Copanlisib及其二盐酸盐的合成的制作方法
发明领域
本发明涉及制备2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺(10)、2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐(11)、2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐水合物i和2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐水合物ii的新方法、和新中间体化合物以及所述新中间体化合物用于制备所述2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺(10)、2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐(11)、2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐水合物i和2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐水合物ii的用途:
2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺,
copanlisib,
(10);
2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并-[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐,
(11)。
本发明还涉及作为化合物的copanlisib二盐酸盐水合物。
发明背景
2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺(10)(其在下文称为“copanlisib”)是具有新作用机制(抑制i类磷脂酰肌醇-3-激酶(pi3ks))的专有癌症药物。这类激酶是有吸引力的靶标,因为pi3ks在用于存活和增殖的表面受体的细胞信号转导中起核心作用。copanlisib在体外和体内均显示对多种组织学类型的肿瘤的广谱活性。
copanlisib可根据作为wo04/029055a1于2004年4月8日公开的国际专利申请pct/ep2003/010377(其全文通过引用并入本文)第26页及以下中给出的方法来合成。
copanlisib在作为wo2008/070150a1于2008年6月12日公开的国际专利申请pct/us2007/024985(其全文通过引用并入本文)中作为实施例13的化合物2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺被公开。
copanlisib可根据wo2008/070150第9页及以下和第42页及以下中给出的方法来合成。所述式(i)的化合物的生物测试数据在wo2008/070150第101至107页中给出。
2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐(11)(其在下文称为“copanlisib二盐酸盐”)在作为wo2012/136553于2012年10月11日公开的国际专利申请pct/ep2012/055600(其全文通过引用并入本文)中作为实施例1和2的化合物2-氨基-n-[7-甲氧基-8-(3-吗啉-4-基丙氧基)-2,3-二氢咪唑并[1,2-c]喹唑啉-5-基]嘧啶-5-甲酰胺二盐酸盐被公开,其可根据所述实施例1和2中给出的方法来合成。
copanlisib可以以一种或多种互变异构形式存在:互变异构体(有时称为质子转移互变异构体)是通过氢原子的迁移、伴随一或多个单键及一或多个毗邻双键的迁移而相关联的两种或更多种的化合物。
copanlisib可例如以互变异构形式(ia)、互变异构形式(ib)或互变异构形式(ic)存在,或可如以下所描绘作为任何这些形式的混合物存在。所有此类互变异构形式意欲包括在本发明的范围内。
copanlisib可以作为溶剂化物存在:用于本发明目的的溶剂化物是处于固体状态的溶剂与copanlisib的复合物。示例性的溶剂化物包括但不限于copanlisib与乙醇或甲醇的复合物。
copanlisib和copanlisib二盐酸盐可以作为水合物存在。水合物是溶剂化物的特定形式,其中溶剂是水,其中所述水是copanlisib或copanlisib二盐酸盐的晶格的结构要素。所述水的量可以化学计量或非化学计量比存在。在化学计量水合物的情况下,copanlisib或copanlisib二盐酸盐的一半-、(半-)、单-、倍半-、二-、三-、四-或五-水合物是可能的。水也可能存在于copanlisib或copanlisib二盐酸盐的晶格的表面上。本发明包括copanlisib或copanlisib二盐酸盐的所有此类水合物,特别是如本文的实验部分中制备和表征的被称为“水合物i”或如本文的实验部分中制备和表征的被称为“水合物ii”的copanlisib二盐酸盐水合物。
如上文所提及,copanlisib在wo2008/070150中描述于第9页及以下,且可根据其中第42页及以下给出的方法来合成,即:
反应方案1:
在反应方案1中,乙酸香兰素酯可经由硝化条件、例如纯发烟硝酸或在另一强酸例如硫酸存在下的硝酸、转化为中间体(iii)。预期中间体(iii)中的乙酸酯会在质子溶剂例如甲醇中、在碱例如氢氧化钠、氢氧化锂或氢氧化钾存在下水解。保护中间体(iv)以产生式(v)的化合物可通过标准方法(greene,t.w.;wuts,p.g.m.;protectivegroupsinorganicsynthesis;wiley&sons:newyork,1999)完成。式(v)的化合物向式(vi)的化合物的转化可使用氨、在非质子溶剂例如thf或二噁烷中在碘存在下实现。式(vi)中硝基的还原可使用乙酸中的铁或氢气、在适宜的钯、铂或镍催化剂存在下完成。式(vii)的化合物向式(viii)的咪唑啉的转化最佳地使用乙二胺、在催化剂例如元素硫存在下在加热下来完成。式(viii)的化合物环化为式(ix)的化合物使用溴化氰、在卤化溶剂例如dcm或二氯乙烷中、在胺碱例如三乙胺、二异丙基乙胺或吡啶存在下完成。式(ix)中保护基的移除将取决于所选基团且可通过标准方法(greene,t.w.;wuts,p.g.m.;protectivegroupsinorganicsynthesis;wiley&sons:newyork,1999)完成。式(x)中酚的烷基化可使用碱例如碳酸铯、氢化钠或叔丁醇钾、在极性非质子溶剂例如dmf或dmso中、伴随具有适当离去基团例如卤化物或磺酸酯基团的侧链的引入来实现。最后,式(i)的酰胺可使用活化酯例如酰氯和酸酐来形成,或替代性地使用羧酸和适当的偶联剂例如pybop、dcc或edci于极性非质子溶剂中形成。
反应方案2:
在反应方案2中,如上文所述制备的式(iv)的化合物可使用氨、在非质子溶剂例如thf或二噁烷中在碘存在下转化为式(xii)的结构。式(xii)中的酚的烷基化可使用碱例如碳酸铯、氢化钠或叔丁醇钾于极性非质子溶剂例如dmf或dmso中、伴随具有适当离去基团例如卤化物或磺酸酯基团的侧链的引入来实现。式(xiii)中硝基的还原可使用乙酸中的铁或氢气在适宜钯、铂或镍催化剂存在下完成。式(xiv)的化合物向式(xv)的咪唑啉的转化最佳地使用乙二胺在催化剂例如元素硫存在下、在加热下来完成。将式(xv)的化合物环化为式(xvi)的化合物使用溴化氰、在卤化溶剂例如dcm或二氯乙烷中、在胺碱例如三乙胺、二异丙基乙胺或吡啶存在下完成。最后,式(i)的酰胺可使用活化酯例如酰氯和酸酐形成,或替代性地使用羧酸及适当的偶联剂例如pybop、dcc或edci于极性非质子溶剂中形成。
上文的两个已知合成路径-反应方案1及2具有许多缺点,这些缺点尤其会在较大规模时造成问题:
•由于安全担忧,易受到氧化的影响的分子的分批硝化对于按比例扩大会成为问题。为此,我们研发了经由微量反应技术的连续方法,如实施例1(参见下文)中所例示。
•使用氨及碘作为试剂将醛基转化为腈是危险的,因为氨及碘可形成高敏感性爆炸物质三碘化氮。
•使用乙二胺环化为咪唑啉环需要硫。由于硫在具有固定反应器及管道的技术系统中的清洁过程中极难清除,因此此环化反应并不适于按比例扩大。
•使用铁及酸难以在较大规模下将硝基还原为相应的胺。标准催化还原经常发生副反应、例如咪唑啉开环,这显著降低产率。
因此期望设计新的合成,其可避免这些缺点且适于生产规模/工业规模。
已极为令人惊讶地发现且提供本发明的基础的是,以下结构类型的化合物,特别是copanlisib,可根据以下方案合成,参见以下反应方案3:
反应方案3:
下文给出如上文反应方案3中所描绘的本发明合成的特定步骤的下列优点:
•步骤a1:硝化反应可以在流动反应器系统中实施。由此可容易地控制放热反应且不会产生失控反应的危险。数千克量的2-硝基香兰素可在数天或几周内容易地制备。分离的材料含有与通过分批硝化所产生的材料类似的量(大约10%)的非期望的位置异构体6-硝基香兰素。
•步骤a3:烷基化由碱如碳酸钾介导,在向反应混合物中添加水后,通过过滤容易地以高产率分离产物。反应混合物的浓度和具有相分离的水后处理不是必需的。
•步骤a4:使用乙二胺及n-溴琥珀酰亚胺(“nbs”)的环化和氧化的一锅式反应。所述新的工序解决两个问题,因为其避免:
a)使用氨/碘来将醛转化为腈(安全担忧),和
b)在咪唑啉合成期间使用硫(按比例扩大问题)。在甲醇和乙腈中进行该工序导致了较少的副产物,使得该工序更容易进行(给予nbs溶液)并使其在规模上更安全。一个额外的意料不到的优点是在这些处理条件下移除了错误的硝基位置异构体。
•步骤a5:使用氢和特定制备的催化剂还原。该催化剂由炭载的铂及铁组成。出乎意料地,用该催化剂没有观察到脱苄基化。产物以优异的产率从异丙醇和水中结晶和分离。已经在3巴下在thf中快速氢化
•步骤a6:二氯甲烷可以被乙腈交换。在甲苯中产物的搅拌导致优异质量的产物。
•步骤a7:通过用炭载钯简单氢化来移除苄基保护基。通过过滤容易地分离产物。
•步骤a8:在正丁醇或正丁醇与其它溶剂例如dmf和水的混合物中的烷基化,允许容易的后处理、和经由产物从正丁醇-叔丁基甲基醚(“mtbe”)中的结晶的分离。从水中重结晶移除了无机杂质且生成优异质量的产物。
•步骤a9:将n-[3-(二甲基氨基)丙基]-n'-乙基碳二亚胺盐酸盐(“edci”)用作偶联剂。通过简单过滤分离copanlisib。
•步骤a11:copanlisib通过其二盐酸盐容易纯化(二盐酸盐是最终产物)。
因此,在第一方面中,本发明涉及经由示于以下反应方案3中的以下步骤制备copanlisib(10)的方法:
反应方案3:
在第一方面的一个实施方案中,本发明涉及制备copanlisib(10)的方法:
其包括以下步骤:
步骤a9:
其中使式(9)的化合物:
与式(9b)的化合物:
任选在催化剂例如n,n-二甲基-4-氨基吡啶存在下、任选在偶联剂例如n-[3-(二甲基氨基)丙基]-n'-乙基碳二亚胺盐酸盐存在下、任选在溶剂例如n,n-二甲基甲酰胺中反应,
由此提供copanlisib(10):
所述式(9)的化合物:
通过以下步骤a8制备:
其中使式(8)的化合物:
与式(8a)的化合物:
任选在碱例如碳酸钾存在下,在溶剂例如正丁醇、n,n-二甲基甲酰胺和水中,任选在加热下例如在回流下反应,
由此提供式(9)的化合物;
所述式(8)的化合物:
通过以下步骤a7制备:
其中使式(7)的化合物:
与还原剂例如氢、任选在催化剂例如金属催化剂存在下、任选溶解于溶剂例如n,n-二甲基甲酰胺中或溶剂例如n,n-二甲基甲酰胺中的悬浮液中进行反应,由此提供式(8)的化合物,所述金属催化剂例如是炭载钯,特别是水润湿的、5%炭载钯;
所述式(7)的化合物:
通过以下步骤a6制备:
其中使式(6)的化合物:
任选在碱例如三乙胺存在下与增环剂(annelatingagent)例如溴化氰(也称为氰化溴)、任选在溶剂例如乙腈或二氯甲烷中反应,
由此提供式(7)的化合物;
所述式(6)的化合物:
通过以下步骤a5制备:
其中使式(5)的化合物:
与还原剂例如氢、任选在催化剂例如双金属催化剂存在下、任选溶解于溶剂例如四氢呋喃中或溶剂例如四氢呋喃中的悬浮液中进行反应,由此提供式(6)的化合物,所述双金属催化剂例如是炭载铂铁,特别是1%pt/0.2%fe/c、任选为水润湿的;
所述式(10)的copanlisib:
任选通过使其与氯化氢、任选盐酸反应而生成copanlisib二盐酸盐(11),
由此提供copanlisib二盐酸盐(11):
在第一方面的一个实施方案中,本发明涉及制备copanlisib二盐酸盐(11)的方法:
其包括以下步骤a11:
其中使式(10)的copanlisib:
与氯化氢、任选盐酸反应,
由此提供copanlisib二盐酸盐(11):
在第一方面的一个实施方案中,本发明涉及制备copanlisib二盐酸盐水合物i的方法:
其包括以下步骤a11:
其中使式(10)的copanlisib:
与氯化氢、任选盐酸反应,
由此提供copanlisib二盐酸盐水合物i。
在第一方面的一个实施方案中,本发明涉及制备copanlisib二盐酸盐水合物ii的方法,
其包括以下步骤a11:
其中使式(10)的copanlisib:
与氯化氢、任选盐酸反应,
由此提供copanlisib二盐酸盐水合物ii。
在第一方面的一个实施方案中,本发明涉及制备copanlisib(10)的方法:
其包括以下步骤a9:
其中使式(9)的化合物:
与式(9b)的化合物:
任选在催化剂例如n,n-二甲基-4-氨基吡啶存在下、任选在偶联剂例如n-[3-(二甲基氨基)丙基]-n'-乙基碳二亚胺盐酸盐存在下、任选在溶剂例如n,n-二甲基甲酰胺中反应,
由此提供copanlisib(10):
在第一方面的一个实施方案中,本发明涉及制备上述式(9b)的化合物的方法:
其包括以下步骤a10:
其中使式(9a)的化合物:
a)与碱例如甲醇钠任选在溶剂例如1,4-二噁烷中在加热下例如在回流下反应,然后,
b)冷却例如至室温后,添加甲酸甲酯,然后
c)添加盐酸胍,随后加热例如在回流下,然后,
d)添加水和碱例如氢氧化钠的水溶液,随后加热,然后,
e)添加无机酸例如盐酸的水溶液,
f)添加胺例如二环己胺并过滤,然后
g)添加强碱例如氢氧化钠的水溶液,然后
h)添加无机酸例如盐酸的水溶液,
由此提供式(9b)的化合物:
在第一方面的一个实施方案中,本发明涉及制备上述式(9)的化合物的方法:
其包括以下步骤a8:
其中使式(8)的化合物:
与式(8a)的化合物:
任选在碱例如碳酸钾存在下、在溶剂例如正丁醇中、任选在加热下例如在回流下反应,
由此提供式(9)的化合物。
在第一方面的一个实施方案中,本发明涉及制备上述式(8)的化合物的方法:
其包括以下步骤a7:
其中使式(7)的化合物:
与还原剂例如氢、任选在催化剂例如金属催化剂存在下、任选溶解于溶剂例如n,n-二甲基甲酰胺中或溶剂例如n,n-二甲基甲酰胺中的悬浮液中、任选在酸例如三氟乙酸存在下进行反应,由此提供式(8)的化合物,所述金属催化剂例如是炭载钯,特别是水润湿的、5%炭载钯。
在第一方面的一个实施方案中,本发明涉及制备上述式(7)的化合物的方法:
其包括以下步骤a6:
其中使式(6)的化合物:
任选在碱例如三乙胺存在下与增环剂例如溴化氰(也称为氰化溴)、任选在溶剂例如乙腈或二氯甲烷中反应,
由此提供式(7)的化合物。
在第一方面的一个实施方案中,本发明涉及制备上述式(6)的化合物的方法:
其包括以下步骤a5:
其中使式(5)的化合物:
与还原剂例如氢、任选在催化剂例如双金属催化剂存在下、任选溶解于溶剂例如四氢呋喃中或溶剂例如四氢呋喃中的悬浮液中进行反应,由此提供式(6)的化合物,所述双金属催化剂例如是炭载铂铁,特别是水润湿的1%pt/0.2%fe/c。
在第一方面的一个具体实施方案中,本发明涉及制备上述式(6)的化合物的方法:
其包括以下步骤a5:
其中使式(5)的化合物:
与氢在双金属催化剂存在下、在四氢呋喃中的悬浮液中进行反应,由此提供式(6)的化合物,所述双金属催化剂为水润湿的1%pt/0.2%fe/c。
在第一方面的一个实施方案中,本发明涉及制备上述式(5)的化合物的方法:
其包括以下步骤a4:
其中使式(4)的化合物:
与乙二胺、任选在n-溴代琥珀酰亚胺存在下、任选在溶剂混合物例如甲醇和乙腈中反应,
由此提供式(5)的化合物。
在第一方面的一个特定实施方案中,本发明涉及制备上述式(4)的化合物的方法:
其包括以下步骤a3,
其中使式(3)的化合物:
任选在溶剂例如n,n-二甲基甲酰胺中、任选在碱例如碳酸钾存在下,
与苄基溴、任选在加热下例如在回流下反应,
由此提供式(4)的化合物。
在第一方面的一个特定实施方案中,本发明涉及制备上述式(3)的化合物的方法:
其包括以下步骤a2,
其中使式(2)的化合物:
与碱例如碳酸钾、在溶剂例如甲醇中反应,
由此提供式(3)的化合物。
在第一方面的一个特定实施方案中,本发明涉及制备上述式(2)的化合物的方法:
其包括以下步骤a1,
其中使式(1)的化合物:
在溶剂例如二氯甲烷中的溶液中与硝酸和硫酸反应,
由此提供式(2)的化合物。
在第一方面的另一个实施方案中,本发明涉及制备copanlisib(10)或copanlisib二盐酸盐(11)或copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的方法,其中如上述方案3中所示的所述步骤a1、a2、a3、a4、a5、a6、a7、a8、a9、a10和a11中的各步骤如上所述进行。
在第一方面的另一个实施方案中,本发明涉及制备copanlisib二盐酸盐(11)的方法,所述copanlisib二盐酸盐(11)呈如实验部分中所制备和表征的copanlisib二盐酸盐水合物i的形式。
在第一方面的另一个实施方案中,本发明涉及如实验部分中所制备和表征的copanlisib二盐酸盐水合物i。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物i。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物i,其具有5.6的xrpd峰最大值[°2θ](铜(cu))。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物i,其具有7.0的xrpd峰最大值[°2θ](铜(cu))。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物i,其具有15.4的xrpd峰最大值[°2θ](铜(cu))。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物i,其具有26.4的xrpd峰最大值[°2θ](铜(cu))。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物i,其具有5.6、7.0、15.4和26.4的xrpd峰最大值[°2θ](铜(cu))。
在第一方面的另一个实施方案中,本发明涉及制备copanlisib二盐酸盐(11)的方法,所述copanlisib二盐酸盐(11)呈如实验部分中所制备和表征的copanlisib二盐酸盐水合物ii的形式。
在第一方面的另一个实施方案中,本发明涉及如实验部分中所制备和表征的copanlisib二盐酸盐水合物ii。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物ii。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物ii,其具有5.7的xrpd峰最大值[°2θ](铜(cu))。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物ii,其具有7.3的xrpd峰最大值[°2θ](铜(cu))。
在第一方面的另一个实施方案中,本发明涉及copanlisib二盐酸盐水合物ii,其具有5.7和7.3的xrpd峰最大值[°2θ](铜(cu))。
根据第二方面,本发明涉及用于制备copanlisib(10)和copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i和copanlisib二盐酸盐水合物ii的中间体化合物。
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
在所述第二方面的一个实施方案中,本发明涉及以下化合物:
根据第三方面,本发明涉及所述第二方面的中间体化合物用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)或copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)或copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在所述第三方面的一个实施方案中,本发明涉及
用于制备copanlisib(10)、copanlisib二盐酸盐(11)、copanlisib二盐酸盐水合物i或copanlisib二盐酸盐水合物ii的用途。
在本发明的上下文中,如任选存在于本发明方法的任何反应步骤中的术语“溶剂”,如本领域技术人员所理解的那样、被理解为意指其他材料溶解于其中以形成溶液的任何物质,例如但不限于:极性溶剂,例如极性质子溶剂例如水、正丁醇、异丙醇、正丙醇、乙醇、甲醇或甲酸或乙酸等;极性非质子溶剂,例如1,4-二噁烷、四氢呋喃、1,2-二甲氧基乙烷、丙酮、乙腈、二甲基甲酰胺、环丁砜、吡啶或二甲亚砜等;或非极性溶剂,例如戊烷、己烷、苯、甲苯、乙醚、甲基乙基酮、二氯甲烷、氯仿、四氯甲烷、乙酸乙酯等;或上文所列溶剂的任何混合物。
应理解上文所提及的实施方案中给出的定义的任何组合可能在本发明的上下文内。
当阅读以下作为本发明的说明所提供的实施例时,会更好地理解本发明。以下实施例绝不构成如在本文中所述及如其所附权利要求中所限定的本发明的限制。
实验部分
所用的缩写:
以下用于实施例中的缩写具有以下含义:
1h-nmr质子核磁共振波谱法
(化学位移(δ)以ppm给出)
ac乙酰基
boc叔丁氧基羰基
bm宽多重峰
br宽峰
bs宽单峰
c-环-
d双重峰
dd双重双重峰
dcm二氯甲烷
dme1,2-二甲氧基乙烷
dipe二异丙基醚
dipean,n-二异丙基乙胺
dmfn,n-二甲基甲酰胺
dmso二甲亚砜
edcin-[3-(二甲基氨基)丙基]-n'-乙基碳二亚胺盐酸盐
eq当量
esi电喷雾电离
hatun-[(二甲基氨基)(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基氧基)¬亚甲基]-n-甲基甲铵六氟磷酸盐
hünig碱n,n-二异丙基乙胺
m多重峰
m.p.以℃计的熔点
ms质谱法
mtbe叔丁基甲基醚
mw分子量
naotbu叔丁醇钠;2-甲基丙-2-醇钠
nmpn-甲基吡咯烷酮
nmr核磁共振波谱法:化学位移(δ)以ppm给出
q四重峰
quin五重峰
rac外消旋
rt室温
r.t.室温
rt以分钟计的保留时间
s单峰
t三重峰
tbaf四丁基氟化铵
tbtun-[(1h-苯并三唑-1-基氧基)(二甲基氨基)亚甲基]-n-甲基甲铵四氟硼酸盐
tea三乙胺
tfa三氟乙酸
thf四氢呋喃
tms三甲基甲硅烷基
ts对甲苯磺酰基;(甲苯磺酰基)
uplc超高效液相色谱。
实施例
实施例1:步骤a1:4-乙酰氧基-3-甲氧基-2-硝基苯甲醛(2)的制备
在0℃下将3.94kg硝酸(65w%)添加至5.87kg浓硫酸中(硝化酸)。将1.5kg乙酸香兰素酯溶解于2.9kg二氯甲烷中(乙酸香兰素酯溶液)。两种溶液在微型反应器中以约8.0ml/min(硝化酸)和约4.0ml/min(乙酸香兰素酯溶液)的流速在5℃下反应。在3℃下将反应混合物直接加入到8kg水中。3小时后将流速增加至10ml/min(硝化酸)和5.0ml/min(乙酸香兰素酯溶液)。另外的9小时后,流动反应完成。在室温下分离各层,并使用2l二氯甲烷萃取水相。将合并的有机相使用2l饱和碳酸氢钠洗涤,然后用0.8l水洗涤。将二氯甲烷溶液在真空中浓缩至约3l,添加3.9l甲醇并通过蒸馏再次移除大致相同的体积。添加另外的3.9l甲醇,并将溶液浓缩至约3.5l的体积。将该4-乙酰氧基-3-甲氧基-2-硝基苯甲醛(2)的溶液直接用于下一步骤。
实施例2:步骤a2:4-羟基-3-甲氧基-2-硝基苯甲醛(2-硝基-香兰素)(3)的制备
向如实施例1(参见上文)中所述那样制备的4-乙酰氧基-3-甲氧基-2-硝基苯甲醛(2)的溶液中添加1.25kg甲醇,随后添加2.26kg碳酸钾。将该混合物在30℃下搅拌3小时。在<30℃下添加7.3kg二氯甲烷和12.8kg盐酸水溶液(10w%)(ph0.5-1)。将该混合物搅拌15分钟,并分离各层。过滤有机层,并用0.5l二氯甲烷洗涤滤饼。用4.1kg二氯甲烷将水层萃取两次。将合并的有机层在真空中浓缩至约4l。添加3.41kg甲苯,并将混合物浓缩至约4l的最终体积。将该混合物冷却至0℃。90分钟后过滤该悬浮液。用冷甲苯洗涤收集的固体并干燥以获得0.95kg(62%)。
nmr光谱还含有位置异构体6-硝基香兰素的信号(约10%):
实施例3:步骤a3:4-(苄基氧基)-3-甲氧基-2-硝基苯甲醛(4)的制备:
将10g的3在25℃下溶解于45mldmf中。向该溶液中装入14g碳酸钾,并且温度确实升至约30℃。在15分钟内,在30℃的温度下,向该悬浮液中加入7.1ml苄基溴。将反应混合物搅拌2小时以完成反应。冷却至25℃后,添加125ml水。将悬浮液过滤,用50ml水洗涤两次,并用水/甲醇(10ml/10ml)洗涤一次,并在40℃下在减压下试验。以这种方式,获得14.2g(97%产率)作为淡黄色固体的4。
实施例4a:步骤a4:2-[4-(苄基氧基)-3-甲氧基-2-硝基苯基]-4,5-二氢-1h-咪唑(5):方法a
将10g的4溶解于100ml甲醇中,并在20-25℃下添加2.5g乙二胺。将该反应混合物在该温度下搅拌1小时,冷却至0℃,并添加n-溴代琥珀酰亚胺(8.1g)于60ml乙腈中的溶液。继续搅拌1.5小时,并将该反应混合物温热至20℃,并再搅拌60分钟。用8.6gnahco3和2.2gna2so3于100ml水中的溶液淬灭反应。10分钟后,添加230ml水,将产物过滤,用40ml水洗涤,并在40℃下在减压下试验。以这种方式,获得8.9g(78%产率)作为白色固体的5。
实施例4b:步骤a4:2-[4-(苄基氧基)-3-甲氧基-2-硝基苯基]-4,5-二氢-1h-咪唑(5):方法b
将28.7kg化合物4在20℃下溶解于231kg二氯甲烷中,并添加8.2kg乙二胺。搅拌60分钟后,以4份(4x5.8kg)添加n-溴代琥珀酰亚胺,控制温度不超过25℃。当添加完成时,在22℃下继续搅拌90分钟。向反应混合物中添加9kg碳酸钾/39kg水,并分离各层。从有机层中经由蒸馏移除150kg溶剂,并添加67kg甲苯。在减压下移除另外的50kg溶剂,并添加40kg甲苯。在35-45℃下搅拌30分钟后,将反应冷却至20℃,并经由过滤分离产物。将产物用甲苯(19kg)洗涤,在减压下试验,并获得26.6kg(81%产率)棕色产物。
实施例5:步骤a5:3-(苄基氧基)-6-(4,5-二氢-1h-咪唑-2-基)-2-甲氧基苯胺(6):
将8.6g化合物5悬浮于55mlthf中,并添加1.4g1%pt/0.2%fe/c/4ml水。将该混合物加热至45℃,并在3巴氢压下氢化30分钟。将催化剂滤出并用thf洗涤两次。经由蒸馏移除thf,并向反应混合物中添加65ml异丙醇/水1/1。经由蒸馏移除残留了thf的溶剂,并添加86ml异丙醇/水1/1。将该悬浮液搅拌1小时,过滤,用异丙醇/水1/1洗涤两次,并在减压下干燥,以获得7.8g(99%产率)白色固体。
实施例6a:步骤a6:8-(苄基氧基)-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺(7):方法a
将10g的6悬浮于65ml乙腈中,并添加6.1ml三乙胺。在5-10℃下,经1小时添加8.4ml在乙腈中的50%氰化溴,并继续搅拌1小时。添加86ml2%naoh,并将该反应混合物加热至45℃并搅拌1小时。将该悬浮液冷却至10℃,过滤并用水/丙酮80/20洗涤。为了进一步改进材料的质量,在20-25℃下将湿产物于50ml甲苯中搅拌。将产物滤出,用甲苯洗涤并在减压下干燥。以这种方式,分离8.8g(81%产率)作为白色固体的7。
实施例6b:步骤a6:8-(苄基氧基)-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺(8):方法b
将20kg化合物6在20℃下溶解于218kg二氯甲烷中,并将该混合物冷却至5℃。在该温度下,向该反应混合物中在15分钟内添加23.2kg三乙胺,随后在60分钟内添加25.2kg氰化溴(在二氯甲烷中3m)。在22℃下搅拌1小时后,将反应浓缩,并在减压下移除188kg溶剂。添加丙酮(40kg)和水(50kg),并经由蒸馏再移除100kg溶剂。添加丙酮(40kg)和水(150kg),并在36℃下继续搅拌30分钟。冷却至2℃后,将该悬浮液搅拌30分钟,分离,用80kg冷水洗涤并在减压下试验。用该工序,获得20.7kg(95%产率)灰白色产物。
实施例7a:步骤a7:方法a:5-氨基-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-8-醇(8)的制备:
将2kg8-(苄基氧基)-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺、203g5%炭载钯(50%水润湿的)和31.8kgn,n-二甲基甲酰胺的混合物在60℃下在3巴氢气下搅拌18小时。将混合物过滤,并将残余物用7.5kgn,n-二甲基甲酰胺洗涤。将滤液(38.2kg)在真空中浓缩(收集并丢弃约27l馏出物)。将剩余的混合物在1小时内从50℃冷却至22℃,在该冷却阶段期间,在30分钟内添加14.4kg水。将所得悬浮液在22℃下搅拌1小时,然后过滤。将收集的固体用水洗涤并在真空中干燥,以获得0.94kg(65%)。
实施例7b:步骤a7方法b:5-氨基-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-8-醇(8)的制备:
将222.8g三氟乙酸添加至600g8-(苄基氧基)-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺和2850gdmf的混合物中。添加18g5%炭载钯(50%水润湿的)。将该混合物在3巴氢气下搅拌过夜。通过过滤移除催化剂并用570gdmf洗涤。将滤液在真空中浓缩(收集并丢弃432g馏出物)。在2小时内添加4095ml的0.5m氢氧化钠水溶液。将所得悬浮液搅拌过夜。使用离心机分离产物。将收集的固体用水洗涤。分离的材料(480.2g;含有约25w%水)可以直接用于下一步骤(实施例8b)。
实施例8a:步骤a8:方法a:7-甲氧基-8-[3-(吗啉-4-基)丙氧基]-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺(9)的制备:
将2.5kg碳酸钾添加至1.4kg5-氨基-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-8-醇、14l正丁醇、1.4ln,n-二甲基甲酰胺和1.4l水的混合物中。添加1.57kg4-(3-氯丙基)吗啉盐酸盐。将所得悬浮液加热至90℃,并在该温度下搅拌5小时。将该混合物冷却至室温。在50℃下添加8.4kg水。在室温下将该混合物搅拌15分钟。相分离后,用12l正丁醇萃取水相。将合并的有机相在真空中浓缩至约11l的体积。在50℃下添加10.7l叔丁基甲基醚。将所得混合物在2小时内冷却至0℃,并在该温度下搅拌1小时。将该悬浮液过滤,并将收集的固体用叔丁基甲基醚洗涤并干燥,以得到1.85kg(86%)。
将分离的1.85kg与额外的0.85kg根据相同方法产生的材料合并。添加10.8l水,并将该混合物加热至60℃。将该混合物在该温度下搅拌10分钟,然后在30分钟内冷却至45℃,然后在1小时内冷却至0℃。将该悬浮液在0℃下搅拌2小时,然后过滤。将固体用冷水洗涤并干燥以获得2.5kg。
hplc:固定相:kinetexc18(150mm,3.0mmid,2.6µm粒径):流动相a:0.5ml三氟乙酸/1l水;流动相b:0.5ml三氟乙酸/l乙腈;在256nm的uv检测;箱温度:40℃;注射体积:2.0µl;流速1.0ml/min;4个步骤中的线性梯度:0%b->6%b(20min),6%b->16%b(5min),16%b->28%b(5min),28%b->80%b(4min),在80%b的保留时间4分钟;纯度:>99.5%(rt=11.0min),相关的潜在副产物:降解产物1,rrt(相对保留时间)为0.60(6.6min)通常<0.05%,5-氨基-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-8-醇rrt0.71(7.8min):通常<0.05%,降解产物2rrt1.31(14.4min):通常<0.05%,7-甲氧基-5-{[3-(吗啉-4-基)丙基]氨基}-2,3-二氢咪唑并[1,2-c]喹唑啉-8-醇rrt1.39(15.3min):通常<0.05%,9-甲氧基-8-[3-(吗啉-4-基)丙氧基]-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺rrt1.43(15.7min):通常<0.05%,降解产物3rrt1.49(16.4min):通常<0.05%,7-甲氧基-8-[3-(吗啉-4-基)丙氧基]-n-[3-(吗啉-4-基)丙基]-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺rrt1.51(16.7min):通常<0.10%,8-(苄基氧基)-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺rrt2.56(28.2min):通常<0.05%,8-(苄基氧基)-7-甲氧基-n-[3-(吗啉-4-基)丙基]-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺rrt2.59(28.5min):通常<0.05%。
实施例8b::步骤a8(方法b):7-甲氧基-8-[3-(吗啉-4-基)丙氧基]-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺(9)的制备:
将13.53g5-氨基-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-8-醇(含有约26w%水)悬浮于110g正丁醇中。将该混合物在真空中浓缩(收集并丢弃13.5g馏出物)。添加17.9g碳酸钾和11.2g4-(3-氯丙基)吗啉盐酸盐。将所得混合物加热至90℃,并在该温度下搅拌4小时。将该反应混合物冷却至50℃,并添加70g水。分离各层。将有机层在真空中浓缩(收集并丢弃54g馏出物)。在65℃下添加90g叔丁基甲基醚。将所得混合物冷却至0℃。将该混合物过滤,并将收集的固体用叔丁基甲基醚洗涤,然后在真空中干燥,以得到13.4g(86%)。
将13.1g分离的材料悬浮于65.7g水中。将该混合物加热至60℃。将所得溶液缓慢冷却至0℃。将沉淀的固体通过过滤分离,用水洗涤并在真空中干燥,以得到12.0g(92%)。
实施例9:步骤a10:2-氨基嘧啶-5-甲酸(9b)的制备
将1kg3,3-二甲氧基丙酸甲酯溶解于7l的1,4-二噁烷中。添加1.58kg甲醇钠溶液(30w%,于甲醇中)。将该混合物加热至回流,并移除约4.9kg馏出液。将所得悬浮液冷却至室温,并添加0.5kg甲酸甲酯。将该反应混合物搅拌过夜,然后添加0.71kg盐酸胍,并在室温下将该反应混合物搅拌2小时。然后将该反应混合物加热至回流,并搅拌2小时。添加13.5l水,随后添加0.72kg氢氧化钠水溶液(45w%)。将该反应混合物在回流下再加热0.5小时,然后冷却至50℃。添加0.92kg盐酸水溶液(25w%),直至达到ph6。添加晶种,并在50℃下再添加0.84kg盐酸水溶液(25w%),直至达到ph2。将该混合物冷却至20℃并搅拌过夜。将该悬浮液过滤,用水将收集的固体洗涤两次,然后用甲醇洗涤两次,获得0.61kg(65%)。
将根据上文工序产生的四批合并(总共2.42kg)。添加12l乙醇,并将所得悬浮液在室温下搅拌2.5小时。将该混合物过滤。用乙醇洗涤收集的固体并在真空中干燥以获得2.38kg。
向800g该物质中添加2.5l二氯甲烷和4l水,随后添加1375ml二环己胺。将该混合物在室温下搅拌30分钟并过滤。丢弃收集的固体。分离滤液的各相,并丢弃有机相。将345ml氢氧化钠水溶液(45w%)添加至水相。用2.5l乙酸乙酯萃取水相。分离各相并丢弃有机相。使用约500ml盐酸(37w%)将水相的ph值调节至ph2。将该混合物过滤,并用水洗涤收集的固体并干燥,获得405g。
将该405g与相当品质的第二批(152g)合并。添加2l乙酸乙酯和6l水,随后添加480ml氢氧化钠水溶液(45w%)。在室温下将该混合物搅拌30分钟。分离各相。用约770ml盐酸水溶液(37w%)将水相的ph调节至ph2。将该混合物过滤,并用水洗涤收集的固体并干燥,以获得535g。
实施例10:步骤a9:copanlisib(10)的制备
将1250g7-甲氧基-8-[3-(吗啉-4-基)丙氧基]-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺、20.3kgn,n-二甲基甲酰胺、531g2-氨基嘧啶-5-甲酸、425gn,n-二甲基氨基吡啶和1000gn-[3-(二甲基氨基)丙基]-n'-乙基碳二亚胺盐酸盐的混合物在室温下搅拌17小时。将该反应混合物过滤。将收集的固体用n,n-二甲基甲酰胺洗涤,然后用乙醇洗涤,并在50℃下干燥,以得到1.6kg(96%)。将分离的材料直接转化为二盐酸盐。
实施例11:步骤a11:copanlisib二盐酸盐(11)的制备
向1.6kgcopanlisib和4.8kg水的混合物中添加684g盐酸水溶液(32w%),同时保持温度在20至25℃之间,直至达到3至4的ph。将所得混合物搅拌10分钟,并检查ph(ph3.5)。将该混合物过滤,并将滤饼用0.36kg水洗涤。将109g盐酸水溶液添加至滤液,直至ph为1.8至2.0。将该混合物搅拌30分钟,并检查ph(ph1.9)。在20至25℃下,在5小时内缓慢添加7.6kg乙醇,20分钟后暂停添加1小时,此时开始结晶。乙醇完全添加后,将所得悬浮液搅拌1小时。过滤该悬浮液。将收集的固体用乙醇-水混合物洗涤,最后用乙醇洗涤,然后在真空中干燥,以得到1.57kgcopansilib二盐酸盐(85%)。
hplc:固定相:kinetexc18(150mm,3.0mmid,2.6µm粒径):流动相a:2.0ml三氟乙酸/1l水;流动相b:2.0ml三氟乙酸/l乙腈;在254nm(在1分钟后切换至282nm)的uv检测;箱温度:60℃;注射体积:2.0µl;流速1.7ml/min;2个步骤中在1分钟等度运行后的线性梯度:0%b->18%b(9min),18%b->80%b(2.5min),在80%b的保持时间2.5分钟;纯度:>99.8%(rt=6.1min),相关的潜在副产物:2-氨基嘧啶-5-甲酸,rrt(相对保留时间)为0.10(0.6min),通常<0.01%,4-二甲基氨基嘧啶rrt0.26(1.6min):通常<0.01%,7-甲氧基-8-[3-(吗啉-4-基)丙氧基]-2,3-二氢咪唑并[1,2-c]喹唑啉-5-胺rrt0.40(2.4min):通常<0.03%,副产物1rrt0.93(5.7min):通常<0.05%,副产物6rrt1.04(6.4min):通常<0.05%,2-氨基-n-{3-(2-氨基乙基)-8-甲氧基-7-[3-(吗啉-4-基)丙氧基]-4-氧代-3,4-二氢喹唑啉-2-基}嘧啶-5-甲酰胺rrt1.12(6.9min):通常<0.10%,2-氨基嘧啶-5-甲酸5-{[(2-氨基嘧啶-5-基)羰基]氨基}-7-甲氧基-2,3-二氢咪唑并[1,2-c]喹唑啉-8-基酯rrt1.41(8.6min):通常<0.01%。
实施例12:步骤a11:copanlisib二盐酸盐(11)的制备的进一步的实例
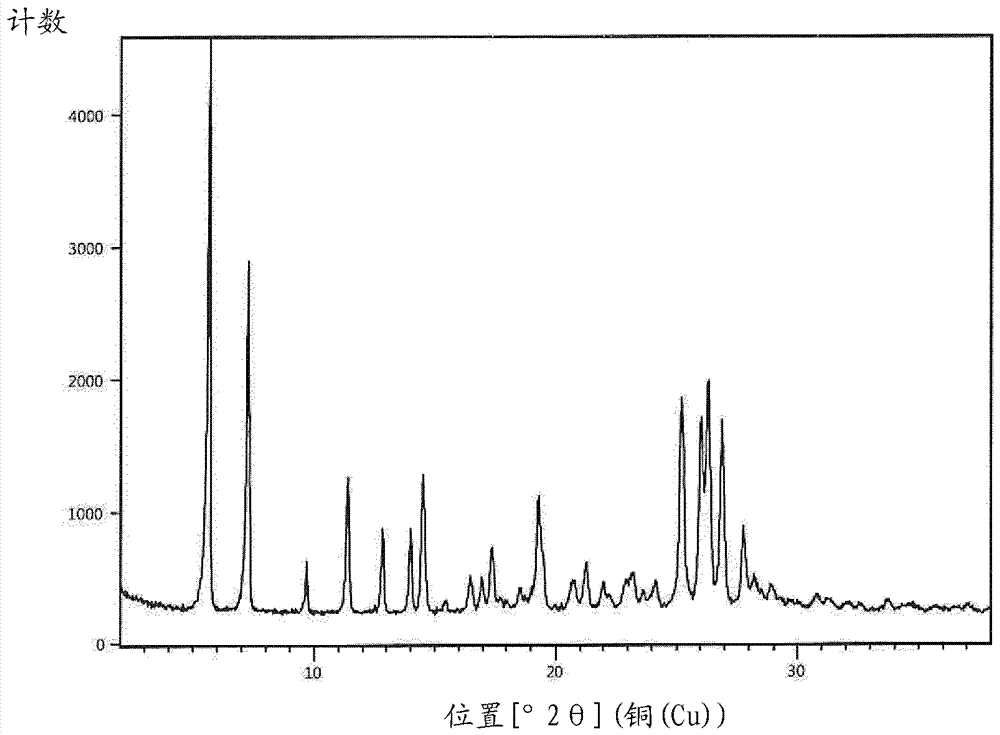
将99ml盐酸(37w%)在24-30℃下添加至300gcopanlisib和1450ml水的混合物中,并在30℃下搅拌10分钟。将该混合物过滤,并将过滤残余物用25ml水洗涤两次。在室温下在18分钟内将6.0l乙醇添加至滤液中。将所得悬浮液加热至76℃,并在76-78℃下搅拌1小时。将该混合物冷却至22℃,并在该温度下搅拌1小时。将该悬浮液过滤,并将收集的固体用120ml水和480ml乙醇的混合物洗涤。将该悬浮液过滤,并将收集的晶体在40℃下在真空中干燥,以得到295g作为水合物ii的copanlisib二盐酸盐。
水(karl-fisher):7.9%
氯化物(离子色谱法):11.7%
xrpd:水合物ii
测量条件:
扫描轴2θ-ω
开始位置[°2θ]2.0000
结束位置[°2θ]37.9900
k-α1[å]1.54060
发生器环境35ma,45kv
衍射仪类型透射衍射仪
入射波束单色器是
旋转否
x-射线衍射图在图1中给出。
实施例13:步骤a11:copanlisib二盐酸盐(11)的制备的进一步的实例
将9.10g盐酸(25w%)添加至15gcopanlisib于37.5g水中的混合物中。将该混合物搅拌10分钟,并过滤。将过滤残余物用7.0g水洗涤。在40℃下在1小时内将滤液添加至70.6g乙醇中。使用额外的2.0g水来冲洗添加设备。将所得悬浮液在1小时内冷却至23℃,并在该温度下搅拌1小时。将该悬浮液过滤,并将收集的晶体用17.9g乙醇和7.5g水的混合物洗涤两次,然后进行空气干燥,以得到17.0g作为水合物ii的copanlisib二盐酸盐。
通过hplc的纯度:99.9%,<0.06%2-氨基-n-{3-(2-氨基乙基)-8-甲氧基-7-[3-(吗啉-4-基)丙氧基]-4-氧代-3,4-二氢喹唑啉-2-基}嘧啶-5-甲酰胺
干燥失重(120℃,30分钟):12.9w%
乙醇(顶空-gc):<0.1%
xrpd:水合物ii
测量条件:
注释配置=反射-透射旋转器台,
扫描轴gonio
开始位置[°2θ]2.0066
结束位置[°2θ]37.9906
阳极材料cu
k-α1[å]1.54060
k-α2[å]1.54443
k-β[å]1.39225
k-a2/k-a1比率0.50000
发生器环境40ma,40kv
入射波束单色器聚焦x-射线镜
旋转是
x-射线衍射图在图2中给出。
实施例14:步骤a11:copanlisib二盐酸盐(11)的制备的进一步的实例
将17gcopanlisib二盐酸盐溶解于66g水中。在40℃下在1小时内将澄清溶液添加至127.5g乙醇中。用2g水冲洗添加设备。将该混合物在40℃下搅拌30分钟,然后在3小时内冷却至0℃。过滤该悬浮液。将收集的晶体用20ml3:1乙醇:水-混合物(v/v)洗涤三次,然后进行空气干燥,以得到15.8g作为水合物ii的copanlisib二盐酸盐。
通过hplc的纯度:99.9%,0.06%2-氨基-n-{3-(2-氨基乙基)-8-甲氧基-7-[3-(吗啉-4-基)丙氧基]-4-氧代-3,4-二氢喹唑啉-2-基}嘧啶-5-甲酰胺
质量损失(热重分析):12.3w%
水(karl-fisher):12.0w%
乙醇(顶空-gc):<0.1%
xrpd:水合物ii
测量条件:
注释扫描2-80trans(stoe-金属板小槽)
扫描轴gonio
开始位置[°2θ]2.0066
结束位置[°2θ]37.9906
阳极材料cu
k-α1[å]1.54060
k-α2[å]1.54443
k-β[å]1.39225
k-a2/k-a1比率0.50000
发生器环境40ma,40kv
入射波束单色器聚焦x-射线镜
旋转是
x-射线衍射图在图3中给出。
实施例15:步骤a11:copanlisib二盐酸盐(11)的制备的进一步的实例
在最高30℃下将7.3g盐酸添加至12gcopanlisib和33g水的混合物中。将所得混合物在25℃下搅拌15分钟,并过滤。将过滤残余物用6g水洗涤。在23℃下在1小时内将11.5g乙醇添加至滤液中。完成添加后,将该混合物在23℃下搅拌1小时。用3小时将额外的59g乙醇添加至该混合物中。完成添加后,将该混合物在23℃下搅拌1小时。过滤所得的悬浮液。将收集的晶体用11.9g乙醇和5.0g水的混合物洗涤三次,并进行空气干燥,以得到14.2g作为水合物i的copanlisib二盐酸盐。
通过hplc的纯度:>99.8%;<0.05%2-氨基-n-{3-(2-氨基乙基)-8-甲氧基-7-[3-(吗啉-4-基)丙氧基]-4-氧代-3,4-二氢喹唑啉-2-基}嘧啶-5-甲酰胺
质量损失(热重分析):14.5w%
水(karl-fisher):14.1%
乙醇(顶空-gc):<0.1%
氯化物(离子色谱法):11.9%
xrpd:水合物i
测量条件:
注释配置=反射-透射旋转器台,
原始数据来源xrd测量(*.xrdml)
扫描轴gonio
开始位置[°2θ]2.0066
结束位置[°2θ]37.9906
阳极材料cu
k-α1[å]1.54060
k-α2[å]1.54443
k-β[å]1.39225
k-a2/k-a1比率0.50000
发生器环境40ma,40kv
入射波束单色器聚焦x-射线镜
旋转是
x-射线衍射图在图4中给出。
实施例16:步骤a11:copanlisib二盐酸盐(11)的制备的进一步的实例
在28℃的最高温度下,将9.1kg盐酸(25w%)添加到14.7kgcopanlisib和41.9kg水的混合物中。将所得混合物在23℃下搅拌80分钟,直至形成澄清溶液。将该溶液转移至第二反应容器,并用6kg水冲洗转移管线。在23℃下在70分钟内缓慢添加14.1kg乙醇。完成添加乙醇后,将该混合物在23℃下搅拌1小时。在23℃下在3.5小时内缓慢添加额外的72.3kg乙醇,并将所得混合物在该温度下搅拌1小时。将该悬浮液过滤,并将收集的固体用31kg乙醇-水混合物(2.4:1(w/w))洗涤两次。将产物用40℃的最高夹套温度在真空中干燥3.5小时,以得到15.0kg作为水合物i的copanlisib二盐酸盐。
通过hplc的纯度:>99.9%;<0.05%2-氨基-n-{3-(2-氨基乙基)-8-甲氧基-7-[3-(吗啉-4-基)丙氧基]-4-氧代-3,4-二氢喹唑啉-2-基}嘧啶-5-甲酰胺
干燥失重:14.7w%
氯化物(滴定):10.8%
水(karl-fisher):14%
xrpd:水合物i
测量条件:
扫描轴gonio
开始位置[°2θ]2.0066
结束位置[°2θ]37.9906
阳极材料cu
k-α1[å]1.54060
k-α2[å]1.54443
k-β[å]1.39225
k-a2/k-a1比率0.50000
发生器环境40ma,40kv
入射波束单色器聚焦x-射线镜
旋转是
x-射线衍射图在图5中给出。
xrpd(表)
- 还没有人留言评论。精彩留言会获得点赞!