参与甘草酸生物合成的糖基转移酶基因及其编码产物与应用的制作方法
本发明涉及甘草酸生物合成相关的糖基转移酶(ugt)基因,并利用体外酶促反应的方法对该基因编码蛋白进行功能活性分析,涉及到甘草酸生物合成途径中ugt基因及其编码产物与应用,属于药用植物基因工程领域。
背景技术:
甘草酸是一种来源于甘草属(glycyrrhiz)植物的三萜皂苷类天然产物,其具有广泛的生物活性,包括抗炎、抗过敏以及抗病毒等。目前甘草酸已被开发成注射液在临床上用于慢性乙肝和急性肝损伤的治疗。据报道,以甘草酸为主要成分制成的天晴甘美注射剂年销量可达16亿人民币。同时,由于甘草酸的甜度是蔗糖的150倍,它也作为甜味剂远销世界各地,其相关出口产品年销量可达4千万美元,市场需求量大。
植物中甘草酸的生物合成需要其生物合成途径中的相关酶进行一系列催化反应,目前,甘草酸生物合成途径中的大多数酶的基因已被成功克隆,并能利用生物技术的方法人工合成甘草次酸,但是,催化甘草酸合成的最后一步反应的ugt基因仍未能被成功克隆。因此,克隆甘草酸ugt基因将对于调节和生产甘草酸具有重要意义。
技术实现要素:
(1)本发明要解决的问题
ugt基因是植物中重要的超家族基因,成员众多,功能多样,克隆和表征其功能往往十分困难。在本发明被公布之前,尚未有任何公开或报道过本专利申请中涉及甘草酸ugt核苷酸及其氨基酸序列。本发明的目的在于提供甘草酸ugt核苷酸及其氨基酸序列。
(2)技术方案
为获得甘草酸ugt核苷酸及其氨基酸序列,我们将克隆得到的甘草(glycyrrhizauralensisfisch)中的ugt核苷酸序列连接于表达载体并在宿主细胞中表达获得蛋白,再经过体外催化实验表征其功能,最终实现了本发明。
本发明具体如下所述。
[1]一种多核苷酸,为以下的(a)、(b)或(c)所示的多核苷酸:
(a)一种核苷酸,其核苷酸序列由seqidno.1所示的第1-1446位核苷酸组成;
(b)一种多核苷酸,其包含与(a)所述核苷酸序列具有35%或更高百分比同一性的核苷酸序列,并且编码的蛋白质具有甘草酸ugt活性;或
(c)一种多核苷酸,其在严格条件下与(a)反向互补的核苷酸杂交,并且编码的蛋白质具有甘草酸ugt活性。
[2]一种蛋白质,为以下的(a)、(b)或(c)所示的蛋白质:
(a)一种蛋白质,其氨基酸序列由seqidno.2所示的氨基酸组成;
(b)一种蛋白质,其氨基酸序列包含与(a)所述的氨基酸序列具有30%或更高百分比同一性的氨基酸序列,并且具有甘草酸ugt活性;或
(c)一种蛋白质,其氨基酸序列包含(a)所述的氨基酸序列中具有一个或几个氨基酸的缺失、置换、添加或插入的氨基酸序列,并且具有甘草酸ugt活性。
[3]一种重组载体,其由[1]所述的多核苷酸或编码[2]所述蛋白质的多核苷酸与已知载体重组而成。
[4]一种转化体,其通过将根据[3]所述的重组载体、[1]所述的多核苷酸或编码[2]中所述蛋白质的多核苷酸导入宿主中制备而成。
[5]在[3]所述的已知载体是已知的细胞系统载体或已知的无细胞系统载体。
[6]在[4]所述的宿主是属于棒状杆菌属(corynebacterium)、泛菌属(pantoea)、肠杆菌属(enterobacter)或酵母菌属(saccharomyces)已知的微生物。
[7]一种生产甘草酸的方法,所述方法是通过培养[4]和[6]中任一项所述的转化体来直接生产甘草酸。
[8]含有[1]所述的多核苷酸或编码[2]所述蛋白质的多核苷酸在甘草遗传育种中的应用。
(3)有益效果
在本发明被公布之前,尚未有任何公开或报道过本专利申请中涉及甘草酸ugt核苷酸及其氨基酸序列,本发明提供了甘草酸ugt的多核苷酸序列,它们编码的蛋白具有甘草酸ugt活性,并可用于调节和生产甘草酸。
本发明所述的“甘草酸ugt”指的是具有催化甘草次酸(cas:471-53-4)生成甘草次酸单葡萄糖醛酸(cas:34096-83-8)或甘草酸(cas:1405-86-3),或是催化甘草次酸单葡萄糖醛酸(cas:34096-83-8)生成甘草酸(cas:1405-86-3)的ugt。所述的cas指的是物质数字识别号码(chemicalabstractsservice),是每一种自然世界中的化学物质的唯一识别码。
本发明所述的百分比同一性是指通过blast(basiclocalalignmentsearchtool)算法来确定的核苷酸或蛋白质之间的相似性,核苷酸之间的百分比同一性可通过blastn算法计算确定,蛋白质之间的百分比同一性可通过blastp算法计算确定,blastp和blastn的程序可参见网站(http://blast.ncbi.nlm.nih.gov/blast.cgi)。
附图说明
图1是甘草酸ugt催化示意图;
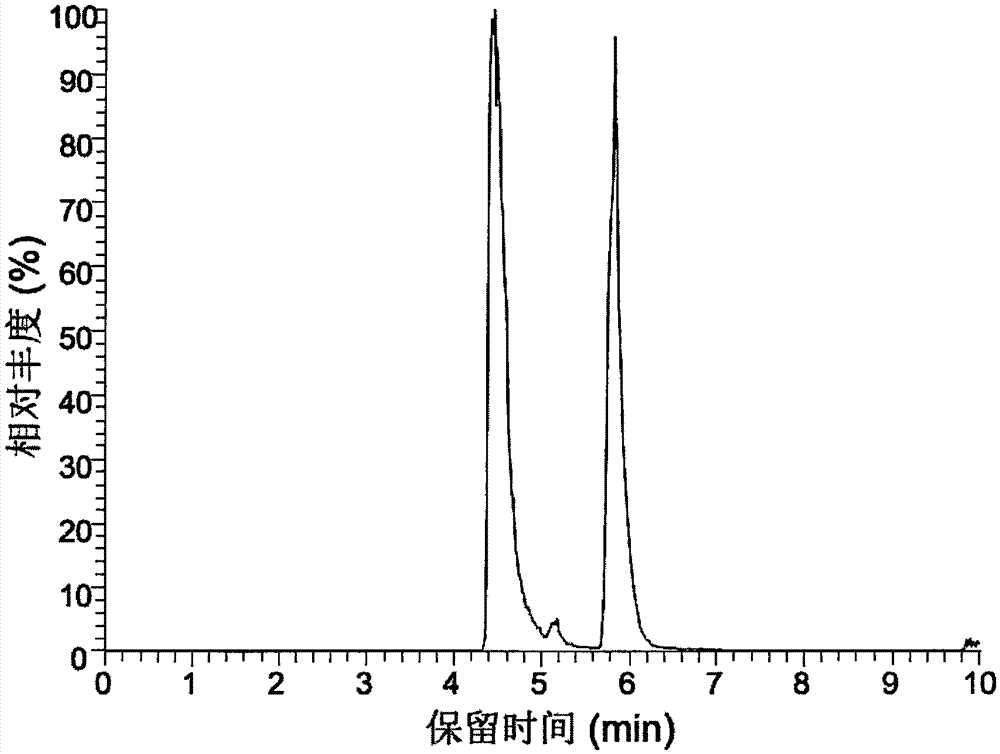
图2是以甘草次酸为底物时seqidno.1编码蛋白催化反应产物lc-ms色谱图(图中保留时间为5.84min的峰是甘草次酸,保留时间为5.18min的峰是甘草次酸单葡萄糖醛酸,保留时间为4.46min的峰是甘草酸);
图3是以甘草次酸单葡萄糖醛酸为底物时seqidno.1编码蛋白催化反应产物lc-ms色谱图(图中保留时间为5.18min的峰是甘草次酸单葡萄糖醛酸,保留时间为4.46min的峰是甘草酸)。
具体实施方式
本发明通过克隆得到的编码甘草酸ugt的多核苷酸并将其连接到表达载体,再将带有编码甘草酸ugt的多核苷酸的重组表达载体分别转入宿主中表达。提取得到的纯化蛋白加入到含有不同底物的体外酶促反应体系中进行反应,催化产物用lc-ms分析。lc-ms结果证明该多核苷酸编码的蛋白酶具有甘草酸ugt活性。具体内容如下所述。
<编码甘草酸ugt的多核苷酸>
本发明提供了编码甘草酸ugt的多核苷酸。
在一个实施方案中,本发明的核苷酸是由seqidno.1核苷酸序列中的第1-1446位核苷酸组成,其编码蛋白的氨基酸序列如seqidno.2所示,并且该蛋白质具有甘草酸ugt活性。
在另一个实施方案中,本发明的多核苷酸是包含与seqidno.1的核苷酸序列具有35%或更高百分比同一性的核苷酸序列的多核苷酸,且该多核苷酸能编码均具有甘草酸ugt活性的蛋白质。该百分比同一性可以是35%、40%、50%、60%、70%、80%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99.1%、99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%、99.9%或更高。
在另一个实施方案中,本发明的多核苷酸是在严格条件下能与seqidno.1反向互补核苷酸序列杂交的多核苷酸,且该多核苷酸能编码均具有甘草酸ugt活性的蛋白质。
“严格条件”是指其中形成所谓的特异性杂交而不形成非特异性杂交的条件。这样的条件是难以清楚定量的。然而,举例而言,该条件是其中基本上相同的多核苷酸具有高度同一性的条件,例如,以上所描述的具有百分比同一性的多核苷酸相互杂交,而具有较低同一性的多核苷酸相互不杂交。特别地,该条件可包括在约45℃下在6×scc(氯化钠/柠檬酸钠)中杂交,然后在50至65℃下在0.2×scc和0.1%sds中洗涤一次或两次或多次。
本发明的多核苷酸可以是dna或rna,且优选为dna。本发明的多核苷酸还可以是编码本发明下文的蛋白质的多核苷酸。
<甘草酸ugt的蛋白>
本发明还提供了具有甘草酸ugt活性的蛋白质。
在一个实施方案中,本发明的蛋白质的氨基酸序列如seqidno.2所示,其具有甘草酸ugt活性。
在另一个实施方案中,本发明的蛋白质是包含与seqidno.2的氨基酸序列具有30%或更高百分比同一性的蛋白质,且该蛋白质均具有甘草酸ugt活性。该百分比同一性可以是30%、40%、50%、60%、70%、80%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99.1%、99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%、99.9%或更高。
在又一个实施方案中,本发明的蛋白质是包含在seqidno.2的氨基酸序列中具有一个或数个氨基酸突变的蛋白质,且该蛋白质均具有甘草酸ugt活性。氨基酸残基突变的例子可包括一个或数个氨基酸残基的缺失、置换、添加和插入。术语“一个或数个”是指对蛋白三维结构和蛋白质的活性未受实质性影响的范围。
在本发明的蛋白质中,可将突变导入到催化结构域中的位点和除催化结构域之外的位点中,且保持原有活性。能够保持目的活性的待突变的氨基酸残基的位置是本领域技术人员已知的。特别地,本领域技术人员能够识别结构和功能之间的关联,因为本领域技术人员能够:(1)比较具有相同类型的活性的多个蛋白质的氨基酸序列(例如,由seqidno.2代表的氨基酸序列与其他甘草酸ugt的氨基酸序列的比较),(2)明确相对保守的区域和非相对保守的区域,(3)从相对保守的区域和非相对保守的区域分别地预测能够起重要功能作用的区域和不能起重要作用的区域。因此,本领域技术人员能够获得甘草酸ugt的氨基酸序列的待突变的氨基酸残基的位置。
氨基酸残基的置换可以是保守置换,也可以是不影响蛋白质活性的非保守置换。如本文所用的,术语“保守置换”指利用具有相似的侧链的氨基酸残基置换某些氨基酸残基。具有相似侧链的氨基酸残基的家族是本领域中已知的。这样的家族的例子可包括具有碱性侧链的氨基酸(例如,赖氨酸、精氨酸、组氨酸),具有酸性侧链的氨基酸(例如,天冬氨酸、谷氨酸),具有不带电荷的极性侧链的氨基酸(例如,天冬酰胺、谷氨酰胺、丝氨酸、苏氨酸、酪氨酸、半胱氨酸),具有非极性侧链的氨基酸(例如,甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、甲硫氨酸、色氨酸),在具有β位置具有支链的氨基酸(例如,苏氨酸,缬氨酸、异亮氨酸),具有芳香族侧链的氨基酸(例如,酪氨酸、苯丙氨酸、色氨酸、组氨酸),具有含羟基(例如,醇式,酚式)侧链的氨基酸(例如,丝氨酸、苏氨酸、酪氨酸),和具有含硫侧链的氨基酸(例如,半胱氨酸、甲硫氨酸)。优选地,氨基酸的保守置换可以是天冬氨酸和谷氨酸之间的置换,精氨酸、赖氨酸和组氨酸之间的置换,色氨酸和苯丙氨酸之间的置换,苯丙氨酸和缬氨酸之间的置换,亮氨酸、异亮氨酸和缬氨酸之间的置换,和甘氨酸和丙氨酸之间的置换。
<重组载体>
本发明提供了重组载体。本发明的重组载体由本发明的多核苷酸或编码本发明中蛋白质的多核苷酸与已知载体重组而成。所述的已知载体可包括细胞系统载体(其在宿主细胞中表达蛋白质)和无细胞系统载体(其利用蛋白质翻译系统)。已知载体还可以是质粒或整合载体。已知载体还可以是dna载体或rna载体。
细胞系统载体的例子包括基于cole质粒,代表为pbr322衍生物,具有p15a起始点的基于pacyc质粒,基于psc的质粒和来源于bac的f因子的小型f质粒以及大肠杆菌(eschericbiacoli)中类似的表达载体。此外,还可包括具有色氨酸启动子诸如trc和tac的表达载体、lac启动子、t7启动子、t5启动子、t3启动子、sp6启动子、阿拉伯糖诱导型启动子、冷休克启动子、四环素诱导型启动子和类似物。
无细胞系统载体的例子可包括具有在细胞系统载体中示例的t7启动子或t3启动子的表达载体和用于在小麦无细胞系统中合成蛋白质的载体,比如基于peu的质粒,其具有sp6启动子或t7启动子。
在利用无细胞系统载体的蛋白质合成中,首先利用转录系统将目的蛋白质的cdna转录而合成mrna。所述转录系统可包括常规地和公开地已知的那些转录系统,其中通过rna聚合酶转录cdna。rna聚合酶的例子可包括t7rna聚合酶。
因此,利用无细胞蛋白质合成系统翻译mrna以合成蛋白质,该系统为翻译系统。在该系统中,包含翻译所需的核糖体、翻译起始因子、翻译延伸因子、解离因子和氨酰trna合成酶。这样的蛋白质翻译系统的例子可包括大肠杆菌提取物、兔网织红细胞和小麦胚芽提取物。
并且,还可包括重构的无细胞蛋白质合成系统,其由以上翻译所需的因子组成,其独立地纯化。
利用细胞系统载体进行的蛋白质合成将在下文<转化体>中描述。
可纯化利用细胞系统载体或无细胞系统载体合成的蛋白质。纯化方法的例子可包括利用盐析方法和多种层析方法的方法。当设计表达载体以表达标签序列诸如目的蛋白质的c末端或n末端的组氨酸标签,可应用利用诸如镍或钴的对该标签具有亲和力的物质利用亲和层析方法的纯化方法。此外,通过适当的纯化方法与离子交换层析、凝胶过滤层析或类似的方法相组合可提高本发明的蛋白质的纯度。
<转化体>
本发明提供了包含本发明的表达载体的转化体。本发明的转化体是通过将本发明的表达载体导入到宿主中获得的一种转化体。用于本发明的宿主优选为细菌或真菌。
细菌可以是革兰氏阳性细菌或革兰氏阴性细菌。
革兰氏阳性菌可包括属于以下属的细菌:杆菌属(bacillus)、李斯特菌属(listeria)、葡萄球菌属(staphylococcus)、链球菌属(streptococcus)、肠球菌属(enterococcus)、梭菌属(clostridium)、棒状杆菌属(corynebacterium)和链球菌属(streptomyces)。优选的是属于杆菌属和棒状杆菌属的细菌。
杆菌属的细菌可包括枯草芽孢杆菌(bacillussubtilis)、炭疽杆菌(bacillusanthracis)和蜡样芽孢杆菌(bacilluscereus)。枯草芽孢杆菌是更优选的。
棒状杆菌的细菌可包括谷氨酸棒状杆菌(corynebacteriumglutamicum)、有效棒状杆菌(corynebacteriumefficiens)和帚石南棒状杆菌(corynebacteriumcallunae)。谷氨酸棒状杆菌是更优选的。
革兰氏阴性细菌的例子可包括属于以下属的细菌:大肠杆菌属(escherichia)、泛菌属(pantoea)、沙门氏菌属(salmonella)、弧菌属(vivrio)、沙雷氏菌属(serratia)和肠杆菌属(enterobacter)。属于大肠杆菌属和肠杆菌属的细菌是优选的。
作为属于大肠杆菌属的大肠杆菌是优选的。
属于泛菌属的细菌的例子可包括菠萝泛菌(pantoeaananaris)、斯氏泛菌(pantoeastewartii)、成团泛菌(pantoeaagglomerans)和柠檬泛菌(pantoeacitrea)。菠萝泛菌和柠檬泛菌是优选的。欧洲专利申请公开0952221中示例的菌株可用作属于泛菌属的细菌。属于泛菌属的细菌的代表性菌株的例子可包括公开于欧洲专利申请公开0952221中的菠萝泛菌aj13355菌株(fermbp-6614)和菠萝泛菌aj13356菌株(fermbp-6615)。
属于肠杆菌属的细菌的例子可包括成团肠杆菌(enterobacteragglomerans)和产气肠杆菌(enterobacteraerogenes)。产气肠杆菌是优选的。欧洲专利申请公开0952221中示例的细菌菌株可用作属于肠杆菌属的细菌。属于肠杆菌属的细菌的代表性菌株的例子可包括成团肠杆菌atcc12287菌株、产气肠杆菌tacc13048菌株、产气肠杆菌nbrc12010菌株(biotechnol.bioeng.,2007,mar.27;98(2):340-348)和产气肠杆菌aj110637(fermbp-10955)。
真菌的例子可包括属于以下属的微生物:酵母(saccharomyces)、裂殖酵母(schizosaccharomyces)、耶罗威亚酵母(yarrowia)、木霉(trichoderma)、曲霉(aspergillus)、镰刀霉(fusarium)和毛霉(mucor)。属于酵母属、裂殖酵母属、亚罗酵母属或木霉属的微生物是优选的。
属于酵母属的微生物的例子可包括卡尔斯伯酵母(saccharomycescarlsbergensis)、酿酒酵母(saccharomycescerevisiae)、分散酵母(saccharomycesdiastaticus)、道格拉氏酵母(saccharomycesdouglasii)、克鲁弗酵母(saccharomyceskluyveri)、诺地酵母(saccharomycesnorbensis)和卵形酵母(saccharomycesoviformis)。酿酒酵母是优选的。
粟酒裂殖酵母(schizosaccharomycespombe)作为属于裂殖酵母属的微生物是优选的。
解脂耶罗威亚酵母(yarrowialypolytica)作为属于耶罗威亚酵母属的微生物是优选的。
属于木霉属的微生物的例子可包括哈茨木霉(trichodermaharzianum)、康氏木霉(trichodermakoningii)、长枝木霉(trichodermalongibrachiatum)、里氏木霉(trichodermareesei)和绿色木霉(trichodermaviride)。里氏木霉是优选的。
此外,用于本发明的宿主不受具体局限,宿主也可以是微生物、昆虫细胞、动物细胞、植物细胞、昆虫组织、动物组织、植物组织、昆虫器官、动物器官、植物器官、昆虫全体、动物全体、植物全体或类似物。
实施例
以下将以核苷酸序列表中的seqidno.1为代表具体描述本发明,但本发明不限于以下实施例。
实施例1:乌拉尔甘草中ugt的克隆
我们通过rna提取试剂盒(日本takara公司)提取甘草(glycyrrhizauralensisfisch)根中总rna,用逆转录试剂盒(日本takara公司)获得cdna,依据ugt基因序列的5’端和3’端设计带有酶切位点的引物,用高保真的hstaq酶(日本takara公司)进行pcr扩增,pcr产物进行测序和拼接,可获得本发明涉及的甘草酸生物合成相关的ugt基因的全长核苷酸序列,其由seqidno.1所代表,其全长开放读码框(orf)的长度为1446bp,编码481个氨基酸,详细氨基酸序列见序列表中的seqidno.2。
实施例2:重组表达载体的构建
我们将实施例1中得到的甘草酸ugt的核苷酸片段(seqidno.1)和表达载体pet-32a(+)同时用限制性内切酶(日本takara公司)进行双酶切,酶切后的产物用琼脂糖凝胶回收试剂盒(日本takara公司)的方法进行纯化并回收,回收的产物用dna连接酶在16℃的金属浴中连接过夜,得到重组质粒。重组质粒再转化大肠杆菌dh5α感受态细胞并涂板于含有氨苄青霉素的固体培养基中筛选,阳性克隆再次送测序验证,测序验证正确的克隆通过质粒纯化试剂盒(日本takara公司)的方法可回收得到带有seqidno.1核苷酸序列的重组表达载体。
实施例3:宿主菌的构建
我们将实施例2中得到的重组表达载体按照感受态细胞转化说明书(日本takara公司)的方法转化大肠杆菌bl21(de3)感受态细胞并涂板于含有氨苄青霉素的固体培养基中筛选,阳性克隆再次送测序验证,测序验证正确的克隆作为宿主菌,其含有带有seqidno.1核苷酸序列的重组表达载体。
实施例4:诱导表达与蛋白纯化
我们将实施例3中得到的宿主菌的菌液2ml分别加入100ml的lb液体培养基中,在37℃条件下震荡培养至对数生长期,此时od600值约为0.6至1.0。其中以不含有甘草酸ugt的空载体pet-32a(+)转化大肠杆菌bl21(de3)的宿主菌作为阴性对照。上述4个宿主菌分别加入0.4mm异丙基硫代半乳糖苷(iptg),18℃,诱导12h以上。5000rpm,4℃离心5min收集菌体,用预冷的蒸馏水洗涤2次,收集菌体。加入2ml蒸馏水重新悬浮菌体,冰浴中超声破碎细菌,条件为:超声10s,间隔10s,共12次。整个操作在冰浴中进行,并不时摇动菌液防止其过热,每隔3次拿出来冰浴一次。4℃,13000rpm离心2min,收集上清。上清液通过蛋白纯化试剂盒(日本takara公司)的方法纯化并获得带有甘草酸gut的纯化蛋白(图2)和不带有甘草酸gut的纯化蛋白。
实施例5:蛋白的催化分析
我们将实施例4中得到的2种纯化蛋白分别加入到以甘草次酸(cas:471-53-4)和甘草次酸单葡糖糖醛酸(cas:34096-83-8)为底物的体外酶促反应体系中进行反应,催化反应缓冲体系包括50mm的tric-hcl(ph8.0)、1mm的尿苷-5-二磷酸葡萄糖醛酸(udp-glca)、1mm的二硫苏糖醇(dtt)和200μm底物甘草次酸,催化反应温度为30℃,催化时间在30min上,含有带有甘草酸gut纯化蛋白的催化反应体系作为催化组,含有不带有甘草酸gut纯化蛋白的催化反应体系作为对照组,催化产物用lc-ms分析。lc-ms仪器为accela600pump高效液相色谱-ltq-orbitrapxl质谱联用仪(美国thermoscientific公司),配有电喷雾离子源(esi)。色谱柱为agilentzorbaxsbc18柱(250mm×4.6mm×5μm)。流动相a为0.1%的甲酸水,b为纯甲醇。线性梯度洗脱程序为:0~3.5min,79%b;3.5~8.2min,79%~98%b;8.2~11min,98%~79%b;11~20min,79%b。流速1.0ml/min,柱温25℃,进样量10μl。质谱esi检测为负离子检测,全离子扫描模式。结果发现,催化组与对照组相比,均生成了新的峰,通过对比标准品与新峰的保留时间、精确分子离子峰与主要裂解方式,确定以甘草次酸(cas:471-53-4)为底物的催化组(图2)产生了甘草次酸单葡萄糖醛酸(cas:34096-83-8)和甘草酸(cas:1405-86-3);以甘草次酸单葡萄糖醛酸(cas:34096-83-8)为底物的催化组(图3)产生了甘草酸(cas:1405-86-3)。因此,通过实验证明,核苷酸(序列如seqidno.1所示)编码的蛋白(序列如seqidno.2)具有催化甘草次酸(cas:471-53-4)生成甘草次酸单葡萄糖醛酸(cas:34096-83-8)或甘草酸(cas:1405-86-3),或是催化甘草次酸单葡萄糖醛酸(cas:34096-83-8)生成甘草酸(cas:1405-86-3)的活性。
实施例6:突变分析
我们通过点突变试剂盒(日本takara公司)的方法将seqidno.2中的部分氨基酸序列进行敲除实验,敲除位点分别包括:seqidno.2序列4位点的“g”(此时该突变体核苷酸序列与seqidno.1核苷酸序列的百分比同一性为99%,该突变体蛋白质序列与seqidno.2蛋白质序列的百分比同一性为99%)、seqidno.2序列4至6位点的“gne”(此时该突变体核苷酸序列与seqidno.1核苷酸序列的百分比同一性为99%,该突变体蛋白质序列与seqidno.2蛋白质序列的百分比同一性为99%)。通过实施例2至5的方法进行实验证明:本实施例中部分氨基酸序列敲除的蛋白仍具有甘草酸ugt活性。
我们也通过点突变试剂盒(日本takara公司)的方法将seqidno.2中的部分氨基酸序列进行置换实验,置换位点分别包括:seqidno.2序列121位点的“d”置换成“a”(此时该突变体核苷酸序列与seqidno.1核苷酸序列的百分比同一性为99%,该突变体蛋白质序列与seqidno.2蛋白质序列的百分比同一性为99%)、seqidno.2序列9至11位点的“elh”置换成“ead”(此时该突变体核苷酸序列与seqidno.1核苷酸序列的百分比同一性为99%,该突变体蛋白质序列与seqidno.2蛋白质序列的百分比同一性为99%)。通过实施例2至5的方法进行实验证明:本实施例中部分氨基酸序列置换的蛋白仍具有甘草酸ugt活性。
我们还通过基因融合的方法将带有麦芽糖结合蛋白mbp标签的核苷酸序列分别添加在seqidno.1核苷酸序列的5’端,获得新的核苷酸序列,命名为rhgs1(此时该突变体核苷酸序列与seqidno.1核苷酸序列的百分比同一性为74%)。通过实施例2至4的方法将rhgs1与载体pma1-c4x进行连接并表达获得新的融合蛋白,命名为rhdb1(此时该突变体蛋白质序列与seqidno.2蛋白质序列的百分比同一性为74%)。通过实施例5的方法进行分析表明:本实施例中部分氨基酸序列添加的蛋白rhdb1仍具有甘草酸ugt活性。
我们还通过基因融合的方法将带有trx·tag标签蛋白的核苷酸序列插入在seqidno.1核苷酸序列的12至13位点之间获得的新核苷酸序列,命名为rhgs2(此时该突变体核苷酸序列与seqidno.1核苷酸序列的百分比同一性为82%)。通过实施例2至4的方法将rhgs2与载体pma1-c4x进行连接并表达获得新的融合蛋白,命名为rhdb2(此时该突变体蛋白质序列与seqidno.2蛋白质序列的百分比同一性为82%).通过实施例5的方法进行分析表明:本实施例中部分氨基酸序列插入的蛋白rhdb2仍具有甘草酸ugt活性。
同时,我们还人工设计了seqidno.3核苷酸序列,其与seqidno.1核苷酸序列的百分比同一性为35%,seqidno.3编码蛋白与seqidno.2氨基酸序列的百分比同一性为30%。通过实施例2至5的方法进行实验证明:本实施例中seqidno.3编码的蛋白仍具有甘草酸ugt活性。
实施例7:序列杂交分析
我们将seqidno.3与seqidno.1的反向互补核苷酸在约45℃下在6×scc(氯化钠/柠檬酸钠)中进行杂交,然后在50至65℃下在0.2×scc和0.1%sds中洗涤一次或两次或多次。结果表明:在该条件下,两核苷酸成功杂交。同时,在实施例6中已证明了seqidno.3编码的蛋白具有甘草酸ugt的活性,所以,该结果证明:如果能与seqidno.1反向互补核苷酸序列杂交,且编码具有甘草酸ugt活性的蛋白质的核苷酸分子也在本发明保护范围。
实施例8:产甘草酸的酵母工程菌的构建
我们通过实施例1的方法克隆已知的bas基因(genbank注册号为gu072921.1)、cyp72a154基因(genbank注册号为ab558153.1)、cyp88d6基因(genbank注册号为ab433179.1)、atcpr1(genbank注册号为nm_001203894.1)和ugdh基因(genbank注册号为aj007702.1),同时我们还克隆了酵母菌中已知的启动子(ppgk1、pyef1、pfba1、ptdh3)、终止子(tadh1、tcyc1、ttpi1、ttdh2)和thmgm1基因。随后,通过基因融合的方法,我们获得了融合片段rh1(ppgk1-thmg1-tadh1)、rh2(ptdh3-seqidno.1-ttpi1-ptef1-ugdh-tcyc1)和rh3(ppgk1-cyp72a154-tadh1-pfba1-cyp88d6-ttdh2-ptdh3-atcpr1-ttpi1-ptef1-bas-tcyc1)。接着,我们运用基因同源重组的方法将获得的三个融合片段分别导入酿酒酵母(saccharomycescerevisiae)by4742菌株基因组中的6dna位点、rdna位点和his3位点构建工程菌。构建的工程菌在酵母膏胨葡萄糖(ypd)琼脂培养基中7天,培养条件为30℃和250rpm。培养得到的产物用实施例5的方法检测,结果证明获得了10.6mg/l的甘草酸。
实施例9:seqidno.1基因在甘草毛状根遗传育种中的应用
本实施例以甘草毛状根的遗传改良为例描述常规的甘草遗传改良方式。依据seqidno.1基因序列的5’端和3’端设计带有酶切位点的引物,分别将seqidno.1基因和载体pbi121进行双酶切并回收产物,用t4dna连接酶在4℃连接过夜,取1μl连接产物转化大肠杆菌dh5α,菌液进行pcr检验,将正确的单克隆菌液进行测序。对测序序列进行分析后,提取正确重组的质粒并转化感受态农杆菌。转化后的农杆菌在yeb固体培养基中筛选并用pcr验证。pcr检验正确的农杆菌用于侵染甘草(glycyrrhizauralensisfisch)毛状根,侵染后的甘草毛状根放在新的ms培养基中25℃培养,之后转入1/2ms液体培养基中培养,pcr鉴定成功侵染的毛状根。结果表明:与对照组相比,成功转入seqidno.1基因过表达的甘草毛状根中甘草酸的含量提高了2.5倍。
- 还没有人留言评论。精彩留言会获得点赞!