一种兰索拉唑的制备工艺的制作方法
本发明属于药物化合物的制备技术领域,具体涉及一种兰索拉唑的制备工艺。
背景技术:
兰索拉唑(lansoprazole),其化学名称为:2-[[[3-甲基-4-(2,2,2-三氟乙氧基)-2-吡啶基]甲基]亚磺酰基]-1h-苯并咪唑,为苯并咪唑类化合物,口服吸收后转移至胃黏膜,在酸性条件下转化为活性代谢体,该活化体特异性地抑制胃壁细胞h+/k+atp酶系统从而阻断胃酸分泌。兰索拉唑以剂量依赖方式抑制基础胃酸分泌以及刺激状态下的胃酸分泌,对胆碱和组胺h2受体无拮抗作用,用作治疗胃溃疡、十二指肠溃疡、反流性食管炎、佐-艾综合征(胃泌素瘤)。
消化系统疾病是常见的多发病之一,其中又以消化性溃疡为主,主要是因吸烟、饮酒、情绪紧张、药物刺激引起,胃肠疾病发病率约占人口的10~12%。因社会的发展、生活节奏加快,胃肠疾病的发病率有逐年升高的趋势,此类疾病的药物市场也将稳步增长。
兰索拉唑是奥美拉唑开发后的第二个新型质子泵抑制剂,由于引入了氟,使其性质有别于奥美拉唑,热力学和氧化稳定性增强,而且大大改善了生物活性,对基础胃酸分泌和由组胺、五肽胃泌素、二丁基环腺酸、胆碱及食物等引起的胃酸形成与分泌有强有力持久的抑制作用,同时对胃黏膜有保护作用。该药口服及注射后吸收迅速,吸收范围和剂量成正比,耐受性和安全性好,因而被广泛应用。
国内兰索拉唑同国外同类产品相比,存在收率低、晶体粒度过小、粒度分布不均匀、纯度较低等方面的差距。另外,就目前的工艺现状而言,在兰索拉唑制备过程中,工艺复杂,生产周期长,结晶过程不易控制。这些问题的存在一方面导致产品质量较差且不稳定,另外还在一定程度上导致生产成本的提高及原材料的浪费。随着市场的扩大,产品规模化生产时存在着原料以及制剂工艺落后、成本偏高、产品性能不稳定等缺点。
因此,有必要对兰索拉唑的制备方法进行深入研究,从而解决其含量、收率以及药品的稳定性等关键问题,使其更适合工业化生产。
技术实现要素:
本发明的目的在于提供一种兰索拉唑的制备工艺。
基于上述目的,本发明采取如下技术方案:一种兰索拉唑的制备工艺,包括以下步骤:(a)将原料a:2-巯基苯并咪唑在碱存在下溶于甲醇中,再加入原料b:2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐于40~50℃反应,反应结束后加水过滤,对沉淀洗涤、干燥得中间体c:[[[3-甲基-4-(2,2,2,-三氟乙氧基)-2-吡啶基]甲基]巯基]-h-苯并咪唑;所述原料a、原料b、碱和甲醇的用量比为1g∶0.5~1.0g∶0.5~1.0g∶6~10ml;
(b)将中间体c溶于乙醇中,再加入双氧水、催化剂及乙醇的混合液,于10~15℃下反应,至hplc检测反应液中中间体c含量小于0.5%,加水过滤,沉淀洗涤后甩干得兰索拉唑;所述催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑);所述双氧水、催化剂、溶解中间体c所需乙醇与原料a的用量比为:0.3~0.6g∶0.02~0.05g∶8~10ml∶1g;双氧水、催化剂及乙醇的混合液中双氧水与乙醇的用量比为1∶0.5~0.8。与钼酸铵、钛等非过渡金属体系催化剂相比,采用这些催化剂时,反应时间较长,也易发生过度氧化而生成砜类物,后处理砜类物比较困难,影响产品收率及含量;本发明采用的催化剂在氧化过程中选择性高,反应液中hplc检测兰索拉唑含量99%以上,并且砜类物含量≤0.5%,得到的兰索拉唑粗品只需简单的后处理便可以得到含量99.6%以上的产品。
还包括步骤(c):将(b)得到的粗品加入乙醇、水和三乙胺的混合液中,加热溶解后,热滤,滤液降温结晶,过滤、洗涤后干燥即可。
步骤(a)中,所述碱选自氢氧化钾、氢氧化钠、甲醇钠、碳酸钾和碳酸钠中的一种或两种以上的混合。
所述碱为氢氧化钠,之所以优选氢氧化钠,其原因在于:当采用甲醇钠和氢氧化钾时,对反应过程会有副作用,产生的副产物使物料颜色偏黄,且与氢氧化钠相比成本较高;采用氢氧化钠作为缩合剂时,副反应小,得到的产品色泽亮且成本低。
步骤(a)中,甲醇的质量浓度为95%~100%;步骤(b)和步骤(c)中,乙醇的质量浓度为95%~100%,双氧水的质量浓度为20%~60%。
双氧水的质量浓度为30%~40%。采用双氧水的质量浓度过低时,反应的时间较长,随着反应的进行,生成的产物易被继续氧化成砜;质量浓度过高时,反应过于剧烈,也易过度氧化,并且高浓度的双氧水在操作时危险性较大,因此,本发明优选质量浓度为30%~40%的双氧水,反应时间适中,能有效避免过度氧化反应,且较安全。
步骤(c)中,兰索拉唑湿粗品的溶解温度为65~75℃,结晶温度为10~15℃,乙醇、三乙胺和水与原料a的重量比例为6.5~8.0ml∶1.5~2.0g∶0.5~1.0g∶1ml。
本发明制备兰索拉唑的反应式如下:
(1)
(2)
利用本发明的方法,可得到晶型粒度好的兰索拉唑产品,产品为白色结晶体,纯度可达到99.6%以上,质量收率达到120%以上。同时提高了收率,降低了成本,缩短了生产周期,更适合工业化生产。
与现有技术相比,本发明的有益效果在于:
1、本发明采用氢氧化钠为缩合剂,甲醇为溶剂,得到的中间体c的质量收率高达133%;而且,原料a在甲醇中溶解度大,用量小,后处理方便,显著降低生产成本,减少环境污染;
2、本发明采用以廉价的过氧化氢替代文献中的氯过氧苯甲酸等氧化剂,不仅降低辅料成本,而且提高了产品收率。
3、本发明采用直接加水析晶的方法,工艺操作简便,设备要求简单易行,缩短反应周期,提高生产效率;
4、本发明在精制过程中采用乙醇、水和三乙胺的混合液加热溶解处理,使工艺操作简单易行,并且物料稳定,兰索拉唑含量达99.6%以上,质量收率高达120%。
附图说明:
图1是本实施例1所得产品的核磁谱图;
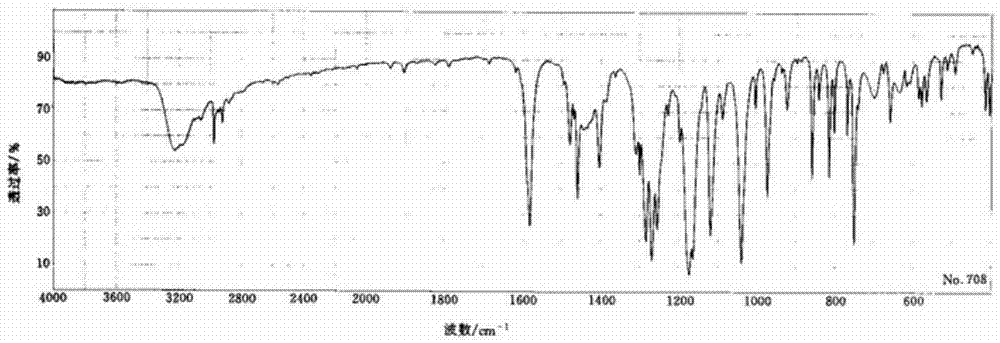
图2是本实施例1所得产品的红外谱图;
图3是兰索拉唑标准品的红外谱图。
具体实施方式
下面实施例只为进一步说明本发明,不以任何形式限制本发明。
实施例1
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入6l无水甲醇,搅拌下加入0.7kg2-巯基苯并咪唑(原料a)和0.5kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,沉淀用纯化水洗涤、甩干,45℃真空干燥,得中间体c;
(b)将中间体c加入10l95%乙醇,控温10~15℃,搅拌溶解后,再加入由0.6kg30%双氧水、0.02kg催化剂和0.3kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,沉淀用纯化水洗涤,甩干,得兰索拉唑粗品。
(c)在洁净的玻璃釜中,先加入6.5l无水乙醇,1.7kg三乙胺,0.5l纯化水,搅拌下加入上述粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.30kg,收率130%,经hplc检测,含量为99.8%,熔点:168.5-170.2℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例2
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入7l无水甲醇,搅拌下加入0.8kg2-巯基苯并咪唑(原料a)和0.6kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,沉淀用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入10l95%乙醇,搅拌下加入上述所得的中间体c,控温10~15℃,搅拌溶解后,再加入由0.6kg30%双氧水、0.05kg催化剂和0.48kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,沉淀用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入6.5l无水乙醇,1.7kg三乙胺,0.6l纯化水,搅拌下加入上述所得粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.27kg:收率127%,经hplc检测,含量为99.9%,熔点:168.3-170.0℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例3
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入8l无水甲醇,搅拌下加入0.8kg2-巯基苯并咪唑(原料a)和0.6kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入10l95%乙醇,搅拌下加入上述所得的中间体c,控温10~15℃,搅拌溶解后,再加入由0.3kg30%双氧水、0.05kg催化剂和0.24kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,沉淀用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入7l无水乙醇,1.7kg三乙胺,0.8l纯化水,搅拌下加入上述所得粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.25kg,收率125%,经hplc检测,含量为100.1%,熔点:167.5-169.2℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例4
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入10l无水甲醇,搅拌下加入0.7kg2-巯基苯并咪唑(原料a)和0.7kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,沉淀用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入10l95%乙醇,搅拌下加入上述所得的中间体c,控温10~15℃,搅拌溶解后,再加入由0.6kg30%双氧水、0.04kg催化剂和0.3kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,沉淀用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入6.5l无水乙醇,1.7kg三乙胺,0.7l纯化水,搅拌下加入上述所得粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.20kg,收率120%,经hplc检测,含量为100.8%,熔点:168.7-170.7℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例5
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入8l95%甲醇,搅拌下加入0.7kg2-巯基苯并咪唑(原料a)和0.5kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,沉淀用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入10l95%乙醇,搅拌下加入上述所得的中间体c,控温10~15℃,搅拌溶解后,再加入由0.4kg30%双氧水、0.05kg催化剂和0.2kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,沉淀用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入8l95%乙醇,1.7kg三乙胺,0.5kg纯化水,搅拌下加入上述所得粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.28kg:收率128%,经hplc检测,含量为99.7%,熔点:168.7-171.2℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例6
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入10l95%甲醇,搅拌下加入0.8kg2-巯基苯并咪唑(原料a)和0.8kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,沉淀用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入10l95%乙醇,搅拌下加入上述所得的中间体c,控温10~15℃,搅拌溶解后,再加入由0.3kg40%双氧水、0.03kg催化剂和0.24kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,沉淀用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入7l无水乙醇,1.7kg三乙胺,0.5kg纯化水,搅拌下加入上述所得粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.30kg,收率131%,经hplc检测,含量为99.6%,熔点:167.5-170.3℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例7
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入7l无水甲醇,搅拌下加入0.7kg2-巯基苯并咪唑(原料a)和0.6kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,沉淀用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入8l无水乙醇,搅拌下加入上述所得的中间体c,控温10~15℃,搅拌溶解后,再加入由0.6kg30%双氧水、0.05kg催化剂和0.3kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,沉淀用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入7l无水乙醇,2.0kg三乙胺,0.5kg纯化水,搅拌下加入上述所得粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.23kg:收率123%,经hplc检测,含量为100.6%,熔点:167.1-169.2℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例8
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入9l无水甲醇,搅拌下加入0.6kg2-巯基苯并咪唑(原料a)和0.5kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入8l无水乙醇,搅拌下加入上述所得的中间体c,控温10~15℃,搅拌溶解后,再加入由0.6kg40%双氧水、0.05kg催化剂和0.3kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入7l无水乙醇,1.7kg三乙胺,0.6kg纯化水,搅拌下加入上述所得粗品,搅拌升温至65~75℃,溶解后,热过滤,滤液转入结晶罐中,降温至10~15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.32kg:收率132%,经hplc检测,含量为99.6%,熔点:167.9-169.4℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例9
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入6l无水甲醇,搅拌下加入0.5kg2-巯基苯并咪唑(原料a)和0.5kg氢氧化钠固体,升温至40℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入8l无水乙醇,搅拌下加入上述所得的中间体c,控温10℃,搅拌溶解后,再加入由0.6kg40%双氧水、0.02kg催化剂和0.3kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入6.5l无水乙醇,1.5kg三乙胺,0.5l纯化水,搅拌下加入上述所得粗品,搅拌升温至65℃,溶解后,热过滤,滤液转入结晶罐中,降温至10℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.32kg:收率132%,经hplc检测,含量为99.6%,熔点:167.9-169.4℃。所得产品晶型好,粒度好,易甩料,易干燥。
实施例10
一种兰索拉唑的制备工艺,包括以下步骤:
(a)在洁净的玻璃釜中,先加入10l无水甲醇,搅拌下加入1.0kg2-巯基苯并咪唑(原料a)和1.0kg氢氧化钠固体,升温至40~50℃,控温,搅拌溶解后,再加入1kg2-氯甲基-3-甲基-4-(2,2,2,-三氟乙氧基)吡啶盐酸盐(原料b)溶解,反应2h,加适量纯化水,降温至室温,甩滤,用纯化水洗涤,甩干,45℃真空干燥,得中间体c。
(b)在洁净的玻璃釜中,先加入10l无水乙醇,搅拌下加入上述所得的中间体c,控温10℃,搅拌溶解后,再加入由0.6kg40%双氧水、0.05kg催化剂和0.48kg乙醇混合的混合液,控温反应2h,经hplc检测,中间体c的残留小于0.5%,加适量纯化水,搅拌10min,甩滤,用纯化水洗涤,甩干,得兰索拉唑粗品;催化剂为:vo2f(dmpz)2(dmpz=3,5-二甲基吡唑)。
(c)在洁净的玻璃釜中,先加入8l无水乙醇,2.0kg三乙胺,1.0l纯化水,搅拌下加入上述所得粗品,搅拌升温至75℃,溶解后,热过滤,滤液转入结晶罐中,降温至15℃,结晶,甩滤,用纯化水洗涤,甩干,45℃真空干燥到恒重。干燥后得兰索拉唑成品1.32kg:收率132%,经hplc检测,含量为99.6%,熔点:167.9-169.4℃。所得产品晶型好,粒度好,易甩料,易干燥。
结构表征:
对实施例1得到的产品进行核磁和红外扫描,同时对兰索拉唑标准样品红外扫描作对照,其结果见图1、图2和图3所示。
对图1分析可知:1hnmrδ:2.23(s,3h),4.30-4.35(m,2h),4.80(s,j=13.6hz,2h),6.64(d,j=5.8hz,1h),7.32-7.36(m,1h),7.45(bs,1h),11.35(d,j=5.6hz,1h);以及将图2与图3的标准样品的红外谱图对照分析,可确认所得产品即为目标产品。
此外,对实施例2-10得到的产品进行同样的核磁和红外分析,结果均证实所得产品即为目标产品。
- 还没有人留言评论。精彩留言会获得点赞!