链基因拷贝数检测的标准曲线来计算10ng样品DNA所含轻链基因拷贝数。
1.7、基因拷贝数的计算:
根据CHO基因组研究的进展,CHO细胞的基因组大小为2.45Gb(Xu X等,Nat Biotechnol,2011,29(8):735-741),则10ng的基因组DNA含有3723个DNA分子,可以按下列公式计算转基因CHO细胞基因组中含有的轻链基因拷贝数:
上述公式中,动物细胞CHO细胞的基因组大小为2.45Gb。
结果见表1。为获得更高的抗体表达,转染抗体的CHO细胞需要经过长期的连续的MTX增加压力的过程,这过程实际上就是抗体轻链基因在细胞基因组中拷贝数增加的过程(Cacciatore JJ等.Biotechnology Advances.2010,(28):673–681)。表1中也可看出采用MTX增加压力的轻链基因拷贝数大于不加入MTX的轻链基因拷贝数。
Bubner B et.al.认为,一般单个样品的扩增反应的Ct值的标准偏差小于0.3,就可以 认为能够精确测定样品的拷贝数(Bubner B等,Plant Cell Rep.2004,23(5):263-271)。由表1可见,本方法每个样品扩增反应的Ct值的标准偏差均小于0.3,说明我们的方法是准确可靠的。
表1定量PCR检测轻链基因的拷贝数
注:表1中,+MTX是指摇瓶中加入MTX,-MTX是指摇瓶中不加入MTX。
实施例2重链基因拷贝数的检测方法
2.1、质粒DNA的获得:
a)设计针对重链基因的正向引物JHc-1F和反向引物JHc-1R:
正向引物JHc-1F的核苷酸序列为:5'-ATGCTAGCAAGCTTCCATGGGCGTCGACAAAGGGACCATCTGTGT-3'(如SEQ ID No.7所示);反向引物JHc-1R的核苷酸序列为:5'-TAGGTACCTCGAGTCATTTACCAGGAGACAG-3'(如SEQ ID No.8所示)。
b)获得重链pHc质粒菌种
以完整抗体分子的重链DNA为模板,进行普通PCR扩增,获得抗体分子重链Hc片段,然后插入到T载体上,转化DH5α感受态细胞,获得重链pLc质粒菌种。
c)经序列测定,获得了分子大小为3718bp的pHc质粒菌种。
2.2、重链标准质粒DNA的制备
将含有抗体重链基因的pHc质粒菌种,接种于10ml的LB液体培养基中,37℃摇床培养过夜,取1ml培养液,10000rpm离心弃上清,菌液沉淀按按Axygen公司“质粒 DNA小量抽提试剂盒”说明书的方法抽提质粒DNA,最终溶于60ml无菌超纯水中。获得的质粒用核酸蛋白分析仪(Bio-Rad公司的SmartSpecTM Plus Spectrophotometer)检测提取的DNA质量。用无菌超纯水将轻链质粒稀释至0.002ng/ml,然后采用5倍倍比稀释,制备得到重链质粒DNA标准品。
2.3、标准曲线的建立
取10μl制备的重链质粒DNA标准品作为DNA模板,正反引物JRTH3F 0.5μl(浓度10μM)和反向引物JRTH3R 0.5μl(浓度10μM)、SYBR Green PCR Mix 15μl(购自Applied Biosystems公司)以及无菌超纯水4μl,进行定量PCR反应。其中采用的实时定量PCR仪为Bio-Rad公司的iQ5Real-Time PCR Detection System系统。定量PCR反应条件为95℃10min;95℃30s、60℃30s、72℃30s,共40个循环。重链的定量PCR扩增曲线见图3。
其中,正向引物JRTH3F的核苷酸序列为:5'-CAAGCTGACCGTGGATAA-3'(如SEQ ID No.1所示);反向引物JRTH3R的核苷酸序列为:5'-GACAGGCTCTTCTGAGTG-3'(如SEQ ID No.2所示)。
根据以下公式计算质粒DNA拷贝数:
计算得到:1ng重链质粒含有的拷贝数为2.45×108个,则重链质粒DNA标准品的拷贝数分别为:4.90×106,9.80×105,1.96×105,3.92×104,7.84×103,1.57×103。
以定量PCR仪软件(仪器自带的iQ5分析软件)自动优化得到的Ct值作为纵坐标,起始模板中标准质粒拷贝数的对数作为横坐标生成标准曲线见图4,PCR的扩增效率91.8%,二者的曲线相关系数R2=1.000,标准曲线的回归方程为:Y=-3.536X+38.388。
依据标准曲线,最低检测的拷贝数是1.57×103,即1.57×103/3723=0.42,也就是能检测到一个拷贝数,能够胜任转基因细胞中检测的需要;最大检测的拷贝数是4.96×106,即4.96×106/3723=1332,也就是最大能检测到1332拷贝数。考虑到轻链重链的一致性和检测曲线的良好线性,应该也可以测到1500拷贝数的,但如果严格地来描述,可以用1300为最大检测值。
2.4、细胞培养
复苏一支转染了抗体重链基因的CHO细胞株,接种于无血清培养基中,37℃,5%CO2摇床中培养,待细胞活力达到98%以上时,分别接种于另外二瓶摇瓶中,一瓶加入MTX (氨甲喋呤,amethopterin),另一瓶不另加入MTX,在相同条件下开始连续培养,分别在培养第10、20、30、35、40代时,取细胞并离心,沉淀保存于-80℃冰箱。
2.5、细胞基因组DNA的制备
按照QIAmp DNA Mini Kit说明书,分别提取未转染抗体分子的CHO细胞和转染抗体轻链基因的CHO细胞株的基因组DNA,溶于无菌超纯水中。获得的基因组DNA用核酸蛋白分析仪(Bio-Rad公司的SmartSpecTM Plus Spectrophotometer)检测提取的DNA质量,并分别用无菌超纯水将基因组DNA稀释至1ng/ml,作为定量PCR检测的阴性对照和样品,并用无菌超纯水作为空白对照。
2.6、重链基因的拷贝数的检测:
各取基因组DNA 10μl作为PCR反应模板,JRTH3F/JRTH3R分别为正反引物,进行定量PCR反应,并进行熔解曲线的试验,结果见图7和8。
实时定量PCR反应体系如下,定量PCR反应的条件为95℃10min;95℃30s、60℃30s、72℃30s,共40个循环。
熔解曲线的试验设置:针对反应产物从55℃开始,以0.5℃为间隔进行升温,至95℃,每个温度保持30s并检测荧光。反应结束后,iQ5分析软件自动以荧光随温度变化率与温度的关系绘图,得到熔解曲线。不同浓度的标准品,样品基因组DNA,阴性对照基因组DNA和空白对照,同时进行扩增,供试样品设多个复孔进行平行检测(≥3)。
从7、8中可以看出,样品PCR扩增反应良好,而水和未转染抗体分子的CHO细胞的基因组没有任何扩增,所有的样品都仅在81℃左右出现一个单一吸收峰,说明PCR扩增的特异性好。同时,所有的样品的Ct值均落在标准曲线的范围以内。反应结束后,分析软件会自动优化得到Ct值,根据Ct值与轻链基因拷贝数检测的标准曲线来计算10ng样品DNA所含轻链基因拷贝数。
2.7、基因拷贝数的计算:
根据CHO基因组研究的进展,CHO细胞的基因组大小为2.45Gb(Xu X et.al.,Nat Biotechnol,2011,29(8):735-741),则10ng的基因组DNA含有3723个DNA分子,可以按下列公式计算转基因CHO细胞基因组中含有的重链基因拷贝数:
结果见表2。为获得更高的抗体表达,转染抗体的CHO细胞需要经过长期的连续的MTX增加压力的过程,这过程实际上就是抗体重链基因在细胞基因组中拷贝数增加的过程(Cacciatore JJ等.Biotechnology Advances.2010,(28):673–681)。表2中也可看出采用MTX增加压力的重链基因拷贝数大于不加入MTX的重链基因拷贝数。
Bubner B et.al.认为,一般单个样品的扩增反应的Ct值的标准偏差小于0.3,就可以认为能够精确测定样品的拷贝数(Bubner B等,Plant Cell Rep.2004,23(5):263-271)。由表2可见,本方法每个样品扩增反应的Ct值的标准偏差均小于0.3,说明我们的方法是准确可靠的。
表2定量PCR检测重链基因的拷贝数
注:表2中,+MTX是指摇瓶中加入MTX,-MTX是指摇瓶中不加入MTX。
重组抗体的表达水平依赖于轻链和重链基因的协同表达,二者之间存在着一个合适的轻链重链比例(Chusainow J等,Biotechnol Bioeng.2009,102(4):1182-1196.;Ho SC1等,J Biotechnol.2013,165(3-4):157-166)。
采用本发明的方法对轻链基因和重链基因的拷贝数进行动态监测,可筛选出轻链和重链基因的拷贝数比较接近、处于比例合适的范围的高表达细胞株,作为商业化的稳定表达细胞株,对于商业化的重组抗体的产品质量稳定性上有非常重要的作用。
- 还没有人留言评论。精彩留言会获得点赞!