可用于胆固醇动员的化合物、组合物和方法与流程
本申请要求2010年10月18日提交的美国临时申请号61/394,136和2011年2月18日提交的美国临时申请号61/444,212的权益,它们各自的公开内容特此通过引用整体并入本文。
技术领域
本发明提供了治疗或预防受试者中的异常状况的化合物、组合物和方法。
背景技术:
在人体中循环的胆固醇由血浆脂蛋白携带,所述血浆脂蛋白是复合脂和蛋白组合物的颗粒,其在血液中运输脂类。两类携带胆固醇的血浆脂蛋白是低密度脂蛋白(“LDL”)和高密度脂蛋白(“HDL”)。认为LDL颗粒负责在体内将胆固醇从肝脏(合成或从饮食来源获得胆固醇的场所)运输到肝外组织。另一方面,认为HDL颗粒辅助将胆固醇从肝外组织运输到肝脏,在肝脏中分解和消除胆固醇。这样的胆固醇从肝外组织向肝脏的运输被称作“胆固醇逆向转运”。
胆固醇逆向转运(“RCT”)途径具有3个主要步骤:(i) 胆固醇流出,即从各种外周细胞库初步去除胆固醇;(ii) 通过卵磷脂:胆固醇酰基转移酶(“LCAT”)的作用,进行胆固醇酯化,由此防止流出的胆固醇重新进入细胞;和(iii) 由HDL摄取胆固醇酯,并将HDL-胆固醇酯复合物递送至肝细胞。
RCT途径由HDL颗粒介导。最近描述了肝脏中的涉及F1-ATP酶和P2Y13受体的途径(Martinez等人. 2003 Nature 421; 75-79),该途径调节HDL-胆固醇除去。最近描述了F1-ATP酶亚基的核苷酸酶活性在肝细胞的细胞表面处的存在,从而允许ATP水解为ADP,所述ADP又刺激P2Y13受体活性,从而导致细胞对HDL的摄入(Jacquet等人. 2005 Cell Mol Life Sci 62; 2508-2515)。近年来,Fabre等人(Fabre等人. Hepatology 52; 1477-1483) 证实了小鼠中P2Y13r和胆固醇逆向转运之间的关联。
技术实现要素:
在一个实施方案中,本发明提供了下式(I)的化合物
及其药学上可接受的盐,其中
R1、R2和R3中的每一个独立地是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R4是-H、-OH、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、-烃基、-O-烃基、-芳基、-O-芳基、-芳烷基、-O-芳烷基、-杂芳基、-O-杂芳基、-杂环基、-O-杂环基、-卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基);
Z1是CH2、S、O、NH、N-烃基、N-芳基、N-杂芳基或N-杂环基;
Z2是CH或N;且
n是1-6的整数。
在另一个实施方案中,本发明提供了下式(II)的化合物
及其药学上可接受的盐,其中
每个R9独立地是-H、-烃基、-芳基、-芳烷基、-杂芳基、-杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烷基、-O-烯基、芳基、-O-芳基、芳烷基、-O--芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、NHC(O)(C2-C10-烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
Q1、Q2和Q3中的每一个独立地是CR10或N;
X是CHR10、S、O或NR9;且
每个m独立地是0-3的整数。
在另一个实施方案中,本发明提供了下式III的化合物:
及其药学上可接受的盐,其中
R1、R2和R3中的每一个独立地是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R5是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基或-O-杂环基;
Z1是CH2、S、O、NH、N-烃基、N-芳基、N-杂芳基或N-杂环基;
Z2是CH或N;且
n是1-6的整数。
在另一个实施方案中,本发明提供了下式IV的化合物:
及其药学上可接受的盐,其中
R1是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
Z1是CH2、S、O、NH、N-烃基、N-芳基、N-杂芳基或N-杂环基;和
Z2是CH或N。
在另一个实施方案中,本发明提供了下式V的化合物:
及其药学上可接受的盐,其中
R1、R2和R3中的每一个独立地是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R4是-H、-OH、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基)、;
R6是-H、-OH、-SH、-S-烃基、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基);
R7是H、烃基、芳基、芳烷基、杂芳基或杂环基;
Z2是CH或N;且
n是1-6的整数。
在一个实施方案中,本发明提供了下式VI的化合物:
及其药学上可接受的盐,其中
R1是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R5是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基或卤素;
R6是-H、-OH、-SH、-S-烃基、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基);
R7是H、烃基、芳基、芳烷基、杂芳基或杂环基;和
Z2是CH或N。
在一个实施方案中,本发明提供了下式VII的化合物:
及其药学上可接受的盐,其中
R1a、R1b和R1c中的每一个独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
每个X独立地是CHR10、S、O或NR9;
每个R9独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;且
m是0-3的整数。
在另一个实施方案中,本发明提供了下式VIII的化合物:
及其药学上可接受的盐,其中
R1是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R11是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
Q1、Q2、Q3和Q4中的每一个独立地是CR10或N;且
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2。
在另一个实施方案中,本发明提供了下式IX的化合物:
及其药学上可接受的盐,其中
R11a、R11b和R11c中的每一个独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)(烷基)、-C(O)O(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)或-SO2NH2;
X1和X2中的每一个独立地是CHR10、S、O、NR9或N-酰基;
每个R9独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;且
每个m独立地是1-3的整数。
在另一个实施方案中,本发明另外提供了下式X的化合物:
及其药学上可接受的盐,其中
每个R9独立地是-H、-烃基、-芳基、-芳烷基、-杂芳基、-杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O--芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
Q1、Q2和Q3中的每一个独立地是CR10或N;
X是CHR10、S、O或NR9;且
每个m独立地是0-3的整数。
在另一个实施方案中,本发明另外提供了下式XI的化合物:
及其药学上可接受的盐,其中
R11a、R11b和R11c中的每一个独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)(烷基)、-C(O)O(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)或-SO2NH2;
X1和X2中的每一个独立地是CHR10、S、O、NR9或N-酰基;
每个R9独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;且
每个m独立地是0-3的整数。
在另一个实施方案中,本发明提供了下式(XII)的化合物
及其药学上可接受的盐,其中
每个R9独立地是-H、-烃基、-芳基、-芳烷基、-杂芳基、-杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烷基、-O-烯基、芳基、-O-芳基、芳烷基、-O--芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、NHC(O)(C2-C10-烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)、-SO2NH2、–S-烷基、-S-芳基、-S-杂芳基、-S-杂环或–S-烃基;
Q1、Q2和Q3中的每一个独立地是CR10或N;
X是CHR10、S、O或NR9;且
每个m独立地是0-3的整数。
在另一个实施方案中,本发明提供了下式XIII的化合物:
及其药学上可接受的盐,其中
R1是独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R11是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
Q1、Q2、Q3和Q4中的每一个独立地是CR10或N;
n是1-4的整数,且
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2。
式(I)-(XIII)中的任一个的化合物或所述化合物的药学上可接受的盐是“本发明的化合物”。
在另一个实施方案中,本发明提供了组合物,其包含有效量的本发明的化合物和药学上可接受的媒介物或载体。
在另一个实施方案中,本发明提供了用于治疗或预防脂蛋白代谢障碍、葡萄糖代谢障碍、心血管障碍或相关的血管障碍、涉及C-反应蛋白的异常调节的病症或相关的病症、老化、阿尔茨海默氏病、帕金森病、胰腺炎(pancreatitis)、胰脏炎(pancreatitius)或异常的胆汁产生(各自为一种“病症”)的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。
在另一个实施方案中,本发明包括用于监测受试者中的心血管障碍的治疗进程的方法,所述方法包括:
a. 测定受试者的血液的高密度脂蛋白中的游离胆固醇水平;
b. 给所述受试者施用本发明的化合物;
c. 在施用所述化合物以后,监测受试者的血液中的游离胆固醇水平一段时间;和
d. 基于步骤a和c的结果的对比,评价受试者中的改善水平。
在另一个实施方案中,本发明包括测定受试者中的P2Y13活性水平的方法,所述方法包括:
a. 测定受试者的血液的高密度脂蛋白中的游离胆固醇水平;
b. 给所述受试者施用本发明的化合物;
c. 在施用所述化合物以后,监测受试者的血液中的游离胆固醇水平一段时间;和
d. 基于步骤a和c的结果的对比,评价受试者中的P2Y13活性水平。
附图说明
图1:化合物Va (黑色三角形;上图)、化合物Ih (R-异构体) (黑色三角形;中图)和化合物Ih (S-异构体) (黑色三角形;中图)对用P2Y13受体转染的1321 N1细胞的活性。未转染的1321 N1细胞用作阴性对照(黑色圆形)。每个数据点是一式三份孔的平均值。
图2:本发明的示例性化合物(黑色三角形)对用P2Y13受体转染的1321 N1细胞的活性,并与阳性参照化合物进行对比。未转染的1321 N1细胞用作阴性对照(黑色圆形)。每个数据点是一式三份孔的平均值。
图3:选择的分子对HepG2细胞的HDL内化的单次剂量效应。与75 μg/ml 3H胆固醇胆甾醇油酸酯[胆甾醇基-1,2-3H(N)] HDL、参照化合物(100 nM)和本发明的示例性化合物(1µM)一起,在37℃温育细胞10 min。将数据表示为内化的放射性相对于对照值(设定为0)的百分比。*p<0.005,**p<0.0001。
图4: 选择的分子对HepG2细胞的HDL内化的剂量响应。与75 μg/ml 3H胆固醇胆甾醇油酸酯[胆甾醇基-1,2-3H(N)] HDL、参照化合物(100 nM)和递增浓度的选择的分子一起,在37℃温育细胞10 min。A:化合物IIa和IIc (0.1、1、10、100和1000 nM)。B:化合物Ih (S-异构体)和化合物Ih (R-异构体) (1、10、100和1000 nM)。将数据表示为内化的放射性相对于对照值(设定为0)的百分比。
图5:本发明的示例性化合物对静脉内注射以后的胆汁酸分泌的单次剂量效应。将小鼠禁食2小时,然后在尾静脉中注射在PBS溶液(100µl)中的选择的分子(10 nmol/kg)。4小时以后,分析胆汁含量。A. 将数据表示为胆汁酸的浓度。B. 将数据表示为每只小鼠的胆汁酸的集合(*p<0.05,**p<0.005,***p<0.0001)。动物组n=5。
图6:化合物IIa对胆汁酸、胆汁磷脂和胆汁胆固醇分泌的剂量-响应效应。将小鼠禁食2小时,然后通过经口管饲法用递增浓度的化合物IIa (0.003、0.03和0.3 mg/kg) 治疗。6小时以后,分析胆汁含量。A. 将数据表示为胆汁酸的浓度。B. 将数据表示为单位小鼠重量的胆汁酸集合。C. 将数据表示为胆汁胆固醇的浓度。D. 将数据表示为单位小鼠重量的胆汁胆固醇集合。E. 将数据表示为胆汁磷脂的浓度。F. 将数据表示为单位小鼠重量的胆汁磷脂集合。动物组n=10. (*p<0.05,**p<0.005)
图7:化合物XIa对胆汁酸、胆汁磷脂和胆汁胆固醇分泌的剂量-响应效应。将小鼠禁食2小时,然后通过经口管饲法用递增浓度的化合物XIa (0.003、0.03和0.3mg/kg) 治疗。6小时以后,分析胆汁含量。A. 将数据表示为胆汁酸的浓度。B. 将数据表示为单位小鼠重量的胆汁酸集合。C. 将数据表示为胆汁胆固醇的浓度。D. 将数据表示为单位小鼠重量的胆汁胆固醇集合。E. 将数据表示为胆汁磷脂的浓度。F. 将数据表示为单位小鼠重量的胆汁磷脂集合。动物组n=10. (*p<0.05,**p<0.005,***p<0.0005,****p<0.0001)
图8:化合物VIIa对胆汁酸、胆汁磷脂和胆汁胆固醇分泌的剂量-响应效应。将小鼠禁食2小时,然后通过经口管饲法用递增浓度的化合物VIIa (0.003、0.03和0.3 mg/kg)治疗。6小时以后,分析胆汁含量。A. 将数据表示为胆汁酸的浓度。B. 将数据表示为单位小鼠重量的胆汁酸集合。C. 将数据表示为胆汁胆固醇的浓度。D. 将数据表示为单位小鼠重量的胆汁胆固醇集合。E. 将数据表示为胆汁磷脂的浓度。F. 将数据表示为单位小鼠重量的胆汁磷脂集合。动物组n=10. (*p<0.05,**p<0.01,***p<0.005)
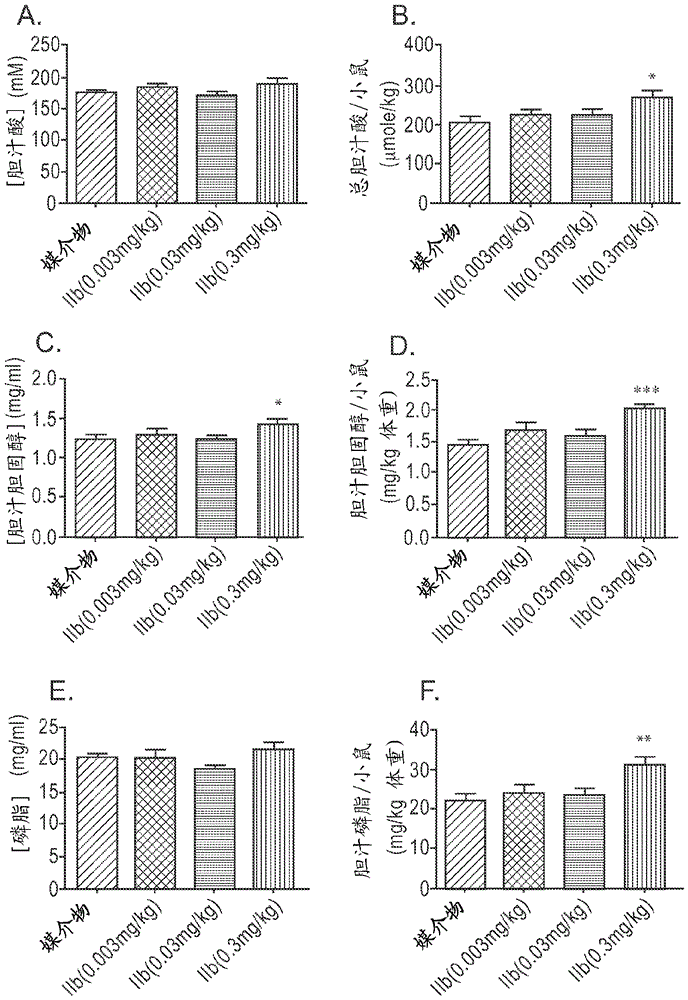
图9:化合物IIb对胆汁酸、胆汁磷脂和胆汁胆固醇分泌的剂量-响应效应。将小鼠禁食2小时,然后通过经口管饲法用递增浓度的化合物IIb (0.003、0.03和0.3 mg/kg) 治疗。6小时以后,分析胆汁含量。A. 将数据表示为胆汁酸的浓度。B. 将数据表示为单位小鼠重量的胆汁酸集合。C. 将数据表示为胆汁胆固醇的浓度。D. 将数据表示为单位小鼠重量的胆汁胆固醇集合。E. 将数据表示为胆汁磷脂的浓度。F. 将数据表示为单位小鼠重量的胆汁磷脂集合。动物组n=10. (*p<0.05,**p<0.005,***p<0.0005)
图10:化合物VIIIa对胆汁酸、胆汁磷脂和胆汁胆固醇分泌的剂量-响应效应。将小鼠禁食2小时,然后通过经口管饲法用递增浓度的化合物VIIIa (0.003、0.03和0.3 mg/kg) 治疗。6小时以后,分析胆汁含量。A. 将数据表示为胆汁酸的浓度。B. 将数据表示为单位小鼠重量的胆汁酸集合。C. 将数据表示为胆汁胆固醇的浓度。D. 将数据表示为单位小鼠重量的胆汁胆固醇集合。E. 将数据表示为胆汁磷脂的浓度。F. 将数据表示为单位小鼠重量的胆汁磷脂集合。动物组n=10. (*p<0.05,**p<0.01,***p<0.005,****p<0.001,*****p<0.0005)
图11:化合物IIc对胆汁酸、胆汁磷脂和胆汁胆固醇分泌的剂量-响应效应。将小鼠禁食2小时,然后通过经口管饲法用递增浓度的化合物IIc (0.003、0.03和0.3 mg/kg) 治疗。6小时以后,分析胆汁含量。A. 将数据表示为胆汁酸的浓度。B. 将数据表示为单位小鼠重量的胆汁酸集合。C. 将数据表示为胆汁胆固醇的浓度。D. 将数据表示为单位小鼠重量的胆汁胆固醇集合。E. 将数据表示为胆汁磷脂的浓度。F. 将数据表示为单位小鼠重量的胆汁磷脂集合。动物组n=10. (*p<0.05,**p<0.005,***p<0.0005)
图12:化合物Ih (外消旋体)对胆汁酸、胆汁磷脂和胆汁胆固醇分泌的影响。将小鼠禁食2小时,然后通过经口管饲法用递增浓度的化合物Ih (外消旋体) (3、30和100 mg/kg)治疗。6小时以后,分析胆汁含量。A: 将数据表示为胆汁酸的浓度。B: 将数据表示为单位小鼠重量的胆汁酸集合。C: 将数据表示为胆汁胆固醇的浓度。D: 将数据表示为单位小鼠重量的胆汁胆固醇集合。E: 将数据表示为胆汁磷脂的浓度。F: 将数据表示为单位小鼠重量的胆汁磷脂集合。动物组n=10. (*p<0.05,**p<0.005,***p<0.0001)。
图13:在治疗1周以后,化合物IIa对胆汁酸分泌的单次剂量效应。通过经口管饲法,每天用化合物IIa (0.03 mg/kg) 治疗小鼠。在处死当天,将小鼠禁食3小时,然后进行经口管饲法(相同浓度)。4小时以后,分析胆汁含量。A: 将数据表示为胆汁酸的浓度。B: 将数据表示为单位小鼠重量的胆汁酸集合(*p<0.05)。动物组n=10。
图14:在Hepa 1-6细胞中的P2Y13抑制(knock-down)。用编码P2Y13 shRNA的慢病毒颗粒(MOI 40)转导Hepa 1-6细胞,所述细胞可通过加入多西环素来诱导。用或不用多西环素(10µM终浓度)处理细胞集合72小时,并通过QPCR测量P2Y13 mRNA表达。*p<0.0001。
图15:在P2Y13抑制的Hepa 1-6细胞上的HDL摄入。用编码P2Y13 shRNA的慢病毒颗粒转导Hepa 1-6细胞,通过加入多西环素诱导所述细胞72小时。与75 μg/ml 3H胆固醇醚HDL和化合物IIa (1µM)一起,在37℃温育细胞10 min。将数据表示为内化的放射性相对于对照值(设定为0)的百分比。*p<0.05,**p<0.005。
图16:在小鼠中活化P2Y13r途径以后,胆汁分泌的增加。将C57Bl/6J小鼠(n = 10) 禁食2 h,然后静脉内注射坎格雷洛®或化合物IIa (10 nmol/kg)。4小时以后,取出胆囊,并分析:胆汁酸含量(A);胆汁胆固醇含量(B)。将C57Bl/6J小鼠(n = 10) 禁食2 h,然后以3、30或300µg/kg,单次口服施用P2Y13r激动剂化合物IIa。6小时以后,使用HPLC和用于胆囊的酶试剂盒,评价胆汁酸含量(C)、胆汁胆固醇含量(D)和肝脏胆汁酸含量(E)。在时间0和治疗6 h以后,收集血浆样品,然后分析:总胆固醇(▲)、未酯化胆固醇(●)、酯化的胆固醇(■)和HDL胆固醇(▼),其中使用酶试剂盒或Lipoprint®(对于HDL胆固醇)进行测定(F)。给C57Bl/6J小鼠(n =5) 静脉内地注射[3H]-胆固醇-标记的小鼠HDL (G)、[3H]-胆甾醇油酸酯-标记的小鼠HDL (H)或[3H]-胆固醇-标记的小鼠LDL (I),并注射坎格雷洛®或化合物IIa (10 nmol/kg)。在2小时以后(对于HDL)或5小时以后(对于LDL),测定肝脏中存在的放射性。通过Charge气溶胶检测,测定得自治疗的动物(100µg/kg) 的粪便的胆固醇含量(J)。*p < 0.05,**p < 0.01。
图17:P2Y13激动剂对ApoE-/-小鼠中的粥样硬化斑块进展的剂量-响应效应。将ApoE-/-小鼠(n = 7) 的左颈动脉的上段结扎。在外科手术当天,给动物饲喂西餐,并通过经口管饲法施用媒介物或递增剂量的化合物IIa。在2:1氯仿/甲醇中提取结扎的颈动脉的脂质。A:通过HPLC,测量未酯化胆固醇(黑色条)和总胆固醇(叠加的灰色条) 的浓度。E和H: ApoE-/-小鼠颈动脉的纵切面的苏木精曙红染色。F、I:ApoE-/-小鼠颈动脉的纵切面的Oil Red O染色。G和J:ApoE-/-小鼠颈动脉的纵切面的CD-68抗体染色。B:内膜/介质比的定量(n = 10)。C:Oil Red O阳性染色定量(n = 10)。D:CD68抗体染色定量(n = 10)。给小鼠施用媒介物(E、F和G)或100µg/kg的化合物IIa (H、I和J),持续2周。
图18:P2Y13沉默对ApoE-/-小鼠中的粥样硬化斑块进展的影响。在结扎左颈动脉之前3天,用编码空载体(模拟)或编码P2Y13r shRNA的载体的5x109腺病毒颗粒感染ApoE-/-小鼠(n = 10)。在外科手术当天,给动物饲喂西餐,并且还通过经口管饲法施用媒介物或100µg/kg的化合物IIa,每天1次,持续2周。A:用抗-P2Y13r或抗-P2Y1r抗体印迹的肝匀浆物的蛋白质印迹。B:在2:1氯仿/甲醇中提取结扎的颈动脉的脂质。通过HPLC,测量未酯化胆固醇(黑色条)和总胆固醇(叠加的灰色条)的浓度。C:使用酶试剂盒,测量小鼠血浆中的酯化胆固醇(黑色条)和总胆固醇(叠加的灰色条) 浓度。D、E和F:使用Superose 6柱和在线酶促检测,通过HPLC分析血浆VLDL胆固醇、LDL胆固醇和HDL胆固醇含量。黑色条代表未酯化胆固醇,灰色条代表酯化的胆固醇。*p < 0.05,** p < 0.001。
图19:P2Y13激动剂对apoE-/-小鼠的主动脉中的粥样硬化斑块进展的影响。给ApoE-/-小鼠(n = 20) 饲喂西餐持续8周,然后通过经口管饲法施用媒介物或100µg/kg化合物IIa持续4周。在2:1氯仿/甲醇中提取主动脉(n = 10) 的脂质。A:通过HPLC和GC/MS,测量总胆固醇的浓度,并与作为基线的正常食物饮食(Chow diet)的平行的apoE-/-小鼠(n = 10)进行对比。B:内膜/介质比的定量(n = 10)。C:F4/80抗体染色定量(n = 10)。D:Oil Red O阳性染色定量(n = 10)。E:Sirius Red染色定量(n = 10)。F:抗-VCAM1抗体染色定量(n = 10)。将定量表示为,染色相对于内膜和介质区域的总和的百分比。*p < 0.05。G至L:用于定量的主动脉切片的染色的典型例子。G和J:apoE-/-小鼠主动脉的横断切片的苏木精曙红染色。H和K:apoE-/-小鼠主动脉的横断切片的Oil Red O染色。I和L:apoE-/-小鼠主动脉的横断切片的VCAM1抗体染色。
图20:在用化合物IIa治疗的兔子中的斑块消退。在高胆固醇饮食2个月以后,新西兰兔(n = 15)形成粥样硬化斑块,通过经口管饲法,以30、100和300µg/kg的化合物IIa进行治疗,每天1次,持续4周。A:通过GC/MS,分析从主动脉提取的脂类的胆固醇浓度。黑色条,未酯化胆固醇;叠加的灰色条,总胆固醇。*p < 0.05。B、C和D:如图2所述,分析血浆VLDL胆固醇、LDL胆固醇和HDL胆固醇。将结果表示为相对于给药前的百分比。E:通过蛋白质印迹分析测得的血浆中的apoB浓度。F:使用得自Biolabo的试剂盒,测量血浆中的甘油三酯。G:使用酶试剂盒,测定肝脏中的胆汁酸浓度。图H和K:得自施用媒介物(H)或300µg/kg的化合物IIa (K)的兔的主动脉的苏木精/伊红染色。图I和L:得自施用媒介物(I)或300µg/kg的化合物IIa (L)的兔的主动脉的平滑肌细胞染色。图J和M:得自施用媒介物(J)或300µg/kg的化合物IIa (M)的兔的主动脉的巨噬细胞和单核细胞染色。
图21:得自用化合物IIa治疗的高胆固醇饮食饲喂的兔的血浆的官能度(functionality)。A:通过SELDI-TOF分析,测量血浆中的ApoA-I浓度。使用得自智人的纯化的ApoA-I (MWSELDI = 28083 Da) 作为参照,用于测定兔ApoA-I (MWSELDI = 27838 Da) 浓度。将结果表示为相对于给药前的百分比。B:使用QPCR技术,测定ApoA-I mRNA水平。观察到治疗的动物的HDL粒度的总体下降。C:使用Lipoprint®系统,根据不同的HDL颗粒的大小,从兔血浆中分离出HDL。将数据表示为每个HDL亚群集合相对于给药前动物的HDL群体的百分比。量化2个主要的HDL颗粒亚群(高和中间——分别是黑色和灰色条)。D:使用Lipoprint®分离技术,用媒介物(灰色线)和用化合物IIa (300µg/kg,黑色线)治疗的兔子的脂蛋白分布的实施例。E和F:使用预载的[3H]-胆固醇-oxLDL巨噬细胞,分别测定血浆和兔HDL的胆固醇流出能力。将结果表示为胆固醇流出的百分比。*p < 0.05,**p < 0.005。
具体实施方式
I. 定义
下述定义与在本文中公开的发明结合使用:
除非另有定义,本文使用的术语“烷基”表示,通过从烷烃除去氢原子而衍生出的直链、支链或环状饱和基团。代表性的直链烷基包括:-甲基、-乙基、-正丙基、-正丁基、-正戊基和正庚基。代表性的支链烷基包括:-异丙基、-仲丁基、-异丁基、-叔丁基、-异戊基、-新戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、1,1-二甲基丙基和1,2-二甲基丙基。代表性的环烷基包括环己基、环戊基和环丙基。
术语“烯基”表示,含有至少一个双键的直链、支链或环状烃基。代表性的烯基包括,但不限于:乙烯、丙烯、1-丁烯、2-丁烯、异丁烯、仲丁烯、1-戊烯、2-戊烯、异戊烯、1-己烯、2-己烯、3-己烯和异己烯。
术语“炔基”表示,含有至少一个三键的直链或支链烃。代表性的炔基包括,但不限于:乙炔、丙炔、1-丁炔、2-丁炔、异丁炔、仲丁炔、1-戊炔、2-戊炔、异戊炔、1-己炔、2-己炔、3-己炔和异己炔。
除非另有定义,本文使用的术语“烃基”表示,通过从烃分子除去氢原子而衍生出的取代基。烃基的非限制性例子包括:烷基、烯基、炔基;由氢和碳组成的环状基团,诸如本文所述的芳基,包括本文所述的芳族和非芳族基团;和本文所述的芳烷基。
除非另有定义,本文使用的术语“芳基”表示芳族基团。芳基的非限制性例子包括:苯基、萘基、吡啶基、菲基、蒽基、呋喃基、吖唑基(azolyl)、咪唑基和吲哚基。在一个实施方案中,所述芳基被下述基团中的一个或多个取代:-卤素、-O-(C1-C6烷基)、-OH、-CN、-COOR'、-OC(O)R'、-N(R')2、-NHC(O)R'或-C(O)NHR' 基团,其中每个R' 独立地是-H或未被取代的–C1-C6烷基。除非另有说明,所述芳基未被取代。
除非另有定义,本文使用的术语“杂芳基”表示芳族基团,其中所述芳族基团含有至少一个非碳的环原子。杂芳基的非限制性例子包括:吡啶基、呋喃基、吖唑基(azolyl)、咪唑基、噻吩基和吲哚基。在一个实施方案中,所述芳基被下述基团中的一个或多个取代:-卤素、-O-(C1-C6烷基)、-OH、-CN、-COOR'、-OC(O)R'、-N(R')2、-NHC(O)R'或-C(O)NHR'基团,其中每个R' 独立地是-H或未被取代的–C1-C6烷基。除非另有说明,所述杂芳基未被取代。
除非另有定义,本文使用的术语“芳烷基”表示,被芳基取代的烷基。芳烷基的非限制性例子包括苯甲基、吡啶甲基、萘基甲基。
除非另有定义,本文使用的术语“杂环基”表示环状基团,其中所述环状基团含有至少一个非碳的环原子。杂环基的代表性例子包括,但不限于:呋喃基、呋咱基、咪唑烷基、咪唑啉基、咪唑基、异噻唑基、异噁唑基、吗啉基、噁二唑基、噁唑烷基、噁唑基、噁唑烷基、嘧啶基、菲啶基、菲咯啉基、哌嗪基、哌啶基、吡喃基、吡嗪基、吡唑烷基、吡唑啉基、吡唑基、哒嗪基、吡啶并噁唑、吡啶并咪唑、吡啶并噻唑、吡啶基、嘧啶基、吡咯烷基、吡咯啉基、奎宁环基、四氢呋喃基、噻二嗪基、噻二唑基、噻吩基、噻吩并噻唑基、噻吩并噁唑基、噻吩并咪唑基、硫代吗啉基、噻吩基、三嗪基、三唑基。在一个实施方案中,所述芳基被下述基团中的一个或多个取代:-卤素、-O-(C1-C6烷基)、-OH、-CN、-COOR'、-OC(O)R'、-N(R')2、-NHC(O)R'或-C(O)NHR'基团,其中每个R' 独立地是-H或未被取代的–C1-C6烷基。除非另有说明,所述杂环基未被取代。
除非另有定义,本文使用的术语“烷氧基”表示-O-(烷基),其中烷基如上所定义。C1-C6烷氧基的代表性例子包括,但不限于:–OCH3、-OCH2CH3、-OCH2CH2CH3、-OCH(CH3)CH3、-OCH2CH2CH2CH3、-OCH2CH(CH3)CH3、-OCH(CH3)CH2CH3、-OC(CH3)3、-OCH2CH2CH2CH2CH3、-OCH2CH(CH3)CH2CH3、-OCH2CH2CH2CH2CH2CH3和-OCH2CH2CH(CH3)CH2CH3。
除非另有定义,本文使用的术语“卤代”和“卤素”表示-F、-Cl、-Br或-I。
除非另有定义,本文使用的术语“受试者”是哺乳动物,例如,人、小鼠、大鼠、豚鼠、狗、猫、马、牛、猪或非人灵长类动物,诸如猴、黑猩猩或狒狒。在一个实施方案中,所述受试者是人。
除非另有定义,本文使用的术语“药学上可接受的盐”是,在本发明的化合物上的碱性基团(诸如氨基)或酸性基团(诸如羧基)的盐。碱性基团的示例性的盐包括,但不限于,硫酸盐、柠檬酸盐、乙酸盐、草酸盐、氯化物、溴化物、碘化物、硝酸盐、硫酸氢盐、磷酸盐、酸式磷酸盐、异烟酸盐、乳酸盐、水杨酸盐、酸式柠檬酸盐、酒石酸盐、油酸盐、鞣酸盐、泛酸盐、酒石酸氢盐、抗坏血酸盐、琥珀酸盐、马来酸盐、龙胆酸盐(gentisinate)、富马酸盐、葡糖酸盐、葡糖醛酸盐(glucaronate)、蔗糖酸盐、甲酸盐、苯甲酸盐、谷氨酸盐、甲磺酸盐、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐、樟脑磺酸盐和扑酸盐(即,1,1'-亚甲基-二-(2-羟基-3-萘甲酸盐))盐。酸性基团的示例性的盐包括,但不限于,锂盐、钠盐、钾盐、钙盐、镁盐、铝盐、铬盐、铁盐、铜盐、锌盐、镉盐、铵盐、胍盐、吡啶鎓盐和有机铵盐。
除非另有定义,本文使用的术语“水合物”和“溶剂化物”描述了本发明的化合物或其盐,其另外包括通过非共价分子间力结合的化学计量的或非化学计量的量的水或其它溶剂。
除非另有定义,本文使用的术语“胆固醇逆向转运”(RCT) 描述了胆固醇从肝外组织向肝脏的运输,所述胆固醇在肝脏处分解和消除。HDL颗粒可以在反向运输过程中起重要作用,起组织胆固醇的清除剂的作用。
除非另有定义,本文使用的术语“改变脂类代谢”是指脂类代谢的至少一方面的可观测(可测量)的变化,包括但不限于血液脂质总含量、血液HDL胆固醇、血液LDL胆固醇、血液VLDL胆固醇、血液甘油三酸酯(TG)、血液Lp(a)、血液Apo A-I、血液Apo E以及血液未酯化脂肪酸(NEFA)。
除非另有定义,本文使用的“改变葡萄糖代谢”是指葡萄糖代谢的至少一方面的可观测(可测量)的变化,包括但不限于血液葡萄糖总含量、血液胰岛素、血液胰岛素对血液葡萄糖比例、胰岛素敏感性以及氧耗量。
当与本发明的化合物结合使用时,“有效量”是可有效地治疗或预防本文所述病症的量。
当与其它治疗剂结合使用时,“有效量”是与本发明的化合物相组合可有效地治疗或预防病症的量。“与……组合”包括在相同组合物内和通过分开的组合物施用;在后一种情况下,其它治疗剂可在本发明的化合物发挥它的预防或治疗效果的时间内有效地治疗或预防病症,或反之亦然。
措辞“基本上不含有它的对应的相反对映异构体”是指,含有下述量的它的对应的相反对映异构体:不超过约10 mol%,在另一个实施方案中不超过约5 mol%,在另一个实施方案中不超过约2 mol%,在另一个实施方案中不超过约1 mol%,在另一个实施方案中不超过约0.5 mol%,和在另一个实施方案中不超过约0.1 mol%。
措辞“基本上不含有其它立体异构体”是指,含有下述量的其它立体异构体:不超过约10 mol%,在另一个实施方案中不超过约5 mol%,在另一个实施方案中不超过约2 mol%,在另一个实施方案中不超过约1 mol%,在另一个实施方案中不超过约0.5 mol%,和在另一个实施方案中不超过约0.1 mol%。
当与参考的数值表示结合使用时,术语“约”是指,所述数值表示±参考的数值表示的最多10%。例如,措辞“约50”覆盖45至55的范围。
本文使用的术语“老年人”表示65岁或以上的人。
本文使用的术语“成年人”表示18岁或以上的人。
本文使用的术语“儿童”表示1-18岁的人。
本文使用的术语“幼儿”表示1-3岁的人。
本文使用的术语“婴儿”表示新生儿至1岁的人。
本文使用的术语“早产婴儿”表示孕龄小于37周出生的婴儿。
本文使用的术语“Apo(a)”表示载脂蛋白(a)。
本文使用的术语“Apo A-I”表示载脂蛋白A-I。
本文使用的术语“Apo B”表示载脂蛋白B。
本文使用的术语“Apo E”表示载脂蛋白E。
本文使用的术语“FA”表示脂肪酸。
本文使用的术语“HDL”表示高密度脂蛋白。
本文使用的术语“IDL”表示中密度脂蛋白。
本文使用的术语“IDDM”表示胰岛素依赖性的糖尿病。
本文使用的术语“LDH”表示乳酸脱氢酶。
本文使用的术语“LDL”表示低密度脂蛋白。
本文使用的术语“Lp(a) ”表示脂蛋白(a)。
本文使用的术语“NIDDM”表示非胰岛素依赖性的糖尿病。
本文使用的术语“NEFA”表示未酯化的脂肪酸。
本文使用的术语“P2Y13”表示GPCR受体。
本文使用的术语“P2Y13r”表示P2Y13受体。
本文使用的术语“P2Y13r”和“P2Y13受体”互换地使用。
本文使用的术语“RXR”表示类视色素X受体。
本文使用的术语“TG”表示甘油三酯。
本文使用的术语“VLDL”表示极低密度脂蛋白。
II.本发明的化合物
在一个实施方案中,本发明提供了下式I的化合物:
及其药学上可接受的盐,其中
R1、R2和R3中的每一个独立地是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R4是-H、-OH、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、-烃基、-O-烃基、-芳基、-O-芳基、-芳烷基、-O-芳烷基、-杂芳基、-O-杂芳基、-杂环基、-O-杂环基、-卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基);
Z1是CH2、S、O、NH、N-烃基、N-芳基、N-杂芳基或N-杂环基;
Z2是CH或N;且
n是1-6的整数。
在有些实施方案中,式I的化合物是这样的化合物:其中Z2是N。在其它实施方案中,Z2是CH。
在有些实施方案中,R1是卤素。在其它实施方案中,R1是氯代。在其它实施方案中,R1是2-卤素。在其它实施方案中,R1是2-氯。在其它实施方案中,R1是H。
在有些实施方案中,Z1是S。在其它实施方案中,Z1是O。在其它实施方案中,Z1是NH。在其它实施方案中,Z1是N-烷基。
在有些实施方案中,R2和R3中的每一个独立地是H或烷基。在其它实施方案中,R2和R3中的每一个是H。在其它实施方案中,R2和R3中的每一个是烷基。在其它实施方案中,R2和R3中的每一个是甲基。
在有些实施方案中,R4是-OH、-COOH、-C(O)O(烷基)或-OC(O)(烷基)。在其它实施方案中,R4是-OH。在其它实施方案中,R4是-COOH。在其它实施方案中,R4是-C(O)O(烷基)或-OC(O)(烷基)。在其它实施方案中,R4是-COOEt。在其它实施方案中,R4是-COOMe。
在有些实施方案中,n是1-6的整数。在其它实施方案中,n是1。在其它实施方案中,n是2。在其它实施方案中,n是3。在其它实施方案中,n是4。在其它实施方案中,n是5。在其它实施方案中,n是6。
在有些实施方案中,R2和R3中的每一个是烷基,且R4是-C(O)O-烷基。在其它实施方案中,R2和R3中的每一个是甲基,且R4是-C(O)O-烷基。在其它实施方案中,R2和R3中的每一个是烷基,且R4是-C(O)OEt。在其它实施方案中,R2和R3中的每一个是甲基,且R4是-C(O)OEt。
在有些实施方案中,R2和R3中的每一个是H,且R4是-C(O)O-烷基。在其它实施方案中,R2和R3中的每一个是H,且R4是-C(O)OEt。
在有些实施方案中,R2和R3中的每一个是烷基,且R4是-C(O)OH。在其它实施方案中,R2和R3中的每一个是甲基,且R4是-C(O)OH。
在有些实施方案中,R2和R3中的每一个是H,且R4是-C(O)OH。
在有些实施方案中,R2和R3中的每一个是烷基,且R4是-OH。在其它实施方案中,R2和R3中的每一个是甲基,且R4是-OH。
在有些实施方案中,R2和R3中的每一个是H,且R4是-OH。
在有些实施方案中,式I化合物的Z1和Z2是下述的:
在有些实施方案中,式I化合物具有结构:
或前述任一种的药学上可接受的盐。
在另一个实施方案中,本发明提供了下式II的化合物:
及其药学上可接受的盐,其中
每个R9独立地是-H、-烃基、-芳基、-芳烷基、-杂芳基、-杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烷基、-O-烯基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、NHC(O)(C2-C10-烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
Q1、Q2和Q3中的每一个独立地是CR10或N;
X是CHR10、S、O或NR9;且
每个m独立地是0-3的整数。
在有些实施方案中,式II化合物是这样的化合物:其中R9是H或烃基。在其它实施方案中,R9是H。在其它实施方案中,R9是烃基。在其它实施方案中,R9是烷基。在其它实施方案中,R9是甲基。在其它实施方案中,R9是乙基。在其它实施方案中,R9是苯基。
在有些实施方案中,式II化合物是这样的化合物:其中R10是H、-OH或烃基。在其它实施方案中,R10是H。在其它实施方案中,R10是-OH。在其它实施方案中,R10是-OMe。在其它实施方案中,R10是-OEt。在其它实施方案中,R10是-NH2。在其它实施方案中,R10是-NHMe。在其它实施方案中,R10是-NMe2。在其它实施方案中,R10是烃基。在其它实施方案中,R10是烷基。在其它实施方案中,R10是甲基。在其它实施方案中,R10是乙基。在其它实施方案中,R10是苯基。
在另一个实施方案中,R9是H,且R10是-OH。
在有些实施方案中,式II化合物是这样的化合物:其中Q1、Q2和Q3中的每一个是N。在其它实施方案中,Q1、Q2和Q3中的每一个是CR10。在其它实施方案中,Q1、Q2中的每一个是N,且Q3是CR10。
在有些实施方案中,式II化合物是这样的化合物:其中X是CH2。在其它实施方案中,X是O。在其它实施方案中,X是NH。在其它实施方案中,X是NMe。在其它实施方案中,X是N-苯甲基。
在有些实施方案中,式II化合物是这样的化合物:其中每个m独立地是0-3的整数。在其它实施方案中,每个m独立地是1-3的整数。在其它实施方案中,m是0。在其它实施方案中,m是1。在其它实施方案中,m是2。在其它实施方案中,m是3。
在其它实施方案中,Q1和Q3是N,Q2是CR10,且R10是-N(烷基)(烷基)。在其它实施方案中,Q1和Q3是N,Q2是CR10,且R10是-N(H)(烷基)。在其它实施方案中,Q1和Q3是N,Q2是CR10,且R10是-N(CH3)2。在其它实施方案中,Q1和Q3是N,Q2是CR10,且R10是-N(H)(CH3)。
在其它实施方案中,每个m是1,X是NR9,且R9是H。在其它实施方案中,每个m是1,且X是O。
在有些实施方案中,式II化合物的Q1、Q2、Q3和X是下述的:
在有些实施方案中,式II的化合物作为单一互变异构体或互变异构体的混合物存在。本领域技术人员会认识到具有互变异构形式的结构,且会理解,单一互变异构体的例证暗示所有可能的互变异构形式的结构。在有些实施方案中,R10是OH,且式II化合物以式II的1、2或3种互变异构形式存在,如方程式I所示。
方程式I
在有些实施方案中,式II的化合物具有下述结构:
或前述任一种的药学上可接受的盐。
在另一个实施方案中,本发明包括下式III的化合物:
及其药学上可接受的盐,其中
R1、R2和R3中的每一个独立地是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R5是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基或-O-杂环基;
Z1是CH2、S、O、NH、N-烃基、N-芳基、N-杂芳基或N-杂环基;
Z2是CH或N;且
n是1-6的整数。
在有些实施方案中,式III化合物是这样的化合物:其中Z2是N。在其它实施方案中,Z2是CH。
在有些实施方案中,R1是卤素。在其它实施方案中,R1是氯代。在其它实施方案中,R1是2-卤素。在其它实施方案中,R1是2-氯。在其它实施方案中,R1是H。
在有些实施方案中,Z1是S。在其它实施方案中,Z1是O。在其它实施方案中,Z1是NH。在其它实施方案中,Z1是N-烷基。
在有些实施方案中,R2和R3中的每一个独立地是H或烷基。在其它实施方案中,R2和R3中的每一个是H。在其它实施方案中,R2和R3中的每一个是甲基。
在有些实施方案中,R5是-OH、-O-烷基或-O-芳烷基。在其它实施方案中,R5是-OH。在其它实施方案中,R5是-O-苯甲基。在其它实施方案中,R5是-OEt。在其它实施方案中,R5是-OMe。
在有些实施方案中,n是1-6的整数。在其它实施方案中,n是1。在其它实施方案中,n是2。在其它实施方案中,n是3。在其它实施方案中,n是4。在其它实施方案中,n是5。在其它实施方案中,n是6。
在有些实施方案中,式III化合物的Z1和Z2是下述的:
在另一个实施方案中,本发明提供了下式IV的化合物:
及其药学上可接受的盐,其中
R1是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
Z1是CH2、S、O、NH、N-烃基、N-芳基、N-杂芳基或N-杂环基;和
Z2是CH或N。
在有些实施方案中,式IV的化合物是这样的化合物:其中Z2是N。在其它实施方案中,Z2是CH。
在有些实施方案中,R1是卤素。在其它实施方案中,R1是氯代。在其它实施方案中,R1是2-卤素。在其它实施方案中,R1是2-氯。在其它实施方案中,R1是H。
在有些实施方案中,Z1是S。在其它实施方案中,Z1是O。在其它实施方案中,Z1是NH。在其它实施方案中,Z1是N-烷基。
在有些实施方案中,式IV化合物的Z1和Z2是下述的:
在有些实施方案中,式IV化合物具有下述结构:
IVa: ,或其药学上可接受的盐。
在一个实施方案中,本发明包括下式V的化合物:
及其药学上可接受的盐,其中
R1、R2和R3中的每一个独立地是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R4是-H、-OH、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基)、;
R6是-H、-OH、-SH、-S-烃基、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基);
R7是H、烃基、芳基、芳烷基、杂芳基或杂环基;
Z2是CH或N;且
n是1-6的整数。
在有些实施方案中,式V的化合物是这样的化合物:其中Z2是N。在其它实施方案中,Z2是CH。
在有些实施方案中,R1是卤素。在其它实施方案中,R1是氯代。在其它实施方案中,R1是2-卤素。在其它实施方案中,R1是2-氯。在其它实施方案中,R1是H。
在有些实施方案中,R2和R3中的每一个独立地是H或烷基。在其它实施方案中,R2和R3中的每一个是H。在其它实施方案中,R2和R3中的每一个是烷基。在其它实施方案中,R2和R3中的每一个是甲基。
在有些实施方案中,R4是-OH、-COOH、-C(O)O(烷基)或-OC(O)(烷基)。在其它实施方案中,R4是-OH。在其它实施方案中,R4是-COOH。在其它实施方案中,R4是-C(O)O(烷基)或-OC(O)(烷基)。在其它实施方案中,R4是-COOEt。在其它实施方案中,R4是-COOMe。
在有些实施方案中,R6是-OH、-O-烷基、-SH、-S-烷基或烷基。在其它实施方案中,R6是-OH。在其它实施方案中,R6是-OMe。在其它实施方案中,R6是H。在其它实施方案中,R6是甲基。在其它实施方案中,R6是SH。在其它实施方案中,R6是-SMe。
在有些实施方案中,R7是H。在其它实施方案中,R7是甲基。在其它实施方案中,R7是乙基。在其它实施方案中,R7是苯甲基。
在有些实施方案中,n是1-6的整数。在其它实施方案中,n是1。在其它实施方案中,n是2。在其它实施方案中,n是3。在其它实施方案中,n是4。在其它实施方案中,n是5。在其它实施方案中,n是6。
在有些实施方案中,式V的化合物具有下述结构:
Va: ,或其药学上可接受的盐。
在一个实施方案中,本发明包括下式VI的化合物:
及其药学上可接受的盐,其中
R1是H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R5是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基或卤素;
R6是-H、-OH、-SH、-S-烃基、-COOH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)或-OC(O)N(烷基)(烷基);
R7是H、烃基、芳基、芳烷基、杂芳基或杂环基;和
Z2是CH或N。
在有些实施方案中,式VI的化合物是这样的化合物:其中Z2是N。在其它实施方案中,Z2是CH。
在有些实施方案中,R1是卤素。在其它实施方案中,R1是氯代。在其它实施方案中,R1是2-卤素。在其它实施方案中,R1是2-氯。在其它实施方案中,R1是H。
在有些实施方案中,R5是OH、O-烷基或O-芳烷基。在其它实施方案中,R5是OH。在其它实施方案中,R5是O-苯甲基。在其它实施方案中,R5是OEt。在其它实施方案中,R5是OMe。
在有些实施方案中,R6是-OH、-O-烷基、-SH、-S-烷基或烷基。在其它实施方案中,R6是-OH。在其它实施方案中,R6是-OMe。在其它实施方案中,R6是-H。在其它实施方案中,R6是甲基。在其它实施方案中,R6是-SH。在其它实施方案中,R6是-SMe。
在有些实施方案中,R7是H。在其它实施方案中,R7是甲基。在其它实施方案中,R7是乙基。在其它实施方案中,R7是苯甲基。
在有些实施方案中,式VI的化合物具有下述结构:
VIa: ; VIb: ,或其药学上可接受的盐。
在一个实施方案中,本发明包括下式VII的化合物:
及其药学上可接受的盐,其中
R1a、R1b和R1c中的每一个独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
每个X独立地是CHR10、S、O或NR9;
每个R9独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;且
m是0-3的整数。
在有些实施方案中,R1a、R1b和R1c中的一个或多个是卤素。在其它实施方案中,R1a、R1b和R1c中的一个或多个是氯代。在其它实施方案中,R1a、R1b和R1c中的每一个是H。
在有些实施方案中,X是CH2。在其它实施方案中,X是O。在其它实施方案中,X是NH。在其它实施方案中,X是NMe。在其它实施方案中,X是N-苯甲基。
在有些实施方案中,R9是H或烃基。在其它实施方案中,R9是H。在其它实施方案中,R9是烃基。在其它实施方案中,R9是烷基。在其它实施方案中,R9是甲基。在其它实施方案中,R9是乙基。在其它实施方案中,R9是苯基。
在有些实施方案中,R10是H、-OH或烃基。在其它实施方案中,R10是H。在其它实施方案中,R10是-OH。在其它实施方案中,R10是-OMe。在其它实施方案中,R10是-OEt。在其它实施方案中,R10是-NH2。在其它实施方案中,R10是-NHMe。在其它实施方案中,R10是-NMe2。在其它实施方案中,R10是烃基。在其它实施方案中,R10是烷基。在其它实施方案中,R10是甲基。在其它实施方案中,R10是乙基。在其它实施方案中,R10是苯基。
在有些实施方案中,m是0-3的整数。在其它实施方案中,m是1-3的整数。在其它实施方案中,m是0。在其它实施方案中,m是1。在其它实施方案中,m是2。在其它实施方案中,m是3。
在有些实施方案中,本发明包括下式VII-1的化合物:
其中R1a、R1b、R1c、m、R9和R10如上面所定义,且X1和X2如上面的X所定义。
在有些实施方案中,式VII-1化合物的X1和X2是下述的:
在有些实施方案中,式VII化合物具有下述结构:
VIIa: ,或其药学上可接受的盐。
在另一个实施方案中,本发明包括下式VIII的化合物:
或其药学上可接受的盐,其中
R1是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R11是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
Q1、Q2、Q3和Q4中的每一个独立地是CR10或N;且
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2。
在有些实施方案中,R1是烷基。在其它实施方案中,R1是2-烷基。
在其它实施方案中,Q1是H、Q2是CR10,且R10是-OH和Q3是CR10,且R10是O-烷基。在其它实施方案中,Q1是CR10,且R10是-O-烷基、Q2是CR10,且R10是-OH和Q3是H。
在其它实施方案中,Q4是N。在其它实施方案中,Q4是CR10。在其它实施方案中,Q4是C(H)。
在有些实施方案中,R10是H、-OH或烃基。在其它实施方案中,R10是H。在其它实施方案中,R10是-OH。在其它实施方案中,R10是-OMe。在其它实施方案中,R10是-OEt。在其它实施方案中,R10是-NH2。在其它实施方案中,R10是-NHMe。在其它实施方案中,R10是-NMe2。在其它实施方案中,R10是烃基。在其它实施方案中,R10是烷基。在其它实施方案中,R10是甲基。在其它实施方案中,R10是乙基。在其它实施方案中,R10是苯基。
在有些实施方案中,R11是H。在其它实施方案中,R11是甲基。在其它实施方案中,R11是乙基。在其它实施方案中,R11是苯甲基。
在有些实施方案中,式VIII化合物的Q1、Q2、Q3和Q4如下所定义:
在有些实施方案中,式VIII化合物具有下述结构:
VIIIc: ,或其药学上可接受的盐。
在另一个实施方案中,本发明包括下式IX的化合物:
及其药学上可接受的盐,其中
R11a、R11b和R11c中的每一个独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)(烷基)、-C(O)O(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)或-SO2NH2;
X1和X2中的每一个独立地是CHR10、S、O、NR9或N-酰基;
每个R9独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;且
每个m独立地是1-3的整数。
在有些实施方案中,X1和X2中的每一个是CH2。在其它实施方案中,X1和X2中的每一个是O。在其它实施方案中,X1和X2中的每一个是NH。在其它实施方案中,X1和X2中的每一个是NMe。在其它实施方案中,X1和X2中的每一个是N-苯甲基。
在有些实施方案中,R9是H或烃基。在其它实施方案中,R9是H。在其它实施方案中,R9是烃基。在其它实施方案中,R9是烷基。在其它实施方案中,R9是甲基。在其它实施方案中,R9是乙基。在其它实施方案中,R9是苯基。
在有些实施方案中,R10是H、-OH或烃基。在其它实施方案中,R10是H。在其它实施方案中,R10是-OH。在其它实施方案中,R10是-OMe。在其它实施方案中,R10是-OEt。在其它实施方案中,R10是-NH2。在其它实施方案中,R10是-NHMe。在其它实施方案中,R10是-NMe2。在其它实施方案中,R10是烃基。在其它实施方案中,R10是烷基。在其它实施方案中,R10是甲基。在其它实施方案中,R10是乙基。在其它实施方案中,R10是苯基。
在有些实施方案中,R11a、R11b和R11c中的每一个是H。在其它实施方案中,R11a、R11b和R11c中的一个或多个是甲基。在其它实施方案中,R11a、R11b和R11c中的一个或多个是乙基。在其它实施方案中,R11a、R11b和R11c中的一个或多个是苯甲基。在其它实施方案中,R11a、R11b和R11c中的一个或多个是乙酰基。在其它实施方案中,R11a、R11b和R11c中的一个或多个是苯甲酰基。
在有些实施方案中,每个m独立地是1-3的整数。在其它实施方案中,m是1。在其它实施方案中,m是2。在其它实施方案中,m是3。
在有些实施方案中,式IX化合物的X1和X2是下述的:
本文公开的化合物可以包括一个或多个手性中心和/或双键,并且因此作为立体异构体(诸如几何异构体、对映异构体或非对映异构体)存在。本发明的化合物在本文中的例证包括所有可能的异构体的混合物或纯化形式。纯化形式可以是几何富集的、对映异构富集的、非对映异构富集的、光学富集的、几何纯的、对映异构纯的、非对映异构纯的、或光学纯的。混合物可以包括异构体的任意组合,诸如对映异构的、非对映异构的、外消旋的和立体异构的混合物。在一个实施方案中,本发明的化合物作为基本上不含有其它立体异构体的单一立体异构体存在。在另一个实施方案中,本发明的化合物作为基本上不含有它的对应的相反对映异构体的单一对映异构体存在。在另一个实施方案中,本发明的化合物作为外消旋体存在。
使用本领域技术人员已知的方法,包括手性高效液相色谱法、选择性结晶和与手性拆分剂或手性助剂反应,可以得到、分离或纯化本发明的化合物,使得它们是外消旋的、基本上不含有其它立体异构体或基本上不含有对应的相反对映异构体。
在一个实施方案中,本发明的化合物是下式X的化合物或所述化合物的药学上可接受的盐:
其中
每个R9独立地是-H、-烃基、-芳基、-芳烷基、-杂芳基、-杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O--芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
Q1、Q2和Q3中的每一个独立地是CR10或N;
X是CHR10、S、O或NR9;且
每个m独立地是0-3的整数。
在有些实施方案中,式X化合物的Q1、Q2、Q3和X是下述的:
在一个实施方案中,本发明的化合物是下式XI的化合物或所述化合物的药学上可接受的盐:
及其药学上可接受的盐,其中
R11a、R11b和R11c中的每一个独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)(烷基)、-C(O)O(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)或-SO2NH2;
X1和X2中的每一个独立地是CHR10、S、O、NR9或N-酰基;
每个R9独立地是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;且
每个m独立地是0-3的整数。
在有些实施方案中,式XI化合物的X1和X2是下述的:
在一个实施方案中,式XI的化合物具有下述结构:
XIa: ,或其药学上可接受的盐。
在另一个实施方案中,本发明提供了下式XII的化合物:
及其药学上可接受的盐,其中
每个R9独立地是-H、-烃基、-芳基、-芳烷基、-杂芳基、-杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烷基、-O-烯基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、NHC(O)(C2-C10-烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)、-SO2NH2、–S-烷基、-S-芳基、-S-杂芳基、-S-杂环或–S-烃基;
Q1、Q2和Q3中的每一个独立地是CR10或N;
X是CHR10、S、O或NR9;且
每个m独立地是0-3的整数。
在有些实施方案中,式XII的化合物是这样的化合物:其中R9是H或烃基。在其它实施方案中,R9是H。在其它实施方案中,R9是烃基。在其它实施方案中,R9是烷基。在其它实施方案中,R9是甲基。在其它实施方案中,R9是乙基。在其它实施方案中,R9是苯基。
在有些实施方案中,式XII的化合物是这样的化合物:其中R10是H、-OH或烃基。在其它实施方案中,R10是H。在其它实施方案中,R10是-OH。在其它实施方案中,R10是-OMe。在其它实施方案中,R10是-OEt。在其它实施方案中,R10是-NH2。在其它实施方案中,R10是-NHMe。在其它实施方案中,R10是-NMe2。在其它实施方案中,R10是烃基。在其它实施方案中,R10是烷基。在其它实施方案中,R10是甲基。在其它实施方案中,R10是乙基。在其它实施方案中,R10是苯基。在其它实施方案中,R10是–S-烷基
在另一个实施方案中,R9是H,且R10是-OH。
在有些实施方案中,式XII的化合物是这样的化合物:其中Q1、Q2和Q3中的每一个是N。在其它实施方案中,Q1、Q2和Q3中的每一个是CR10。在其它实施方案中,Q1、Q2中的每一个是N,且Q3是CR10。
在有些实施方案中,式XII的化合物是这样的化合物:其中X是CH2。在其它实施方案中,X是O。在其它实施方案中,X是NH。在其它实施方案中,X是NMe。在其它实施方案中,X是N-苯甲基。
在有些实施方案中,式XII的化合物是这样的化合物:其中每个m独立地是0-3的整数。在其它实施方案中,每个m独立地是1-3的整数。在其它实施方案中,m是0。在其它实施方案中,m是1。在其它实施方案中,m是2。在其它实施方案中,m是3。
在其它实施方案中,Q1和Q3是N、Q2是CR10,且R10是-N(烷基)(烷基)。在其它实施方案中,Q1和Q3是N、Q2是CR10,且R10是-N(H)(烷基)。在其它实施方案中,Q1和Q3是N、Q2是CR10,且R10是-N(CH3)2。在其它实施方案中,Q1和Q3是N、Q2是CR10,且R10是-N(H)(CH3)。
在其它实施方案中,每个m是1、X是NR9,且R9是H。在其它实施方案中,每个m是1和X是O。
在有些实施方案中,式XII化合物的Q1、Q2、Q3和X是下述的:
在有些实施方案中,式XII化合物作为单一互变异构体或作为互变异构体的混合物存在。本领域技术人员会认识到具有互变异构形式的结构,且会理解,单一互变异构体的例证暗示所有可能的互变异构形式的结构。在有些实施方案中,R10是OH,且式XII化合物以式XII的1、2或3种互变异构形式存在,如方程式II所示。
方程式II
在某些实施方案中,式XII化合物具有式II的结构,且式XII的示例性化合物包括化合物IIa–IIjj或其药学上可接受的盐。
在有些实施方案中,式XII的化合物具有下述结构:
或其药学上可接受的盐。
在另一个实施方案中,本发明提供了下式XIII的化合物:
及其药学上可接受的盐,其中
R1是独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2;
R11是H、烃基、芳基、芳烷基、杂芳基、杂环基、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)或-SO2NH2;
Q1、Q2、Q3和Q4中的每一个独立地是CR10或N;
n是1-4的整数,且
每个R10独立地是-H、-OH、-NH2、-NH(烷基)、-N(烷基)(烷基)、烃基、-O-烃基、芳基、-O-芳基、芳烷基、-O-芳烷基、杂芳基、-O-杂芳基、杂环基、-O-杂环基、卤素、-OCF3、-C(O)O(烷基)、-OC(O)(烷基)、-C(O)NH2、-C(O)NH(烷基)、-C(O)N(烷基)(烷基)、-NHC(O)(烷基)、N(烷基)C(O)(烷基)、-OC(O)O(烷基)、-OC(O)NH2、-OC(O)NH(烷基)、-OC(O)N(烷基)(烷基)、-CHNH、-CHN(烷基)或-SO2NH2。
在有些实施方案中,R1是烷基。在其它实施方案中,R1是2-烷基。
在其它实施方案中,Q1是H、Q2是CR10,且R10是-OH和Q3是CR10,且R10是O-烷基。在其它实施方案中,Q1是CR10,且R10是-O-烷基、Q2是CR10,且R10是-OH和Q3是H。
在其它实施方案中,Q4是N。在其它实施方案中,Q4是CR10。在其它实施方案中,Q4是C(H)。
在有些实施方案中,R10是H、-OH或烃基。在其它实施方案中,R10是H。在其它实施方案中,R10是-OH。在其它实施方案中,R10是-OMe。在其它实施方案中,R10是-OEt。在其它实施方案中,R10是-NH2。在其它实施方案中,R10是-NHMe。在其它实施方案中,R10是-NMe2。在其它实施方案中,R10是烃基。在其它实施方案中,R10是烷基。在其它实施方案中,R10是甲基。在其它实施方案中,R10是乙基。在其它实施方案中,R10是苯基。
在有些实施方案中,R11是H。在其它实施方案中,R11是甲基。在其它实施方案中,R11是乙基。在其它实施方案中,R11是苯甲基。
在有些实施方案中,式XIII的化合物是这样的化合物:其中n独立地是1-4的整数。在其它实施方案中,n是1。在其它实施方案中,n是2。在其它实施方案中,n是3。在其它实施方案中,n是4。
在有些实施方案中,式XIII化合物的Q1、Q2、Q3和Q4如下所定义:
在某些实施方案中,式XIII的化合物具有式VIII的结构,且式XIII的示例性化合物包括化合物VIIIa VIIIc或其药学上可接受的盐。
在有些实施方案中,式XIII的化合物具有下述结构:
,
或其药学上可接受的盐。
III. 用本发明的化合物治疗或预防病症
根据本发明,本发明的化合物可用于治疗或预防一种或多种病症。
在一个实施方案中,本发明提供了用于治疗或预防一种或多种病症的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。
在另一个实施方案中,本发明提供了一种或多种本发明的化合物在药物制备中的用途,所述药物可用于治疗或预防一种或多种病症。
在另一个实施方案中,本发明提供了用于治疗或预防一种或多种病症的一种或多种本发明的化合物。
通过施用一种或多种本发明的化合物可治疗或可预防的病症的非限制性例子包括:(i) 脂蛋白代谢障碍,包括,血脂异常、异常脂蛋白血症、脂蛋白过度产生或缺乏、总胆固醇升高、低密度脂蛋白浓度升高、甘油三酯浓度升高、胆汁中的脂质消除、代谢障碍、胆汁中的磷脂消除、胆汁中的氧化固醇消除和过氧化物酶体增殖物活化的受体-相关的障碍;(ii) 葡萄糖代谢障碍,包括胰岛素抵抗、葡萄糖耐量降低、空腹血糖水平降低、糖尿病、脂肪营养不良、向心性肥胖、外周脂肪萎缩、糖尿病肾病、糖尿病视网膜病变、肾病和败血症;(iii) 心血管障碍或相关的血管障碍,包括高血压、冠状动脉疾病、心肌梗塞、心律失常、心房颤动、心脏瓣膜疾病、心力衰竭、心肌病、心包炎和阳萎;和(iv) 涉及C-反应蛋白的异常调节的病症或相关的病症,包括炎症、缺血性坏死、结肠癌和血栓性障碍;和(v) 老化、阿尔茨海默氏病、帕金森病、胰腺炎(pancreatitis)、胰脏炎(pancreatitius)和异常的胆汁产生。
本发明另外提供了用于鉴别一种或多种病症的生物标志物的方法,所述方法包括:将有效量的本发明的化合物施用给有此需要的受试者,并测量所述受试者的血液中的未酯化胆固醇的水平。在一个实施方案中,所述病症是心血管相关病症。在另一个实施方案中,所述生物标志物是从动脉血管向干燥的胆固醇逆向转运的存在或胆汁酸中的胆固醇消除或二者。
本发明另外提供了游离胆固醇作为P2Y13活化的生物标志物的用途,所述P2Y13活化以HDL的功能化(functionalisation)(即,它的胆固醇逆向转运剂效能增加)为后果。因此,本发明另外提供了用于测定P2Y13活化程度的方法,所述方法包括:将有效量的本发明的化合物施用给有此需要的受试者,并测量所述受试者的血液中的游离胆固醇的量。
本发明的化合物及其组合物可以口服给药。本发明的化合物及其组合物还可以通过任何其它常规途径给药,例如通过静脉内输注或快速推注、经上皮或皮肤粘膜衬里(例如口腔粘膜、直肠和肠粘膜等)吸收,而且可以与另一种生物活性剂一起用药。给药可以是全身性的或局部的。各种递送系统是已知的,例如脂质体包囊、微粒、微囊、胶囊等,而且均可以用于施用本发明化合物。在某些实施方案中,给受试者施用一种以上本发明化合物。给药方法包括但不限于真皮内、肌肉内、腹膜内、静脉内、皮下、鼻内、硬膜外、口服、舌下、鼻内、大脑内、阴道内、透皮、直肠、吸入或局部给药,尤其是局部用于耳、鼻、眼或皮肤。给药模式可以由从业人员决定,而且部分取决于医病部位。大多数情况下,给药使得本发明化合物释放进血流中。
在具体实施方案中,可能需要将一种或多种本发明化合物局部施用到需要治疗的区域。这可以如下实现:例如,但不限于,在手术期间通过局部输注;局部施用(如在术后与创伤敷料结合使用);通过注射,借助于导管、借助于栓剂、或借助于植入物,所述植入物为多孔的、无孔的或凝胶状物质,包括膜(如硅橡胶膜)或纤维。在一个实施方案中,可在粥样硬化斑块组织的部位(或前部位)处直接注射给药。
在某些实施方案中,可以通过任何合适途径(包括心室内、鞘内和硬膜外注射)将本发明的化合物导入中枢神经系统。心室内导管(例如连接于蓄池如Ommaya蓄池)可以便利心室内注射。
也可以利用肺部给药,例如通过使用吸入器或雾化器和用雾化剂的制剂,或者通过用碳氟化合物或合成的肺表面活性剂灌注。在某些实施方案中,本发明化合物可配制为栓剂,其含有传统粘合剂和媒介物如甘油三酯。
在另一个实施方案中,本发明的化合物和所述化合物的组合物可以在囊泡、尤其是脂质体中递送(参见Langer, 1990, Science 249:1527-1533; Treat等人, in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein和Fidler (编), Liss, New York, 第353-365页(1989); Lopez-Berestein, 文献同上, 第317-327页;一般性参阅以上文献)。
在另一个实施方案中,可以在控释系统中递送本发明的化合物和所述化合物的组合物。在一个实施方案中,可以使用泵(参见Langer, 出处同上; Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201; Buchwald等人, 1980, Surgery 88:507 Saudek等人, 1989, N. Engl. J. Med. 321:574)。在另一个实施方案中,可以使用聚合材料(参见Medical Applications of Controlled Release, Langer和Wise (编), CRC Pres., Boca Raton, Florida (1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen和Ball (编), Wiley, New York (1984); Ranger and Peppas, 1983, J. Macromol. Sci. Rev. Macromol. Chem. 23:61;也参见Levy等人, 1985, Science 228:190; During等人, 1989, Ann. Neurol. 25:351; Howard等人, 1989, J. Neurosurg. 71:105)。在另一个实施方案中,可以将控释系统放置在要治疗的目标区域(例如肝脏)附近,因此只需要全身剂量的一部分(参见,例如,Goodson, in Medical Applications of Controlled Release, 出处同上, 第2卷, 第115-138页(1984))。可以使用Langer综述(1990, Science 249:1527-1533)论述的其它控释系统。
本发明的组合物包含治疗有效量的本发明的化合物(任选地超过一种本发明的化合物)以及合适量的药学上可接受的媒介物,从而提供给受试者的给药形式。
本发明的组合物可以为以下形式:溶液、悬浮液、乳液、片剂、丸剂、弹丸剂、胶囊剂、包含液体的胶囊剂、散剂、持续释放制剂、栓剂、乳剂、气雾剂、喷雾剂、混悬剂或任意其它适用形式。在一个实施方案中,药学上可接受的媒介物为胶囊(参见例如,美国专利号5,698,155)。合适的药用媒介物的其它实例参见E.W. Martin的“Remington’s Pharmaceutical Sciences”,其关于药物组合物和施用它们的方法的教导通过引用整体并入。
在有些实施方案中,本发明的化合物和所述化合物的组合物按照常规方法配制为适合静脉内施用给人类的药用组合物。用于静脉内给药的本发明的化合物和所述化合物的组合物可以为在无菌等渗水性缓冲液中的溶液。所述组合物也可以包含增溶剂。用于静脉内给药的组合物任选地包括局部麻醉剂如利多卡因。各种成分可以分别提供,或者混合在一起成为单位剂型,例如作为在气密密封的容器如安瓿或药囊中的冻干粉或无水浓缩物。在本发明化合物通过静脉内输注给药时,其可以分散于例如装有无菌药用级水或盐水的输注瓶中。当本发明化合物注射给药时,可以提供1安瓿无菌注射用水或盐水,以便可以在施用前混合各成分。
用于口服递送的本发明的化合物和所述化合物的组合物可以为下述形式:片剂、锭剂、水性或油性混悬剂、颗粒剂、散剂、乳剂、胶囊剂、糖浆剂或酏剂。用于口服递送的本发明的化合物和所述化合物的组合物也可以配制在食品和食品混合物中。口服施用的组合物可以包含一种或多种任选的试剂,例如,甜味剂诸如果糖、阿司帕坦或糖精;矫味剂诸如薄荷、冬青油或樱桃;着色剂;和防腐剂,以提供药学上适口的制剂。可以包衣所述组合物以延迟在胃肠道中的崩解和吸收,由此提供在延长的时间段内的持续作用。包围渗透活性的驱动化合物的选透性膜也适用于口服施用的本发明的化合物和所述化合物的组合物。还可以使用延时材料诸如单硬脂酸甘油酯或硬脂酸甘油酯。口服组合物可以包括标准的媒介物诸如甘露醇、乳糖、淀粉、硬脂酸镁、糖精钠、纤维素和碳酸镁。
可有效地治疗本文公开的特定病症的本发明的化合物的量可以取决于所述病症的性质,且可以通过标准的临床技术确定。可以采用体外或体内测定来帮助鉴定最佳剂量范围。要在组合物中采用的精确剂量还可以取决于给药途径或病症的严重程度,且可以根据从业人员的判断和每位受试者的情况来决定。但是,适合口服给药的剂量范围通常是约0.001 mg至2000 mg本发明的化合物/kg体重。在有些实施方案中,所述口服剂量是0.01 mg至100 mg/kg体重、0.1 mg至50 mg/kg体重、0.5 mg至20 mg/kg体重、或1 mg至10 mg/kg体重。在有些实施方案中,所述口服剂量是5 mg本发明的化合物/kg体重。本文所述的剂量的量是指施用的总量;也就是说,如果施用超过一种本发明的化合物,所述剂量可以与施用的本发明的化合物的总量相对应。按质量计,口服组合物可以包含10% 至95%的活性成分。
在某些实施方案中,将本发明的化合物或所述化合物的组合物施用给受试者,诸如人,作为针对本文所述病症的预防措施。本发明的组合物可以作为预防措施施用给具有病症的遗传易感性的受试者,所述病症例如心血管疾病、血脂异常、异常脂蛋白血症、葡萄糖代谢障碍、阿尔茨海默氏病、X综合征、P2Y13-相关的病症、败血症、血栓性障碍、肥胖、胰腺炎(pancreatitis)、高血压、肾病、癌症、炎症或阳萎。这样的遗传易感性的例子包括但不限于:载脂蛋白E的ε4等位基因,该等位基因增加阿尔茨海默氏病的可能性;脂蛋白脂肪酶基因编码区或启动子的功能丧失或无效突变(例如编码区中的导致置换D9N和N291S的突变;关于增加心血管疾病、血脂异常和异常脂蛋白血症的风险的脂蛋白脂肪酶基因中的遗传突变的综述,参见Hayden和Ma, 1992, Mol. Cell Biochem. 113:171-176);和家族性混合型高脂血症和家族性高胆固醇血症
本发明的化合物或所述化合物的组合物可以作为预防措施施用给具有病症的非遗传易感性的受试者,所述病症例如心血管疾病、血脂异常、异常脂蛋白血症、葡萄糖代谢障碍、阿尔茨海默氏病、X综合征、P2Y13-相关的病症、败血症、血栓性障碍、肥胖、胰腺炎(pancreatitis)、高血压、肾病、癌症、炎症或阳萎。这样的非遗传易感性的例子包括但不限于:心脏搭桥手术和经皮腔内冠状动脉血管成形术,这些手术可以导致再狭窄,即加速形式的动脉粥样硬化;女性糖尿病,其可以导致多囊性卵巢疾病;和可以导致阳萎的心血管疾病。因此,本发明化合物的组合物可用于预防一种疾病或障碍,并同时治疗另一种疾病或障碍(例如预防多囊性卵巢疾病,同时治疗糖尿病;预防阳萎,同时治疗心血管疾病)。
本发明提供了用于治疗或预防心血管疾病或其症状的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。本文使用的术语“心血管疾病”表示心脏或循环系统的疾病。心血管疾病可以与异常脂蛋白血症或血脂异常或二者相关联。心血管疾病包括但不限于:动脉硬化;动脉粥样硬化;中风;缺血;血管外周疾病(PVD);短暂性脑缺血发作(TIA)、电击状动脉粥样硬化;器官移植物动脉粥样硬化;内皮功能障碍,特别是影响血管弹性的那些功能障碍;外周血管病;冠心病;心肌梗塞;脑梗死和再狭窄。心血管疾病的症状的非限制性例子包括:心绞痛、呼吸短促、头晕、恶心、疲劳、不规则的心搏和阳萎。在有些实施方案中,心血管疾病的治疗会治疗心血管疾病的一种或多种症状。在有些实施方案中,心血管疾病的治疗会治疗阳萎。
本发明提供了用于治疗或预防血脂异常的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
血脂异常包括但不限于:高脂血症和在血液中低水平的高密度脂蛋白(HDL) 胆固醇。在某些实施方案中,所述高脂血症是家族性高胆固醇血症;家族性混合型高脂血症;脂蛋白脂肪酶水平或活性的降低或缺乏,包括脂蛋白脂肪酶突变引起的降低或缺乏;高甘油三酯血症;高胆固醇血症;血液中高水平的酮体(例如β-OH丁酸);血液中高水平的Lp(a)胆固醇;血液中高水平的低密度脂蛋白(LDL)胆固醇;血液中高水平的极低密度脂蛋白(VLDL)胆固醇和血液中高水平的未酯化脂肪酸。
本发明另外提供了用于改变受试者的脂类代谢(例如,降低受试者血液中的LDL,降低受试者血液中的游离甘油三酯,增加受试者血液中的HDL与LDL之比,和抑制皂化的或未皂化的脂肪酸合成)的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
本发明提供了用于治疗或预防异常脂蛋白血症的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
异常脂蛋白血症是导致或表现为异常的循环脂蛋白水平的病症。就血液中的脂蛋白水平异常高而言,将本发明的化合物施用给受试者以恢复正常水平。相反,就血液中的脂蛋白水平异常低而言,将本发明的化合物施用给受试者以恢复正常水平。正常脂蛋白水平是本领域技术人员众所周知的。
异常脂蛋白血症包括但不限于:血液中高水平的LDL;血液中高水平的载脂蛋白B (apo B);血液中高水平的Lp(a);血液中高水平的apo(a);血液中高水平的VLDL;血液中低水平的HDL;脂蛋白脂肪酶水平或活性的降低或缺乏,包括脂蛋白脂肪酶突变引起的降低或缺乏;低α脂蛋白血症;与糖尿病有关的脂蛋白异常;与肥胖有关的脂蛋白异常;与阿尔茨海默氏病有关的脂蛋白异常;和家族性混合型高脂血症。
本发明另外提供了用于降低受试者血液中的apo C-II水平、降低受试者血液中的apo C-III水平、升高受试者血液中的HDL相关的蛋白(包括、但不限于apo A-I、apo A-II、apo A-IV和apo E)的水平、升高受试者血液中的apo E水平、或促进甘油三酯从受试者血液中清除的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
本发明提供了用于治疗或预防葡萄糖代谢障碍的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
葡萄糖代谢障碍可以包含异常的葡萄糖贮存和/或利用。就葡萄糖代谢的一种或多种指标(即 , 血液胰岛素、血液葡萄糖) 异常高而言,将本发明的化合物施用给受试者以恢复正常水平。相反,就葡萄糖代谢的一种或多种指标异常低而言,将本发明的化合物施用给受试者以恢复正常水平。葡萄糖代谢的正常指标是本领域技术人员众所周知的。
葡萄糖代谢障碍包括但不限于:葡萄糖耐量降低;糖尿病视网膜病变、糖尿病肾病、胰岛素抵抗;胰岛素抵抗相关的癌症,诸如乳腺癌、结肠癌或前列腺癌;糖尿病,包括、但不限于非胰岛素依赖性的糖尿病(NIDDM)、胰岛素依赖性的糖尿病(IDDM)、妊娠糖尿病(GDM)和青春晚期糖尿病(MODY);胰腺炎;高血压;多囊性卵巢疾病;和血液中高水平的胰岛素或葡萄糖或二者。
本发明另外提供了用于改变受试者的葡萄糖代谢(例如增加受试者的胰岛素敏感性或氧耗量)的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
本发明提供了用于治疗或预防P2Y13-相关的病症的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
P2Y13-相关的障碍的例子包括,但不限于:类风湿性关节炎;多发性硬化;银屑病;炎性肠病;乳腺癌、结肠癌或前列腺癌;血液中低水平的HDL;血液、淋巴和/或脑脊液中低水平的apo E;血液、淋巴或脑脊液中低水平的apo A-I;血液中高水平的VLDL;血液中高水平的LDL;血液中高水平的甘油三酯;血液中高水平的apo B;血液中高水平的apo C-III和肝素后肝脏脂肪酶与脂蛋白脂肪酶活性之比降低。可以升高淋巴或脑流体或二者中的HDL。
本发明提供了用于治疗或预防脂肪肝(包括酒精性和非酒精性脂肪肝)的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
本发明提供了用于治疗或预防肾病的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
肾病包括:肾小球疾病(包括但不限于急性和慢性肾小球肾炎、迅速进行性肾小球肾炎、肾病综合征、局部增殖性肾小球肾炎、与系统性疾病有关的肾小球病变,所述系统性疾病例如系统性红斑狼疮、肺出血肾炎综合征、多发性骨髓瘤、糖尿病、瘤形成、镰刀形细胞病和慢性炎性疾病)、肾小管疾病(包括但不限于急性肾小管坏死和急性肾衰竭、多囊性肾病、髓质海绵肾、髓质囊性病、肾性糖尿病和肾小管酸中毒)、肾小管间质疾病(包括但不限于肾盂肾炎、药物和毒素诱发的肾小管间质性肾炎、高钙性肾病和低钾性肾病)、急性迅速进行性肾衰竭、慢性肾衰竭、肾结石或肿瘤(包括但不限于肾细胞癌和肾母细胞瘤)。在另一个实施方案中,所述肾病为血管性疾病,包括但不限于高血压、肾硬化、微血管病性溶血性贫血、动脉粥样硬化栓塞性肾病、弥漫性肾皮质坏死和肾梗死。
本发明提供了用于治疗或预防神经变性疾病或障碍、帕金森病、阿尔茨海默氏病、X综合征、败血症、血栓性障碍、肥胖、胰腺炎(pancreatitis)、高血压、炎症或阳萎的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物。在一个实施方案中,本发明的化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
治疗或预防阿尔茨海默氏病还可以包括治疗或预防与阿尔茨海默氏病有关的一种或多种脂蛋白异常。
治疗或预防X综合征或代谢综合征还可以包括治疗或预防其症状,包括、但不限于:葡萄糖耐量降低、高血压和血脂异常或异常脂蛋白血症。
治疗或预防败血症还可以包括治疗或预防脓毒性休克。
治疗或预防血栓性障碍还可以包括治疗或预防血液中高水平的纤维蛋白原或促进纤维蛋白溶解。
本发明的化合物还可用于促进受试者的体重减轻。
本发明的化合物可在医学应用中用于治疗或预防多种疾病和障碍,例如,但不限于:心血管疾病、中风和外周血管病;血脂异常;高胆固醇血症、动脉粥样硬化、血管外周疾病(PVD)、中风、TIA、电击状动脉粥样硬化、器官移植物动脉粥样硬化;异常脂蛋白血症;葡萄糖代谢障碍;糖尿病肾病、糖尿病视网膜病变、胰岛素抵抗、代谢综合征障碍(例如,X综合征);过氧化物酶体增殖物活化的受体-相关的障碍;败血症;血栓性障碍;肥胖;胰腺炎;高血压;肾病;炎症;炎症性肌肉疾病,诸如风湿性多肌痛、多肌炎、肌病和纤维织炎;炎症性障碍,诸如哮喘、脉管炎、溃疡性结肠炎、克罗恩氏病、川崎病、韦格纳氏肉芽肿病、(RA)、系统性红斑狼疮(SLE)、多发性硬化(MS)和自身免疫性慢性肝炎;关节炎,诸如类风湿性关节炎、幼年型类风湿性关节炎和骨关节炎;骨质疏松症、软组织风湿病,诸如腱炎;滑囊炎;自身免疫病,诸如全身性红斑狼疮;硬皮病;强直性脊柱炎;痛风;假痛风;非胰岛素依赖性的糖尿病;多囊性卵巢疾病;高脂血症,诸如家族性高胆固醇血症(FH)、家族性混合型高脂血症(FCH);脂蛋白脂肪酶缺乏,诸如高甘油三酯血症、低α脂蛋白血症和高胆固醇血症;与糖尿病有关的脂蛋白异常;与肥胖有关的脂蛋白异常。本发明的化合物和组合物可用于治疗或预防:血液中高水平的甘油三酯、高水平的低密度脂蛋白胆固醇、高水平的载脂蛋白B、高水平的脂蛋白Lp(a) 胆固醇、高水平的极低密度脂蛋白胆固醇、高水平的纤维蛋白原、高水平的胰岛素、高水平的葡萄糖和低水平的高密度脂蛋白胆固醇。本发明的化合物和组合物也可用于治疗没有重量增加的非胰岛素依赖性糖尿病(NIDDM)。本发明的化合物还可以用于降低家畜的肉中的脂肪含量和降低蛋的胆固醇含量。
本发明提供了新颖的化合物,其特别适用于治疗或预防多种疾病和病症,包括,但不限于:老化、阿尔茨海默氏病和与阿尔茨海默氏病有关的脂蛋白异常、帕金森病、癌症、心血管疾病、糖尿病肾病、糖尿病视网膜病变、葡萄糖代谢障碍、血脂异常、异常脂蛋白血症、增强的胆汁产生、高血压、阳萎、炎症、胰岛素抵抗、胆汁中的脂质消除、调节性C反应蛋白、肥胖、胆汁中的氧化固醇消除、胰腺炎(pancreatitis)、胰脏炎(pancreatitius)、阳萎;胃肠疾病;肠易激综合征;炎性肠病;过氧化物酶体增殖物活化的受体-相关的障碍、胆汁中的磷脂消除、肾病、败血症、代谢综合征障碍(例如,X综合征)和血栓性障碍。
心血管疾病如动脉粥样硬化可能需要外科手术,例如血管成形术。血管成形术常常将被称为“支架”的增强型金属管形结构放入受损的冠状动脉内。对于更严重的疾病,可能需要进行开心手术如冠状动脉分流术。这些外科手术需要使用侵袭性外科手术装置或植入物,且伴有再狭窄和血栓形成的高风险。因此,本发明的化合物可以用作外科手术装置(例如导管)或植入物(例如支架)上的涂层,以降低与在心血管疾病的治疗中使用的侵袭性操作有关的再狭窄和血栓形成的风险。因此,本发明另外提供了外科手术装置或植入物,其具有包含有效量的本发明的化合物的涂层。
本发明的化合物可以施用给非人动物用于兽医学应用,用于治疗或预防本文公开的疾病或障碍。
在一个具体实施方案中,非人类动物为家养宠物。在另一个实施方案中,非人类动物为家畜动物。在一个优选实施方案中,非人类动物为哺乳动物,例如,牛、马、绵羊、猪、猫、狗、小鼠、大鼠、兔或豚鼠。在另一个实施方案中,非人类动物为家禽,诸如鸡、火鸡、鸭、鹅或鹌鹑。
除了兽医学应用之外,本发明的化合物和组合物可用于降低家畜的脂肪含量,以产生更瘦的肉。可替换地,通过将所述化合物施用给母鸡、母鹌鹑或母鸭,本发明的化合物和组合物可用于降低蛋的胆固醇含量。对于非人类动物用途,本发明的化合物和组合物可以通过动物饲料施用,或者作为兽用顿服药物组合物口服施用。因此,本发明另外提供了用于降低家畜的脂肪含量或蛋的胆固醇含量的方法,所述方法包括:将有效量的本发明的化合物施用给有此需要的受试者。
A. 癌症的治疗或预防
本发明的化合物可用于治疗或预防癌症。因此,本发明提供了用于治疗或预防癌症的方法,所述方法包括:将有效量的本发明的化合物施用给有此需要的受试者。在一个实施方案中,所述方法另外包括:施用有效量的另一种抗癌剂。本文公开的本发明的化合物可用于治疗或预防的癌症的例子包括,但不限于在下面表1中公开的癌症及其转移灶。
表1.
在一个实施方案中,所述癌症是肺癌、乳腺癌、结直肠癌、前列腺癌、白血病、淋巴瘤、非霍奇金淋巴瘤、皮肤癌、脑癌、中枢神经系统癌、卵巢癌、子宫癌、胃癌、胰腺癌、食管癌、肾癌、肝癌或头颈癌症。在另一个实施方案中,所述癌症是转移性癌症。
在另一个实施方案中,所述癌症是脑癌或黑素瘤。在一个实施方案中,所述脑癌是转移性脑癌或神经胶质瘤。在一个实施方案中,所述神经胶质瘤是纤维状细胞性星形细胞瘤、星形细胞瘤、间变性星形细胞瘤或多形性胶质母细胞瘤。在一个实施方案中,所述癌症是同源重组缺陷型,诸如BRCA-I或BRCA-2缺陷型,或缺乏Fanconi家族的一种或多种蛋白。在一个实施方案中,所述缺乏由基因突变造成。在另一个实施方案中,BRCA-I或BRCA-2蛋白的异常低表达会造成由缺乏引起的表型。在另一个实施方案中,Fanconi家族的一种或多种蛋白的异常低表达会造成由缺乏引起的表型。
在另一个实施方案中,所述癌症是:白血病,例如但不限于急性白血病、急性淋巴细胞白血病、急性粒细胞性白血病,例如成髓细胞白血病、前髓细胞白血病、粒单核细胞白血病、单核细胞白血病和红白血病和骨髓增生异常综合征;慢性白血病例如但不限于慢性髓细胞(粒细胞)白血病、慢性淋巴细胞白血病、多毛细胞白血病;真性红细胞增多症;淋巴瘤例如但不限于何杰金病、非何杰金病;多发性骨髓瘤例如但不限于无症状多发性骨髓瘤(smoldering multiple myeloma)、无分泌功能骨髓瘤、骨硬化骨髓瘤、浆细胞性白血病、孤立性浆细胞瘤和髓外浆细胞瘤;Waldenström氏巨球蛋白血症;未定性的单克隆丙种球蛋白病;良性单株丙种球蛋白病;重链病;树突细胞癌,包括浆细胞样树突细胞癌, NK芽殖淋巴瘤(也称作皮肤的NK/T-细胞淋巴瘤和颗粒状(CD4+/CD56+) 皮肤病肿瘤);嗜碱性粒细胞性白血病;骨骼和结缔组织肉瘤,例如但不限于骨骼肉瘤、骨肉瘤、软骨肉瘤、尤因氏肉瘤、恶性巨细胞瘤、骨骼纤维肉瘤、脊索瘤、骨膜肉瘤、软组织肉瘤、血管肉瘤、纤维肉瘤、卡波济氏肉瘤、平滑肌肉瘤、脂肪肉瘤、淋巴管肉瘤、神经鞘瘤、横纹肌肉瘤、滑膜肉瘤;脑部肿瘤例如但不限于神经胶质瘤、星形细胞瘤、脑干神经胶质瘤、室管膜瘤、少突胶质细胞瘤、非神经胶质瘤、听神经瘤、颅咽管瘤、成神经管细胞瘤、脑膜瘤、松果体细胞瘤、成松果体细胞瘤、原发性脑淋巴瘤;乳腺癌包括但不限于导管癌、腺癌、小叶(小细胞)癌、导管内癌、髓样乳腺癌、粘蛋白乳腺癌、管状乳腺癌、乳头状乳腺癌、佩吉特氏病和炎性乳腺癌;肾上腺癌例如但不限于嗜铬细胞瘤和肾上腺皮质癌;甲状腺癌例如但不限于乳头状或滤泡状甲状腺癌、髓样甲状腺癌和间变性甲状腺癌;胰腺癌例如但不限于胰岛素瘤、促胃液素瘤、胰高血糖素瘤、胰腺瘤、生长激素抑制素分泌瘤和类癌瘤或胰岛细胞瘤;垂体癌例如但不限于库欣氏病、催乳素分泌瘤、肢端肥大症和尿崩病(diabetes insipius);眼癌例如但不限于眼黑素瘤例如虹膜黑素瘤、脉络膜黑素瘤、睫状体黑素瘤和视网膜母细胞瘤;阴道癌例如鳞状细胞癌、腺癌和黑素瘤;外阴癌例如鳞状细胞癌、黑素瘤、腺癌、基底细胞癌、肉瘤和佩吉特病;子宫颈癌例如但不限于鳞状细胞癌和腺癌;子宫癌例如但不限于子宫内膜癌和子宫肉瘤;卵巢癌例如但不限于卵巢上皮癌、交界瘤、生殖细胞瘤和间质瘤;食管癌例如但不限于鳞状癌、腺癌、腺样囊性癌、粘液表皮样癌、腺鳞状上皮癌、肉瘤、黑素瘤、浆细胞瘤、疣状癌和燕麦细胞(小细胞)癌;胃癌例如但不限于腺癌、蕈伞样(息肉状)、溃疡、表面扩散、弥漫性扩散、恶性淋巴瘤、脂肉瘤、纤维肉瘤和癌肉瘤;结肠癌;直肠癌;肝癌例如但不限于肝细胞癌和肝胚细胞瘤、胆囊癌例如腺癌;胆管上皮癌例如但不限于乳头状、结节状和弥漫性癌;肺癌例如非小细胞肺癌、鳞状细胞癌(表皮样癌)、腺癌、大细胞癌和小细胞肺癌;睾丸癌例如但不限于生殖肿瘤,精原细胞瘤,间变性、经典(典型)的精细胞非精原细胞瘤,胚胎癌,畸胎瘤癌,绒毛膜癌(卵黄囊肿瘤);前列腺癌例如但不限于前列腺上皮内瘤形成、腺癌、平滑肌肉瘤和横纹肌肉瘤;阴茎癌;口腔癌例如但不限于鳞状细胞癌;基底癌;唾液腺癌例如但不限于腺癌、粘液表皮样癌和腺囊癌;咽癌例如但不限于鳞状细胞癌和疣;皮肤癌例如但不限于基底细胞癌、鳞状细胞癌和黑素瘤、表浅扩散的黑素瘤、结节性黑素瘤、着色斑恶性黑素瘤、肢端着色斑性黑素瘤;肾癌例如但不限于肾细胞癌、腺癌、肾上腺样瘤、纤维肉瘤、移行细胞癌(肾盂或uterer);威尔曼瘤;膀胱癌例如但不限于移行细胞癌、鳞状细胞癌、腺癌、癌肉瘤。此外,癌症包括粘液肉瘤、成骨性肉瘤、内皮肉瘤、淋巴管内皮细胞瘤、间皮瘤、滑膜瘤、成血管细胞瘤、上皮癌、囊腺癌、支气管原癌、汗腺癌、皮脂腺癌、乳头状癌和乳头状腺癌(有关这类病症的综述,参见Fishman等 人, 1985, Medicine, 第2版, J.B. Lippincott Co., Philadelphia和Murphy 等 人, 1997, Informed Decisions: The Complete Book of Cancer Diagnosis, Treatment, and Recovery, Viking Penguin, Penguin Books U.S.A., Inc., United States of America)。
在一个具体的实施方案中,所述癌症是与γ-分泌酶的切口切割有关的癌症,包括、但不限于,白血病、非小细胞肺癌、卵巢癌、乳腺癌或脑癌。
在有些实施方案中,所述癌症是结肠癌。在有些实施方案中,所述癌症是结肠癌、乳腺癌、肺癌或黑素瘤。
在另一个实施方案中,需要治疗的受试者以前已经接受或目前正在接受癌症治疗。所述治疗包括、但不限于:化学疗法、辐射疗法、外科手术或免疫疗法,诸如施用癌症疫苗。在有些实施方案中,本发明的化合物用于预防以前的癌症的转移和侵袭。
本发明的化合物还可用于治疗或预防由病毒造成的癌症。这样的病毒包括:人乳头瘤病毒,其可以导致宫颈癌(参见, 例如,Hernandez-Avila等人, Archives of Medical Research (1997) 28:265-271);爱泼斯坦-巴尔病毒(EBV),其可以导致淋巴瘤(参见, 例如 , Herrmann等人, J. Pathol. (2003) 199(2):140-5);乙型肝炎或丙型肝炎病毒,其可以导致肝癌(参见, 例如, El-Serag, J. Clin. Gastroenterol. (2002) 35(5 Suppl. 2):S72-8);人T细胞白血病病毒(HTLV)-I,其可以导致T-细胞白血病(参见 , 例如 ,Mortreux等人, Leukemia (2003) 17(l):26-38);人疱疹病毒-8感染,其可以导致卡波济氏肉瘤(参见 , 例如 , Kadow等人, Curr. Opin. Investig. Drugs (2002) 3(11):1574-9);和人免疫缺陷病毒(HIV)感染,其可以导致癌症作为免疫缺陷的后果(参见 , 例如 , Dal Maso等人, Lancet Oncol (2003) 4(2):110-9)。这些参考文献中的每一篇通过引用并入本文。
本发明的化合物还可用于预防癌症或预防癌症的进展,包括、但不限于在表1中列出的癌症。这样的预防性用途应用:其中已经发生非赘生性细胞生长诸如增生、组织转化、或更具体地发育异常的应用。除了表征为增生、组织转化或发育异常的异常细胞生长的存在以外,或者可替换地,由得自受试者的细胞样品在体内或在体外表现的转化表型或恶性表型的一种或多种特征的存在,可以指示本发明的化合物的预防性或治疗性施用的需求。这样的转化表型的特征包括形态学变化、更松散的基底附着、缺少接触抑制、缺少贴壁依赖、蛋白酶释放、增加的糖运输、减少的血清需求、胎儿抗原的表达、250,000道尔顿细胞表面蛋白的消失等。在一个具体实施方案中,根据本发明的方法可治疗或可预防白斑、上皮的出现的良性增生性或发育异常性病变或鲍文病、原位癌。
在另一个实施方案中,根据本发明的方法可治疗或可预防纤维囊性疾病(囊性增生、乳房发育不良、特别是腺病(良性上皮细胞增生))。
在其它实施方案中,通过施用有效量的本发明的化合物,可以治疗具有下述恶性肿瘤易感因素中的一个或多个的受试者:与恶性肿瘤有关的染色体易位(例如,慢性髓性白血病的费城染色体,滤泡性淋巴瘤的t(14;18));家族性息肉病或加德纳综合症;良性单克隆丙种球蛋白病;与具有呈现孟德尔(遗传)遗传模式的癌症或癌前期疾病的人的第一度亲属关系(例如,家族性结肠息肉病、加德纳综合症、遗传性外生骨疣、多内分泌腺腺瘤病、具有淀粉样蛋白产生的甲状腺髓样癌和嗜铬细胞瘤、黑斑息肉综合征、Von Recklinghausen神经纤维瘤、成视网膜细胞瘤、颈动脉体瘤、皮肤黑素癌、眼内黑素癌、着色性干皮病、共济失调毛细血管扩张、Chediak-Higashi综合征、白化病、Fanconi氏再生障碍性贫血和布卢姆综合征);以及暴露于致癌物(例如,吸烟、暴露于二手烟和吸入或接触某些化学试剂)。
在一个方面,用于治疗或预防癌症的本发明方法另外包括施用其它抗癌剂。
在一个实施方案中,本发明提供了用于治疗或预防癌症的方法,所述方法包括:给有此需要的受试者施用有效量的本发明的化合物和其它抗癌剂。本发明的化合物和其它抗癌剂可同时施用。在该实施方案中,本发明的化合物和其它抗癌剂可以在相同的组合物内施用,或者可以从不同的组合物经由相同或不同的给药途径施用。在另一实施方案中,本发明的化合物在其它抗癌剂发挥其预防或治疗作用期间施用,反之亦然。
在另一个实施方案中,本发明的化合物和其它抗癌剂以当所述药剂用作癌症治疗的单一治疗时通常采用的剂量施用。
在一个实施方案中,本发明的化合物和其它抗癌剂以少于当所述药剂用作癌症治疗的单一治疗时通常采用的剂量施用。
在另一个实施方案中,本发明的化合物和其它抗癌剂协同作用,并以少于当所述药剂用作癌症治疗的单一治疗时通常采用的剂量施用。所施用的本发明的化合物或其它抗癌剂的剂量以及给药方案取决于各种参数,包括、但不限于:正治疗的癌症、受试者的总体健康状况和主治医师的考虑。本发明的化合物可在其它抗癌剂施用于有此需要的受试者之前(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周之前)、同时、或之后(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周之后)施用。在不同的实施方案中,本发明的化合物和其它抗癌剂相隔1分钟、相隔10分钟、相隔30分钟、相隔小于1小时、相隔1小时至2小时、相隔2小时至3小时、相隔3小时至4小时、相隔4小时至5小时、相隔5小时至6小时、相隔6小时至7小时、相隔7小时至8小时相隔8小时至9小时、相隔9小时至10小时、相隔10小时至11小时、相隔11小时至12小时、相隔不超过24小时或相隔不超过48小时施用。在一个实施方案中,本发明的化合物和其它抗癌剂相隔3小时内施用。在另一个实施方案中,本发明的化合物和其它抗癌剂相隔1分钟至24小时施用。
在一个实施方案中,有效量的本发明的化合物和有效量的其它抗癌剂存在于同一组合物中。在一个实施方案中,该组合物可用于口服给药。在另一个实施方案中,该组合物可用于静脉内给药。
在一个实施方案中,所述组合物包含在一起可有效地治疗或预防癌症的量的本发明的化合物和其它抗癌剂。
在另一个实施方案中,所述组合物包含有效量的替莫唑胺、丙卡巴肼、达卡巴嗪、白介素-2、伊立替康或多柔比星、生理上可接受的载体、稀释剂、赋形剂或媒介物和有效量的本发明的化合物。
在一个实施方案中,本发明的化合物和其它抗癌剂的量为联用化疗剂组合物的重量的至少约0.01%。当计划用于口服给药时,该量可在组合物的重量的约0.1%至约80%的范围内变化。一些口服组合物可包含占组合物的约4%至约50%的组合量的本发明的化合物和其它抗癌剂。制备本发明的其它组合物,以使肠胃外剂量单位包含组合物重量的约0.01%至约2%。
通过施用本发明的化合物和其它抗癌剂可治疗的或预防的癌症包括、但不限于在表1中所述癌症的列表。
在一个实施方案中,所述癌症是脑癌。在具体实施方案中,所述脑癌是纤维状细胞性星形细胞瘤、星形细胞瘤、间变性星形细胞瘤、多形性胶质母细胞瘤或转移性脑瘤。
在一个实施方案中,所述癌症是黑素瘤。在一个具体实施方案中,所述黑素瘤是转移性黑素瘤。
本发明的化合物和其它抗癌剂可累加或协同作用。本发明的化合物和其它抗癌剂的协同组合可允许使用更低剂量的所述药剂中的一种或两种,或更低频率地给患有癌症的受试者施用所述药剂。使用更低剂量的本发明的化合物和其它抗癌剂中的一种或两种或以更低频率施用所述药剂的能力,可降低与给受试者施用该药剂有关的任何毒性,而不降低该药剂在癌症治疗中的效力。此外,协同效应可导致这些药剂在癌症治疗中提高的效力,和/或与任一药剂单独使用有关的任何不利的或不希望的副作用的减少。
在一个实施方案中,有效量的本发明的化合物和有效量的其它抗癌剂的施用会抑制癌症对其它抗癌剂的抗性。在一个实施方案中,所述癌症是肿瘤。
可用于本发明的方法和组合物中的合适的其它抗癌剂包括、但不限于:替莫唑胺、拓扑异构酶I抑制剂、丙卡巴肼、达卡巴嗪、吉西他滨、卡培他滨、甲氨蝶呤、泰素、泰索帝、巯嘌呤、硫鸟嘌呤、羟基脲、阿糖胞苷、环磷酰胺、异环磷酰胺、亚硝基脲、顺铂、卡铂、丝裂霉素、达卡巴嗪、丙卡巴肼(procarbizine)、依托泊苷、替尼泊苷、喜树碱(campathecins)、博来霉素、多柔比星、伊达比星、柔红霉素、更生霉素、普卡霉素、米托蒽醌、L-门冬酰胺酶、多柔比星、表柔比星、5-氟尿嘧啶、紫杉烷s诸如多西他赛和紫杉醇、亚叶酸、左旋咪唑、伊立替康、雌莫司汀、依托泊苷、氮芥、BCNU、亚硝基脲诸如卡莫司汀和洛莫司汀、长春花生物碱诸如长春碱、长春新碱和长春瑞滨、铂复合物诸如顺铂、卡铂和奥沙利铂、甲磺酸伊马替尼、六甲蜜胺、托泊替康、酪氨酸激酶抑制剂、酪氨酸磷酸化抑制剂、除莠霉素A、染料木黄酮、erbstatin和lavendustin A。
在一个实施方案中,所述其它抗癌剂是、但不限于在表2中列出的药物。
表2.
可用于本发明组合物和方法中的其它抗癌剂包括、但不限于:阿西维辛;阿柔比星;盐酸阿考达唑;阿克罗宁;阿多来新;阿地白介素;六甲蜜胺;安波霉素;乙酸阿美蒽醌;氨鲁米特;安吖啶;阿那曲唑;安曲霉素;门冬酰胺酶;曲林菌素;阿扎胞苷;阿扎替派;阿佐霉素;巴马司他;苯佐替派;比卡鲁胺;盐酸比生群;二甲磺酸双奈法德;比折来新;硫酸博来霉素;布喹那钠;溴匹立明;白消安;放线菌素C;卡普睾酮;卡醋胺;卡贝替姆;卡铂;卡莫司汀;盐酸卡柔比星;卡折来新;西地芬戈(cedefmgol);苯丁酸氮芥;西罗霉素;顺铂;克拉屈滨;甲磺酸克立那托;环磷酰胺;阿糖胞苷;达卡巴嗪;更生霉素;盐酸柔红霉素;地西他滨;右奥马铂;地扎胍宁;甲磺酸地扎胍宁;地吖醌;多西他赛;多柔比星;盐酸多柔比星;屈洛昔芬;柠檬酸屈洛昔芬;丙酸屈他雄酮;达佐霉素;依达曲沙;盐酸依氟鸟氨酸;依沙芦星;恩洛铂;恩普氨酯;依匹哌啶;盐酸表柔比星;厄布洛唑;盐酸依索比星;雌莫司汀;雌莫司汀磷酸钠;依他硝唑;依托泊苷;磷酸依托泊苷;氯苯乙嘧胺;盐酸法倔唑;法扎拉滨;芬维A胺;氮尿苷;磷酸氟达拉滨;氟尿嘧啶;氟西他滨;磷喹酮;福司曲星钠;盐酸吉西他滨;羟基脲;盐酸伊达比星;异环磷酰胺;伊莫福新;白介素-2 (包括重组白介素-2或rIL2)、干扰素alfa-2α;干扰素alfa-2β;干扰素alfa-n1;干扰素alfa-n3;干扰素β-1α;干扰素γ-1β;异丙铂;盐酸伊立替康;乙酸兰瑞肽;来曲唑;醋酸亮丙瑞林;盐酸利阿唑;洛美曲索钠;洛莫司汀;盐酸洛索蒽醌;马索罗酚;美坦辛;盐酸氮芥;乙酸甲地孕酮;乙酸美仑孕酮;美法仑;美诺立尔;巯嘌呤;甲氨蝶呤;甲氨蝶呤钠;氯苯氨啶;美妥替哌;米丁度胺;米托克星;丝裂红素;米托洁林;米托马星;丝裂霉素;米托司培;米托坦;盐酸米托蒽醌;麦考酚酸;诺考达唑;诺拉霉素;奥马铂;奥昔舒仑;紫杉醇;培门冬酶;培利霉素;五氮芥;硫酸培洛霉素;培磷酰胺;哌泊溴烷;哌泊舒凡;盐酸吡罗蒽醌;普卡霉素;普洛美坦;卟吩姆钠;泊非霉素;泼尼莫司汀;盐酸丙卡巴肼;嘌罗霉素;盐酸嘌罗霉素;吡唑呋喃菌素;利波腺苷;罗谷亚胺;沙芬戈;盐酸沙芬戈;司莫司汀;辛曲秦;磷乙酰天冬氨酸钠;司帕霉素;盐酸锗螺胺;螺莫司汀;螺铂;链黑霉素;链佐星;磺氯苯脲;他利霉素;替可加兰钠;替加氟;盐酸替洛蒽醌;替莫泊芬;替尼泊苷;替罗昔隆;睾内酪;硫咪嘌呤;硫鸟嘌呤;塞替派;噻唑羧胺核苷;替拉扎明;枸橼酸托瑞米芬;乙酸曲托龙;磷酸曲西立滨;三甲曲沙;葡糖醛酸三甲曲沙;曲普瑞林;盐酸妥布氯唑;乌拉莫司汀;乌瑞替派;伐普肽;维替泊芬;硫酸长春碱;硫酸长春新碱;长春地辛;硫酸长春地辛;硫酸长春匹定;硫酸长春甘酯;硫酸长春罗新;酒石酸长春瑞滨;硫酸长春罗定;硫酸长春利定;伏氯唑;折尼铂;净司他丁;和盐酸佐柔比星。
可用于本发明方法和组合物的另外的抗癌药包括、但不限于:20-表-1、25-二氢维生素D3;5-乙炔基尿嘧啶;阿比特龙;阿柔比星;酰基富烯;腺环戊醇(adecypenol);阿多来新;阿地白介素;ALL-TK拮抗剂;六甲蜜胺;氨莫司汀;amidox;氨磷汀;氨基酮戊酸;氨柔比星;安吖啶;阿那格雷;阿那曲唑;穿心莲内酯(rographolide);血管发生抑制剂;拮抗剂D;拮抗剂G;安雷利克斯(antarelix);抗背部化形态发生蛋白-1;抗雄激素;前列腺癌;抗雌激素;抗瘤酮;反义寡核苷酸;甘氨酸阿非科林;细胞凋亡基因调节剂;细胞凋亡调节剂;脱嘌呤核酸;ara-CDP-DL-PTBA;精氨酸脱氨酶;asulacrine;阿他美坦;阿莫司汀;axinastatin 1;axinastatin 2;axinastatin 3;阿扎司琼;阿扎毒素;重氮酪氨酸;浆果赤霉素III衍生物;balanol;巴马司他;BCR/ABL拮抗剂;苯并二氢卟酚(benzochlorins);苯甲酰星形孢菌素(benzoylstaurosporine);β内酰胺衍生物;β-alethine;亚阿克拉霉素B;桦木酸;bFGH抑制剂;比卡鲁胺;比生群;bisaziridinylspermine;双奈法德;bistratene A;比折来新;breflate;溴匹立明;布度钛;丁基硫堇硫氧胺;卡泊三醇;钙感光蛋白C;喜树碱衍生物;金丝雀痘IL-2;羧酰胺-氨基-三唑;羧基酰氨基三唑;CaRest M3;CARN 700;cartilaga衍生的抑制剂;卡折来新;酪蛋白激酶抑制制(ICOS);栗树精胺;杀菌肽B;西曲瑞克;氢卟酚;氯喹喔啉磺胺;西卡前列素;顺卟啉;克拉屈滨;氯米芬类似物;克霉唑;collismycin A;collismycin B;考布他汀A4;考布他汀类似物;conagenin;crambescidin 816;克立那托;念珠藻环肽8;念珠藻环肽A衍生物;curacin A;环戊烷蒽醌(cyclopentanthraquinones);cycloplatam;cypemycin;阿糖胞苷ocfosfate;细胞溶解因子;磷酸己烷雌酚;达昔单抗;地西他滨;脱氢代代宁B;地洛瑞林;右异环磷酰胺;右雷佐生;右维拉帕米;亚胺醌;膜海鞘素B;didox;二乙基去甲精胺(diethylnorspermine);二氢-5-氮胞苷;二氢紫杉醇;dioxamycin;二苯基螺莫司汀;多西他赛;二十二烷醇;多拉司琼;去氧氟尿苷;屈洛昔芬;屈大麻酚;多卡米星SA;依布硒啉;依考莫司汀;依地福新;依决洛单抗;依氟鸟氨酸;榄香烯;乙嘧替氟;表柔比星;爱普列特;雌氮芥类似物;雌激素激动剂;雌激素拮抗剂;依他硝唑;磷酸鬼臼亚乙苷;依西美坦;法屈唑;法扎拉滨;芬维A胺;非格司亭;非那雄胺;flavopiridol;氟卓斯汀;fluasterone;氟达拉滨;盐酸fluorodaunorunicin;福酚美克;福美司坦;福司曲星;福莫司汀;替沙林钆;硝酸镓;加洛他滨;加尼瑞克;明胶酶抑制剂;吉西他滨;谷胱甘肽抑制剂;hepsulfam;神经生长因子;六亚甲基二乙酰胺;金丝桃素;伊班膦酸;伊达比星;艾多昔芬;伊决孟酮;伊莫福新;伊洛马司他;咪唑并吖啶酮;咪喹莫特;免疫刺激肽;类胰岛素生长素-1受体抑制剂;干扰素激动剂;干扰素;白细胞介素;碘苄胍;碘阿霉素;4-蕃薯宁;伊立替康;伊罗普拉;伊索拉定;isobengazole;isohomohalicondrin B;伊他司琼;jasplakinolide;kahalalide F;片螺素N-三乙酸盐;兰瑞肽;leinamycin;来格司亭;硫酸蘑菇多糖;leptolstatin;来曲唑;白血病抑制因子;白细胞α干扰素;亮丙瑞林+雌激素+孕酮;亮丙瑞林;左旋咪唑;利阿唑;线性多氨基类似物;亲脂性二糖肽;亲脂性铂化合物;lissoclinamide 7;络铂;蚯蚓磷脂;洛美曲索;氯尼达明;洛索蒽醌;洛伐他汀;洛索立宾;勒托替康;德克萨斯卟啉镥;lysofylline;裂解肽;美坦辛;制甘糖霉素A;马立马司他;马索罗酚;maspin;基质裂解素抑制剂;基质金属蛋白酶抑制剂;美诺立尔;麦尔巴隆;美替瑞林(meterelin);甲硫氨酸酶;甲氧氯普胺;MIF抑制剂;米非司酮;米特福辛;mirimostin;错配双链RNA;米托胍腙;二溴卫矛醇;丝裂霉素类似物;米托萘胺;mitotoxin成纤维细胞生长因子-皂草素;米托蒽醌;莫法罗汀;沙格司亭;单克隆抗体;人绒膜促性腺激素;单磷脂A+ 分枝杆菌细胞壁sk;莫哌达醇;多抗药性基因抑制剂;多肿瘤抑制剂1基本疗法;芥子抗癌剂;mycaperoxide B;分枝杆菌细胞壁提取物;myriaporone;N-乙酰地那林;N-取代的苯甲酰胺;那法瑞林;nagrestip;纳洛酮+潘他唑新;napavin;naphterpin;那托司亭;奈达泊汀;奈莫柔比星;奈立膦酸;中性内肽酶;尼鲁米特;nisamycin;氧化氮调节剂;硝基氧抗氧化剂;nitrullyn;O6-苄基鸟嘌呤;薁曲肽;okicenone;寡核苷酸;薁纳司酮;昂丹司琼;恩丹西酮;oracin;口服细胞因子诱导剂;薁马铂;薁沙特隆;oxaliplain;oxaunomycin;紫杉醇类似物;紫杉醇衍生物;palauamine;棕榈酰利索新;帕米膦酸;人参炔三醇;帕诺米芬;副球菌素;帕折普汀;培加帕酶;培地辛;戊聚糖多硫酸钠,戊制菌素;pentrozole;全氟溴烷;派磷酰胺;紫苏子醇;phenazinomycin;苯乙酸;磷酸酶抑制剂;毕西巴尼;盐酸毛果碱;吡柔比星;吡曲克辛;placetin A;placetin B;溶酶原激活物抑制剂;铂复合物;铂化合物;铂三氨基复合物;泊非美钠;甲基丝裂霉素;丙基双-吖啶酮;前列腺素J2;蛋白酶体抑制剂;蛋白A基的免疫调节剂;蛋白激酶C抑制剂;蛋白激酶C抑制剂;微小藻类;蛋白酪氨酸磷酸酶抑制剂;嘌呤核苷磷酸化酶抑制剂;嘌呤;吡唑啉吖啶;吡醇羟乙酯血红蛋白聚氧乙烯交联物;raf拮抗剂;雷替曲塞;拉莫司琼;ras法呢酯蛋白转移酶抑制剂;ras抑制剂;ras-GAP抑制剂;脱甲基化瑞替普汀;羟乙磷酸铼Re 186;利索新;核酶;RII维甲胺;罗谷亚胺;rohitukine;罗莫肽;罗喹美克;rubiginone B1;ruboxyl;沙芬戈;saintopin;SarCNU;肌植醇A;沙格司亭;Sdi 1模拟物;司莫司汀;衰老衍生抑制剂1;有义链寡核苷酸;信号转导抑制剂;信号转导调节剂;单链抗原结合蛋白;西佐喃;索布佐生;硼卡钠;苯乙酸钠;solverol;生长调节素结合蛋白;索纳明;斯帕福斯酸;穗霉素D;螺莫司汀;splenopentin;海绵素1;角鲨烯胺;干细胞抑制剂;干细胞分裂抑制剂;stipiamide;溶基质素抑制剂;sulfmosine;超活跃的血管活性肠肽拮抗剂;suradista;苏拉明;苦马豆素;合成的糖胺聚糖;他莫司汀;他莫昔芬methiodide;牛磺莫司汀;他佐罗汀;替可加兰钠(tecogalan sodium);替加氟;tellurapyrylium;端粒酶抑制剂;替莫卟吩;替莫唑胺;表鬼臼毒噻吩糖苷;tetrachlorodecaoxide;tetrazomine;菌体胚素;噻可拉林;血小板生成素;血小板生成素模拟物;胸腺法新;胸腺生成素受体激动剂;胸腺曲南;促甲状腺激素;乙基初卟啉锡(tin ethyl etiopurpurin);替拉扎明;二氯二茂钛;topsentin;托瑞米芬;全能干细胞因子;翻译抑制剂;维A酸;三乙酰基尿苷;曲西立滨;三甲曲沙;曲普瑞林;托烷司琼;妥罗雄脲;酪氨酸激酶抑制剂;酪氨酸磷酸化抑制剂;UBC抑制剂;乌苯美司;尿生殖窦衍生生长抑制因子;尿激酶受体拮抗剂;伐普肽;variolin B;载体系统;红细胞基因疗法;维拉雷琐;藜芦明;verdins;维特卟吩;长春瑞宾;vinxaltine;vitaxin;伏罗唑;扎诺特隆;折尼铂;亚苄维C和净司他丁斯酯等。
在另一个实施方案中,所述其它抗癌剂是干扰素-α。在另一个实施方案中,所述其它抗癌剂是白介素-2。在一个实施方案中,所述其它抗癌剂是烷化剂,诸如氮芥、亚硝基脲、烷基磺酸盐、三氮烯或含铂剂。在一个实施方案中,所述其它抗癌剂是三氮烯烷化剂。在一个实施方案中,所述其它抗癌剂是O-6-苯甲基鸟嘌呤。在另一个实施方案中,所述其它抗癌剂是O-6-苯甲基鸟嘌呤和替莫唑胺。在另一个实施方案中,所述其它抗癌剂是O-6-苯甲基鸟嘌呤和丙卡巴肼。在另一个实施方案中,所述其它抗癌剂是O-6-苯甲基鸟嘌呤和达卡巴嗪。
本发明的化合物可以施用给这样的受试者,所述受试者已经经历或目前正经历一种或多种其它抗癌疗法,包括、但不限于,外科手术、辐射疗法或免疫疗法,诸如癌症疫苗。
在一个实施方案中,本发明提供了用于治疗或预防癌症的方法,所述方法包括:给有此需要的受试者施用有效量的(1) 本发明的化合物和(2) 其它抗癌疗法,包括、但不限于,外科手术、辐射疗法或免疫疗法,诸如癌症疫苗。
在一个实施方案中,所述其它抗癌疗法是辐射疗法。在另一个实施方案中,所述其它抗癌疗法是外科手术。在另一个实施方案中,所述其它抗癌疗法是免疫疗法。
在一个具体实施方案中,本发明的用于治疗或预防癌症的方法包括:施用有效量的本发明的化合物和辐射疗法。辐射疗法可在本发明的化合物的同时、之前或之后施用,在一个实施方案中,在本发明的化合物施用之前或之后的至少一小时、五小时、12小时、一天、一周、一个月,在另一个实施方案中,在本发明的化合物施用之前或之后的几个月(例如,多至三个月)。在其它的抗癌疗法为辐射疗法的情况下,取决于所治疗的癌症类型,可利用任何辐射疗法方案。例如,而不是当作限制,可施用X射线辐射;尤其是,高能兆伏(大于1MeV能量的放射线)可用于深度肿瘤,并且电子束和中电压X射线辐射可用于皮肤癌。还可以施用发射γ-射线的放射性同位素,如镭、钴和其它元素的放射性同位素。
另外,本发明提供了治疗癌症的方法,所述方法包括:施用本发明的化合物作为化学疗法或辐射疗法的替代方案,其中化学疗法或辐射疗法在所治疗的受试者中会引起负面的副作用。所治疗的受试者可任选地用其它抗癌疗法诸如外科手术、辐射疗法或免疫疗法进行治疗。
本发明的化合物还可以体外或离体施用,例如用于治疗某些癌症,包括、但不限于白血病和淋巴瘤,所述治疗包括自体干细胞移植。这可包括这样的过程:其中收集受试者的自体造血干细胞,并清除所有癌细胞,随后通过施用本发明的化合物或辐射或二者,根除受试者的残留的骨-骨髓细胞群,并将生成的干细胞灌注回受试者中。在骨髓功能恢复以及受试者康复过程中,可接着提供支持性的护理。
B. 治疗或预防神经变性疾病
本发明提供了用于治疗或预防神经变性疾病的方法,所述方法包括:将有效量的本发明的化合物施用给有此需要的受试者。在一个实施方案中,所述化合物存在于组合物中,所述组合物另外包含药学上可接受的媒介物。
神经变性疾病的例子包括,但不限于:亚历山大氏病、阿耳珀病、阿尔茨海默氏病、肌萎缩性侧索硬化(“ALS”)、共济失调性毛细血管扩张。Batten病(也称作Spielmeyer-Vogt-Sjogren-Batten病)、牛海绵状脑病、卡纳万病、科凯恩综合征、皮质基底变性、克雅病、亨廷顿病、HIV-相关的痴呆、肯尼迪氏病、克拉伯氏病、Lewy体痴呆、马-约病(脊髓小脑性共济失调3型)、多发性硬化(“MS”)、多系统萎缩、发作性睡病、神经包柔螺旋体病、帕金森病、佩-梅二氏病、皮克病、原发性侧索硬化、朊病毒疾病、进行性核上麻痹、雷夫苏姆病、Sandhoff氏病、谢耳德病、恶性贫血引起的亚急性脊髓混合变性、脊髓小脑性共济失调、脊髓性肌萎缩、Steele-Richardson-Olszewskidisease和脊髓痨。在一个实施方案中,所述神经变性疾病是阿尔茨海默氏病。神经变性疾病的其它例子包括,但不限于:弥散性Lewy体病、多系统变性(Shy-Drager综合征)、运动神经元疾病包括肌萎缩性侧索硬化、退行性共济失调s、皮质基底变性、关岛ALS-帕金森-痴呆综合症、亚急性硬化性全脑炎、亨廷顿病、共核蛋白病、原发性进行性失语症、纹状体黑质变性、马-约病/脊髓小脑性共济失调3型和橄榄体脑桥小脑变性、日拉斯德拉图拉泰病、延髓麻痹和假性延髓麻痹、脊髓和脊髓延髓肌萎缩(肯尼迪氏病)、原发性侧索硬化、家族性痉挛性截瘫、韦-霍二氏综合征、、库-韦二氏病、Tay-Sach氏病、Sandhoff病、家族性、痉挛性疾病、Wohifart-Kugelberg-Welander病、痉挛性轻截瘫、进行性多灶性白质脑病、朊病毒疾病(包括克雅氏病、Gerstmann-Straussler-Scheinker病、库鲁病和家族致命性失眠症)、年龄相关的痴呆和与记忆丧失有关的其它病症,诸如血管性痴呆、弥散性白质疾病(宾斯旺格病)、内分泌或代谢起源的痴呆、头创伤和弥散性脑损伤的痴呆、拳击员痴呆和额叶痴呆、脑缺血或梗塞包括栓子阻塞和血栓阻塞以及任何类型的颅内出血(包括、但不限于、硬膜外的、硬膜下的、蛛网膜下的和大脑内的出血)和颅内的和脊柱内的病变(包括、但不限于、挫伤、穿透、剪切、压迫和撕裂)。
在一个方面,本发明的用于治疗或预防神经变性疾病的方法可以另外包括:施用其它抗神经变性疾病剂。
在一个实施方案中,本发明提供了用于治疗或预防神经变性疾病的方法,所述方法包括:给需要治疗或预防神经变性疾病的受试者施用有效量的本发明的化合物和其它抗神经变性疾病剂。本发明的化合物和其它抗神经变性疾病剂可以单独施用。本发明的化合物和其它抗神经变性疾病剂还可以同时施用。在该实施方案中,本发明的化合物和其它抗神经变性疾病剂可以在相同的组合物内施用,或者可以从不同的组合物经由相同或不同的给药途径施用。在另一实施方案中,本发明的化合物在其它抗神经变性疾病剂发挥其预防或治疗作用期间施用,或反之亦然。
在另一个实施方案中,本发明的化合物或其它抗神经变性疾病剂以当所述药剂用作神经变性疾病治疗的单一治疗时通常采用的剂量施用。
在一个实施方案中,本发明的化合物或其它抗神经变性疾病剂以少于当所述药剂用作神经变性疾病治疗的单一治疗时通常采用的剂量施用。
在另一个实施方案中,本发明的化合物和其它抗神经变性疾病剂协同作用,并以少于当所述药剂用作神经变性疾病治疗的单一治疗时通常采用的剂量施用。所施用的本发明的化合物或其它抗神经变性疾病剂的剂量以及给药方案取决于各种参数,包括、但不限于:正治疗的神经变性疾病、受试者的总体健康状况和主治医师的考虑。本发明的化合物可在其它抗神经变性疾病剂施用于需要治疗或预防神经变性疾病的受试者之前(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周之前)、同时、或之后(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周之后)施用。在不同的实施方案中,本发明的化合物和其它抗神经变性疾病剂相隔1分钟、相隔10分钟、相隔30分钟、相隔小于1小时、相隔1小时至2小时、相隔2小时至3小时、相隔3小时至4小时、相隔4小时至5小时、相隔5小时至6小时、相隔6小时至7小时、相隔7小时至8小时相隔8小时至9小时、相隔9小时至10小时、相隔10小时至11小时、相隔11小时至12小时、相隔不超过24小时或相隔不超过48小时施用。在一个实施方案中,本发明的化合物和其它抗神经变性疾病剂相隔3小时内施用。在另一个实施方案中,本发明的化合物和其它抗神经变性疾病剂相隔1分钟至24小时施用。
在一个实施方案中,有效量的本发明的化合物和有效量的其它抗神经变性疾病剂存在于同一组合物中。在一个实施方案中,该组合物可用于口服给药。在另一个实施方案中,该组合物可用于静脉内给药。
在一个实施方案中,所述组合物包含在一起可有效地治疗或预防神经变性疾病的量的本发明的化合物和其它抗神经变性疾病剂。
本发明的化合物和其它抗神经变性疾病剂可累加或协同作用。本发明的化合物和其它抗神经变性疾病剂的协同组合可允许使用更低剂量的所述药剂中的一种或两种,和/或更低频率地给患有神经变性疾病的受试者施用所述药剂。使用更低剂量的本发明的化合物和其它抗神经变性疾病剂中的一种或两种和/或以更低频率施用所述药剂的能力,可降低与给受试者施用该药剂有关的任何毒性,而不降低该药剂在神经变性疾病治疗中的效力。此外,协同效应可导致这些药剂在神经变性疾病治疗中提高的效力,和/或与任一药剂单独使用有关的任何不利的或不希望的副作用的减少。
在一个实施方案中,有效量的本发明的化合物和有效量的其它抗神经变性疾病剂的施用会抑制神经变性疾病对其它抗神经变性疾病剂的抗性。
可用于本发明的方法和组合物中的合适的其它抗神经变性疾病剂包括,但不限于:抗阿尔茨海默病药剂,诸如胆碱酯酶抑制剂(例如,他克林、盐酸多奈哌齐、利斯的明或加兰他敏)或部分谷氨酸盐拮抗剂(例如,美金刚);抗帕金森病药剂,诸如左旋多巴、卡比多巴、托卡朋、溴隐亭、培高利特、普拉克索、罗匹尼罗、司来吉兰或金刚烷胺;抗-ALS剂,诸如利鲁唑;和抗-MS剂,诸如干扰素β-1a、干扰素β-1b、乙酸格拉替雷、米托蒽醌或那他珠单抗。
C . 联合治疗
可以与本发明的化合物相组合用于治疗或预防病症(例如,与γ-分泌酶活性有关的疾病,或预防与γ-分泌酶活性有关的疾病)的其它药剂包括,但不限于:小分子、合成药物、肽(包括环肽)、多肽、蛋白、核酸(例如,DNA和RNA核苷酸,包括、但不限于,反义核苷酸序列、三螺旋、RNAi和编码生物活性的蛋白、多肽或肽的核苷酸序列)、抗体、合成的或天然的无机分子、模仿剂、和合成的或天然的有机分子。这样的药剂的具体例子包括,但不限于:免疫调节剂(例如,干扰素)、抗炎剂(例如,肾上腺皮质素、皮质类固醇(例如,倍氯米松、布地奈德、氟尼缩松、氟替卡松、曲安西龙、甲泼尼龙、泼尼松龙、泼尼松、氢化可的松)、糖皮质激素、类固醇和非甾醇抗炎药(例如,阿司匹林、布洛芬、双氯芬酸和COX-2抑制剂)、镇痛剂、白三烯拮抗剂(例如,孟鲁司特、甲基黄嘌呤、扎鲁司特和齐留通)、β2-激动剂(例如,沙丁胺醇、维生素A(biterol)、非诺特罗、异他林(isoetharie)、奥西那林、吡布特罗、沙丁胺醇、特布他林(terbutalin)福莫特罗、沙美特罗和沙丁胺醇特布他林)、抗胆碱能药(例如,异丙托溴铵和氧托溴铵)、柳氮磺吡啶、青霉胺、氨苯砜、抗组胺剂、抗疟剂(例如,羟基氯喹)、抗病毒剂(例如,核苷类似物(例如,齐多夫定、阿昔洛韦、丙氧鸟苷(gangcyclovir)、阿糖腺苷、碘苷、曲氟尿苷和利巴韦林)、膦甲酸、金刚烷胺、金刚乙胺、沙奎那韦、茚地那韦、利托那韦和AZT)和抗生素(例如,更生霉素(以前称作放线菌素)、博来霉素、红霉素(erythomycin)、青霉素、普卡霉素和安曲霉素(AMC))。
V . 治疗性或预防性施用和本发明的组合物
由于它们的活性,本发明的化合物有利地可用于兽药和人药中。
当施用给受试者时,本发明的化合物可以作为组合物中的组分给药,所述组合物包含药学上可接受的载体、稀释剂、赋形剂或媒介物。本发明的组合物(其包含本发明的化合物)可以口服给药。本发明的化合物还可以通过任何其它方便的途径给药,例如,通过输注或快速推注,通过上皮或皮肤粘膜衬里(例如口腔粘膜、直肠粘膜或肠粘膜)的吸收,并且可与其它的生物活性剂一同给药。给药可以是全身的或局部的。各种递送系统是已知的,例如包囊在脂质体、微粒、微胶囊和胶囊中。
给药方法包括、但不限于皮内、肌内、腹膜内、静脉内、皮下、鼻内、硬膜外、口服、舌下、脑内、阴道内、透皮、直肠给药,通过吸入给药,或局部给药,特别是对耳、鼻、眼或皮肤的局部给药。在有些情况下,给药将使本发明的化合物释放入血流中。
在一个实施方案中,本发明的化合物口服施用。在其它实施方案中,理想地可将本发明的化合物局部给药。这可以通过例如以下的非限制性方式实现:在手术期间通过局部输注;局部施用(如在术后与创伤敷料结合使用);通过注射,借助于导管、借助于栓剂或灌肠剂、或借助于植入物,所述植入物为多孔的、无孔的或凝胶状物质,包括膜(如硅橡胶膜)或纤维。
在某些实施方案中,可能希望通过任何适当的途径将本发明的化合物引入到中枢神经系统或胃肠道中,所述途径包括心室内注射、鞘内注射、硬膜外注射和灌肠剂。心室内注射可以通过心室内导管变得更为方便,所述导管如附着于蓄池如Ommaya蓄池的心室内导管。
还可使用经肺给药,例如通过使用喷雾器的吸入器、和与雾化剂一起配制,或通过在碳氟化合物或合成的肺表面活性剂中的灌注。在某些方案中,本发明的化合物可以与常规的粘合剂和赋形剂(如甘油三酯)配制成栓剂。
在另一个实施方案中,本发明的化合物可以在囊泡、特别是脂质体中递送(参见Langer, Science 249:1527-1533 (1990)和Liposomes in Therapy of Infectious Disease and Cancer 317-327和353-365 (1989))。
在另一个实施方案中,本发明的化合物可以在控释系统或缓释系统中递送(参见 ,例如 ,Goodson, Medical Applications of Controlled Release, 出处同上, 第2卷, 第115-138页(1984))。可使用在Langer, Science 249: 1527-1533 (1990) 的综述中讨论的其它的控释或缓释系统。在一个实施方案中,可使用泵(Langer, Science 249: 1527- 1533 (1990); Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald等人, Surgery 88:507 (1980);和Saudek等人, N. Engl . J Med. 321:574 (1989))。在另一个实施方案中,可使用聚合材料(参见 Medical Applications of Controlled Release (Langer和Wise编, 1974); Controlled Drug Bioavailability, Drug Product Design and Performance (Smolen和Ball编, 1984); Ranger and Peppas, J . Macromol. Sd. Rev. Macromol. Chem. 2:61 (1983); Levy等人, Science 228:190 (1935); During等人, Ann. Neural. 25:351 (1989);和Howard等人, J . Neurosurg. 71:105 (1989))。
在另一个实施方案中,可将控释或缓释系统置于本发明的化合物的目标(如脊柱、脑、皮肤、肺或胃肠道)附近,因此仅需要全身剂量的一部分。
本发明的组合物可以任选包括适当量的药学上可接受的赋形剂,以提供适合施用给受试者的形式。
这样的药用赋形剂可以是液体如水和油,包括石油、动物油、植物油或合成来源的油,如花生油、大豆油、矿物油、芝麻油等。所述药用赋形剂可以是盐水、金合欢树胶、明胶、淀粉糊、滑石粉、角蛋白、胶体二氧化硅、脲等。另外,可使用辅助剂、稳定剂、增稠剂、润滑剂和着色剂。在一个实施方案中,所述药学上可接受的赋形剂在施用给受试者时是无菌的。当本发明的化合物经静脉内给药时,水是有用的赋形剂。还可以使用盐水溶液和葡萄糖水溶液和甘油溶液作为液体赋形剂,特别是对于可注射的溶液而言。合适的药用赋形剂还包括淀粉、葡萄糖、乳糖、蔗糖、明胶、麦芽、米、面粉、白垩、硅胶、硬脂酸钠、单硬脂酸甘油酯、滑石粉、氯化钠、脱脂奶粉、甘油、丙二醇、乙二醇、水、乙醇等。如果需要,本发明的组合物还可包括少量的润湿剂或乳化剂或pH缓冲剂。
本发明的组合物可以是以下形式:溶液、悬浮剂、乳剂、片剂、丸剂、弹丸剂、胶囊剂、含有液体的胶囊剂、粉末剂、缓释剂、栓剂、乳剂、气雾剂、喷雾剂、混悬剂、或任何其它适于使用的剂型。在一个实施方案中,组合物是胶囊形式(参见例如美国专利号5,698,155)。合适的药用赋形剂的其它例子参见Remington 's Pharmaceutical Sciences 1447-1676 (Alfonso R. Gennaro编, 1995年第19版),其通过引用并入本文。
在一个实施方案中,本发明的化合物根据常规过程配制为适于对人口服给药的组合物。用于口服递送的组合物可以是例如片剂、锭剂、水悬浮剂或油悬浮剂、颗粒、粉末剂、乳剂、胶囊剂、糖浆剂或酏剂的形式。口服施用的组合物可以含有一种或多种试剂,例如,甜味剂如果糖、阿斯巴甜或糖精;调味剂,如薄荷、冬青油或樱桃;着色剂;和防腐剂,以提供药学适口的制剂。另外,其中在片剂或药丸形式中,组合物可以被包衣以延迟在胃肠道中的崩解和吸收,从而提供在延长时段内的持续作用。包围渗透活性的驱动化合物的选透性膜也适用于口服施用的组合物。在这些后述平台中,得自胶囊周围环境的流体可以被驱动化合物吸入,其溶胀以移动药剂或药剂组合物通过孔。这些递送平台可以提供与立即释放制剂的尖峰曲线相比基本上为0级的递送曲线。还可以使用延时材料如单硬脂酸甘油酯或硬脂酸甘油酯。口服组合物可包括标准的赋形剂,如甘露醇、乳糖、淀粉、硬脂酸镁、糖精钠、纤维素和碳酸镁。在一个实施方案中,赋形剂为药用级的。
在另一个实施方案中,本发明的化合物可以配制用于静脉内给药。一般地,用于静脉内给药的组合物包括无菌等渗水性缓冲液。如果必要,该组合物也可包括增溶剂。静脉内给药用组合物可任选包括局部麻醉剂如利多卡因,以减少在注射部位的疼痛。
通常,各成分可以分别供给、或在单位剂型中混合在一起供给,例如作为在标明活性剂用量的气密密封的容器如安瓿或药囊中的冻干粉末或无水浓缩物。当本发明的化合物通过输注给药时,它们可以使用例如含有无菌药用级的水或盐水的输液瓶进行分配。当本发明的化合物通过注射给药时,可以提供注射用无菌水或盐水的安瓿,使得各成分在给药前可以混合。
本发明的化合物可以通过控释或缓释装置或通过本领域普通技术人员公知的递送装置进行给药。例子包括、但不限于:在美国专利号3,845,770、3,916,899、3,536,809、3,598,123、4,008,719、5,674,533、5,059,595、5,591,767、5,120,548、5,073,543、5,639,476、5,354,556和5,733,556中所述的那些,它们中的每一篇通过引用整体并入本文。这些剂型可使用例如以下的物质用于提供一种或多种活性成分的控释释放或持续释放:羟丙基甲基纤维素、其它聚合基质、凝胶、渗透膜、渗透系统、多层包衣、微粒、脂质体、微球体、或其组合,以提供不同比例的所需的释放曲线。本领域技术人员公知的适当的控释或缓释制剂包括本文所述的那些,可以容易地选择用于式I至XI的活性成分。因此,本发明提供了适于口服给药的单个单位剂型,例如但不限于适于控释或缓释的片剂、胶囊、软胶囊和囊片。
活性成分的控释或缓释释放可以被各种条件刺激,所述条件包括、但不限于:pH改变、温度改变、酶的浓度或可用性、水的浓度或可用性、或其它生理条件或化合物。有效用于治疗或预防神经变性疾病的本发明的化合物的量可以通过标准临床技术测定。另外,可以任选地采用体外或体内测定以帮助鉴定最佳的剂量范围。使用的精确剂量还依赖于给药途径、治疗的病症的严重性,并且可根据从业人员的判断和每名受试者的情况,考虑例如公开的临床研究进行判断。然而,合适的有效剂量可以为每4小时约10微克到约5克,尽管它们通常为每4小时约500毫克或更低。在一个实施方案中,有效剂量为每4小时约0.01 mg、0.5 mg、约1 mg、约50 mg、约100 mg、约200 mg、约300 mg、约400 mg、约500 mg、约600 mg、约700 mg、约800 mg、约900 mg、约1 g、约1.2 g、约1.4 g、约1.6 g、约1.8 g、约2.0 g、约2.2 g、约2.4 g、约2.6 g、约2.8 g、约3.0 g、约3.2 g、约3.4 g、约3.6 g、约3.8 g、约4.0 g、约4.2 g、约4.4 g、约4.6 g、约4.8 g和约5.0 g。可以经不同的时段施用相等的剂量,包括、但不限于:约每2小时、约每6小时、约每8小时、约每12小时、约每24小时、约每36小时、约每48小时、约每72小时、约每周、约每两周、约每三周、约每月和约每两月。本文所述的有效剂量是指总的给药量;也就是说,如果施用超过一种的本发明的化合物,则有效剂量相当于总的给药量。
组合物可以分别根据常规的混合、造粒或包衣方法制备,并且,按重量或体积计,在一个实施方案中,本发明的组合物可以包含约0.1%至约99%的本发明的化合物;在另一个实施方案中,可以包含约1%至约70%的本发明的化合物。
可根据各种因素选择使用本发明的化合物的剂量方案,所述因素包括:受试者的类型、物种、年龄、体重、性别和医学状况;所治疗的疾病的严重程度;给药途径;受试者的肾或肝功能;以及使用的本发明的具体化合物。本发明的化合物可以以单一日剂量施用,或者可以在每天两次、三次或四次的分份剂量中施用总日剂量。此外,通过局部使用适合的鼻内载体的鼻内形式,或者通过透皮途径使用本领域人员熟知的那些透皮皮肤贴剂的形式,可以施用本发明的化合物。为了以透皮递送系统的形式施用,剂量方案中的剂量给药可以是连续的而不是间断的。其它示例性的局部制剂包括霜剂、膏剂、洗剂、气雾剂和凝胶剂,其中本发明的化合物的浓度在约0.1%至约15%(w/w或w/v)范围内。在用于人类之前,可在体外或体内测定本发明的化合物的期望的治疗或预防性的活性。动物模型系统可用于证实安全性和功效。
在某些实施方案中,将本发明的化合物或其药物组合物施用给在下述年龄范围内的人:约0个月至约6个月、约6至约12个月、约6至约18个月、约18至约36个月、约1至约5岁、约5至约10岁、约10至约15岁、约15至约20岁、约20至约25岁、约25至约30岁、约30至约35岁、约35至约40岁、约40至约45岁、约45至约50岁、约50至约55岁、约55至约60岁、约60至约65岁、约65至约70岁、约70至约75岁、约75至约80岁、约80至约85岁、约85至约90岁、约90至约95岁、或约95至约100岁。
在有些实施方案中,将本发明的化合物或其药物组合物施用给婴儿。在其它实施方案中,将本发明的化合物或其药物组合物施用给幼儿。在其它实施方案中,将本发明的化合物或其药物组合物施用给儿童。在其它实施方案中,将本发明的化合物或其药物组合物施用给成年人。在其它实施方案中,将本发明的化合物或其药物组合物施用给老年人。
在某些实施方案中,将本发明的化合物或其药物组合物施用给这样的受试者:所述受试者处于免疫受损状态或免疫免疫抑制状态,或处于变成免疫受损或免疫免疫抑制的风险中。在某些实施方案中,将本发明的化合物或其药物组合物施用给接受免疫抑制疗法或从免疫抑制疗法中恢复的受试者。
在有些实施方案中,将本发明的化合物或其药物组合物施用给易感常规抗-γ-分泌酶疗法的不良反应的患者。在有些实施方案中,将γ-分泌酶抑制剂或其药物组合物施用给这样的患者:所述患者已经被证实是除了γ-分泌酶抑制剂以外的抗-γ-分泌酶疗法所难治的,但是不再处于这些疗法中。在这些患者中包括难治的患者和对于常规疗法而言太年轻的患者。
在有些实施方案中,在施用本发明的化合物或其药物组合物之前,给其施用本发明的化合物或其药物组合物的受试者尚未接受治疗。
IV.包含本发明的化合物的试剂盒
本发明提供了试剂盒,其可以简化本发明的化合物向受试者的施用。
本发明的典型试剂盒包括本发明的化合物,例如,在单位剂型中。在一个实施方案中,单位剂型为容器,其可以是无菌的,包含有效量的本发明的化合物和药学上可接受的载体、稀释剂、赋形剂或媒介物。该试剂盒可另外包括标签或印刷的说明书,其指导本发明的化合物用于治疗或预防病症的用途。该试剂盒还可以另外包括其它预防剂或治疗剂,例如在单位剂型中,例如,含有有效量的其它预防剂或治疗剂的容器。在一个实施方案中,该试剂盒包括容器,所述容器含有有效量的本发明的化合物和有效量的其它预防剂或治疗剂。其它预防剂或治疗剂的例子包括、但不限于如上所列的那些。
在本文中引用的每篇参考文献特此通过引用整体并入。
本发明的化合物的合成
式I的化合物
路线图1:
通过使化合物2(其中E是合适的离去基团)与化合物1(其中R2和R3如在式I中所定义)反应,可以制备3型的卤代-酯。合适的离去基团是本领域众所周知的,但不限于卤化物,诸如氯化物、溴化物和碘化物;芳基磺酰氧基或烷基磺酰氧基、被取代的芳基磺酰氧基(例如,甲苯磺酰氧基或甲磺酰氧基);被取代的烷基磺酰氧基(例如,卤代烷基磺酰氧基);(C6)芳氧基或被取代的(C6)芳氧基;和酰氧基。化合物2由市售获得(例如Aldrich Chemical Co.,Milwaukee,威斯康辛州),或可以用众所周知的方法制备,例如丁二醇的卤化或磺化。化合物1也由市售获得(例如Aldrich Chemical Co.,Milwaukee,Wisconsin),或可以由众所周知的方法获得,例如列于以下文献中的方法:Larock Comprehensive Organic Transformations; Wiley-VCH: New York, 1999,第1754-1755和1765页。J. Mulzer在Comprehensive Organic Functional Transformations, Pergamon, Oxford 1995,第148-151页中对1型的酯的烷基化作了综述,在美国专利号5,648,387第6栏以及Ackerly, 等人, J. Med. Chem. 1995, 第1608页中介绍了使化合物1与化合物2反应的示范性合成操作。所述反应可以在有合适碱存在下进行。在某些实施方案中,合适碱具有大于约25的pKa,在另一个实施方案中,合适碱具有大于约30的pKa。适碱包括,但不限于:烷基金属碱例如二异丙基氨基化锂、甲基锂、正丁基锂、叔丁基锂、仲丁基锂、苯基锂、苯基钠和苯基钾;氨基化金属碱例如氨基化锂、氨基化钠、氨基化钾、四甲基哌啶化锂、二乙基氨基化锂、二环己基氨基化锂、六甲基二硅叠氮化钠和六甲基二硅叠氮化锂;氢化物碱例如氢化钠和氢化钾。特别有用的是氨基化金属碱,例如二异丙基氨基化锂。在本发明的某些实施方案中,为了使化合物1与化合物2反应,可以在惰性气氛下,将约1至约2当量的合适的碱溶液加入到搅拌下的包含酯1以及合适的有机溶剂的溶液中,溶液保持在下述范围内的某一恒定温度:约–95℃至约室温,在某些实施方案中,约–78℃至约–20℃。在有些实施方案中,可以在加入前用合适的有机溶剂稀释所述碱。在有些实施方案中,可以以约1.5 mol/h的加入速率加入所述碱。适合化合物1与化合物2的反应的有机溶剂包括,但不限于:二氯甲烷、乙醚、四氢呋喃、二甲基甲酰胺、二甲亚砜、苯、甲苯、二甲苯、烃溶剂(例如,戊烷、己烷和庚烷)、及其混合物。加入碱后,将反应混合物搅拌约1小时至约2小时,加入化合物2,其可以溶解在合适的有机溶剂中,在某些实施方案中,以使反应混合物温度保持在初始反应混合物温度的约1-2℃范围内的速率加入。加入化合物2后,可以将反应混合物温度调节至约–20℃至约室温的稳定范围内,包括调节至约室温,并将反应混合物搅拌直到检测出反应基本完成,所述检测用适当的分析方法,诸如薄层色谱法(TLC)或高效液相色谱法(HPLC)。然后将反应混合物淬灭,并可以通过后处理分离出化合物3。
进一步,可以使化合物3通过格里尼亚反应与被取代的醛反应,得到醇4。关于格里尼亚反应的示例性操作,参见March, J. Advanced Organic Chemistry; Reactions Mechanisms, and Structure, 第4版, 1992, 第920-929页。在某些实施方案中,如在Drake, N. L.; Cooke, G. B. Org . Synth. Coll. Vol. II, 1943, 406中所述,在乙醚或四氢呋喃中,用Zn或Mg处理化合物3。通过多种方法,可以从醇4开始合成卤化物5。一种方法包括:将醇转化成离去基团,诸如磺酸酯,例如但不限于,例如,甲苯磺酸酯、对溴苯磺酸酯、甲磺酸酯或对硝基苯磺酸酯(nosylate)。然后可以在诸如THF或乙醚等溶剂中用X-源(其中X-是I-、Br-或Cl-)处理该中间体。用于将乙烯醇和苯醇转化成硫醇的一般方法包括:首先将醇转化成离去基团(例如,甲苯磺酸酯),然后用卤化物亲核体处理。示例性的操作参见:Forrest, O. A.; Gregory, C. F. J. Am. Chem. Soc, 2005, 127 , 10482-10483 (NBS/THF),和Possel, O.; van Leusen, A. M. Tetrahedron. Lett ,1977, 48 , 4229-4232 (HCl/MeOH)。
卤化物5可以与不同的杂环偶联,以生成酯6。具体地,如在Aubert, D.; Touch, P. D.; Ferrand, C.; Ramonville, S.; Maffrand, J. 美国专利 4,529,596. 1985中所述,在有碳酸钾存在下,与4,5,6,7-四氢噻吩并[3,2-c ]吡啶(Z1 = S)反应。为了生成本发明的化合物(Z1 = CH2),如在Godar E.M., Mariella R.P., J . Org. Chem ., 1960, 25 , 557-559中所述,使卤化物5与6, 7-二氢-5H -[1]吡啶-3-甲酸偶联。为了生成酯6(Z1= O),可以如在Koike, H.; Asai, F.; Sugidachi, Kimure, T.; Inoue, T.; Nishino, S.; Tsuzaki, Y. 美国专利 5,288,726, 1994中所述,使卤化物5与4,5,6,7-四氢呋喃并[3,2-c]吡啶反应。类似地,可以如在Krichevskii, E. S.; Alekseeva, L. M.; Granik, V. G. Chem . Hetercycl. Compd. 1990, 1235-1238中所述,使卤化物5与吡咯并[3,2-c]吡啶-1-甲酸叔丁基酯反应,以生成本发明的化合物(Z1 = N)。
酯6可以发生随后的官能化,如在例如Carey, F.; Giuliano, R. Organic Chemistry, McGraw-Hill Science/Engineering/Math;第8版(2010), 第20章中所述。作为一个具体实例,通过随后在有氢氧化钾存在下水解酯基团,得到7型的酸(关于一般方法,参见Vogel’s Practical Organic Chemistry, 第4版, Longman Inc.: New York 1978, 第491页)。
路线图2:
路线图2概述了合成本发明的化合物的一般方法。Rieke金属1是商购可得的,或可以制备,并进行与被取代的苯甲醛的反应,如在Zhu L., Wehmeyer R.M., Rieke R.D., J . Org . Chem., 1991, 56 , 1445-1453中所述。可以用硼氢化钠还原酮中间体2,以形成醇3。可以用合适的离去基团处理醇,用于随后的偶联,如上面关于路线图1中的衍生物6的合成所述。合适的离去基团是本领域众所周知的,例如,但不限于卤化物,诸如氯化物、溴化物和碘化物;芳基磺酰氧基或烷基磺酰氧基、被取代的芳基磺酰氧基(例如,甲苯磺酰氧基或甲磺酰氧基);被取代的烷基磺酰氧基(例如,卤代烷基磺酰氧基);(C6)芳氧基或被取代的(C6)芳氧基;和酰氧基。在本发明的某些实施方案中,在有过量的三乙胺存在下,在低温(诸如0-5℃),用甲磺酰氯处理4型化合物,以形成甲磺酸酯。通过在THF中与过量的三乙胺一起加热,使甲磺酸酯4与适当的杂环进一步偶联,以形成衍生物5。通过众所周知的法定(compendial)反应或通过羧酸衍生物的互变,将腈衍生物5转化为不同的其它官能团。
式II的化合物
路线图1:
路线图2:
路线图3:
路线图4
路线图1解释了式4的嘧啶的合成。通过式2的酮酯和式3的脒的缩合,可以完成合成。可以如在Hull R. 等人, 1946, Journal of the Chemical Society第357-362页中所述,进行缩合反应。通过在Raimundo, Brian C., 等人, 2004, Journal of Medicinal Chemistry 47(12), 第3111-3131页中描述的方法,从商购可得的式1的酯(例如Matrix Scientific, Columbia, SC),或者通过在Bashford K. E. 等人, 2003, Tetrahedron Letters 44, 第1627-1629页和Oikawa Y. 等人, 1978, Journal of Organic Chemistry 43(10), 第2087-2088页中描述的方法,从对应的酸(例如Sigma-Aldrich Corp., St. Louis, MO),可以制备式2的酮酯。通常可以用t-Boc、CBZ或苯甲基保护基(但不限于此)保护存在的胺。式3的脒(其中m是0-3的整数) 通常是商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO),但是也可以通过与同样商购可得的2-甲基-2-硫代假脲硫酸盐(例如Sigma-Aldrich Corp., St. Louis, MO)反应从对应的胺制备。一种这样的非限制性操作参见:Bonafoux D. 等人, 2009, Bioorganic & Medicinal Chemistry Letters 19(3), 第912-916页。
路线图2描述了通过使用卤化剂(包括、但不限于PCl5、PBr5、磷酰氯和磷酰溴),将式4的嘧啶转化成对应的式5卤代化合物。该转化的一个实例参见:Altenbach R. J., 等人, 2008, Journal of Medicinal Chemistry 51(20), 第6571-6580页。可以使式5的卤代化合物与一系列碳、氮、氧或硫亲核体反应,以置换卤素并产生更复杂的式6嘧啶类似物。用于置换的试剂是商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO),且通常需要额外碱的存在,所述碱包括、但不限于三乙胺、二异丙基乙胺、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠。在文献中熟知与氮亲核体(如Ghoneim K. M. 等人, 1986, Journal of the Indian Chemical Society, 63(10), 第914-917页)、氧亲核体(如Dubey P. K. 等人, 2006, Indian Journal of Hetercyclic Chemistry 15(4), 第405-406页)、碳亲核体(如Gillespie R.J. 等人, 2009, Bioorganic & Medicinal Chemistry 17(18), 第6590-6605页)和硫亲核体(如Backer H.J. 等人, 1942, Recueil des Travaux Chimiques des Pays-Bas et de la Belgique 61, 第291-298页) 的这些类型的置换反应的例子。
路线图3描述了式4的嘧啶向对应的式7的O-羰基嘧啶的转化。通过在有碱存在下加入对应的酰基氯,可以从式4的嘧啶制备式7的O-羰基嘧啶,所述碱包括、但不限于三乙胺、二异丙基乙胺、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠。所述酰基氯是商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO),或可以通过将商购可得的酸与氯化剂一起加热来制备,其包括但不限于亚硫酰氯或草酰氯。嘧啶的羰基化的一个例子描述在:Shabbir S. 等人, 2010, Tetrahedron 66(35), 第7204-7212页。
路线图4解释了式6和式7的嘧啶向式(II)的最终结构的转化。可以首先使用所用保护基的标准方法,除去保护基,产生式8的嘧啶(关于常见保护基的除去,参见Greene T. W. 和Wuts P. G., 1999, Protective Groups in Organic Synthesis第3版, Wiley-Interscience, New York)。然后可以在有碱存在下用商购可得的试剂(例如Sigma-Aldrich Corp., St. Louis, MO) 烷基化嘧啶,所述碱包括、但不限于三乙胺、二异丙基乙胺、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠。在这些类型的烷基化中,可以使用在Levy D. E. 等人, 2008, Bioorganic & Medicinal Chemistry Letters, 18(7), 第2395-2398页中描述的操作。在式8的嘧啶向式(II)的最终结构的转化中可以使用的第二种常用的方法是还原胺化。可以在有弱酸(包括、但不限于醋酸、三氟醋酸或盐酸)和还原剂(包括、但不限于硼氢化钠、氰基硼氢化钠和三乙酰氧基硼氢化钠) 存在下,用商购可得的醛或酮(例如Sigma-Aldrich Corp., St. Louis, MO) 处理式8的嘧啶,生成期望的式(II)的最终结构。在下述文献中提供的操作,概述了在所述还原胺化反应中可以使用的方法:Taibakhsh M. 等人, 2011, Synthesis, 第490-496页,和Abdel-Magid A. F. 等人, 1996, Journal Organic Chemistry, 61, 第3849-3862页。
另外,通过在下述文献中公开的方法,可以合成式II化合物:Abdel-Magid A. F. 等人, 1996, “Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures(1)”, Journal Organic Chemistry, 61, 第3849-3862页; Altenbach R. J., 等人, 2008, “Structure-Activity Studies on a Series of a 2-Aminopyrimidine-Containing Histamine H4 Receptor Ligands”, Journal of Medicinal Chemistry 51(20), 第6571-6580页; Backer H. J. 等人, 1942, “Several Sulfanilamido-4-methylpyrimidines”,Recueil des Travaux Chimiques des Pays-Bas et de la Belgique 61, 第291-298页; Bashford K. E. 等人, 2003, “The Bohlmann-Ratz Route to Functionalised Pyridine Scaffolds and Their Use in Library Synthesis’, Tetrahedron Letter, 44, 第1627-1629页; Bonafoux D. 等人, 2009, “2-Aminoimidazoles Inhibitors of TGF-Beta Receptor 1”, Bioorganic & Medicinal Chemistry Letters 19(3), 第912-916页; Dubey P. K. 等人, 2006, “Synthesis of Threobromine Incorporated Pyrimidine”,Indian Journal of Hetercyclic Chemistry 15(4), 第405-406页; Ghoneim K. M. 等人, 1986, “Synthesis and Evaluation of some 2-, 4- and 2,4-di-substituted-6-Methylpyrimidine Derivatives for Antimicrobial Activity”, Journal of the Indian Chemical Society, 63(10, 第914-917页; Gillespie R. J. 等人, 2009, “Preparation of Pyrimidine Carboxamides as Purine Receptor, Particularly Adenosine Receptor Antagonists”, Bioorganic & Medicinal Chemistry 17(18), 第6590-6605页; Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 第3版, Wiley-Interscience, New York; Hull R. 等人, 1946, “Synthetic Antimalarials. III. Some Derivatives of Mono- and Dialkylpyrimidines”, Journal of the Chemical Society第357-362页; Levy D. E. 等人, 2008, “Aryl-indolyl Maleimides as Inhibitors of CaMKII-Delta. Part 2.: SAR of the Amine Tether”, Bioorganic & Medicinal Chemistry Letters, 18(7), 第2395-2398页; Oikawa Y. 等人, 1978, “麦尔酮酸in Organic Synthesis. 2. Q General and Versatile Synthesis of Beta-Keto Esters”, Journal of Organic Chemistry 43(10), 第2087-2088页; Raimundo, Brian C. 等人, 2004, “Integrating Fragment Assembly and Biophysical Methods in the Chemical Advancement of Small-Molecule Antagonists of IL-2: An Approach for Inhibiting Protein-Protein Interactions”, Journal of Medicinal Chemistry, 47(12), 第3111-3130页; Shabbir S. 等人, 2010, “Pyrimidine Based Carboxylic Acid Terminated Aromatic and Semiaromatic Hyperbranched Polyamide-esters: Synthesis and Characterization”, Tetrahedron 66(35), 第7204-7212页; Taibakhsh M. 等人, 2011, “Catalyst-Free One-Pot Reductive Alkylation of Primary and Secondary Amines and N,N-Dimethylation of Amino Acids Using Sodium Borohydride in 2,2,2-Trifluoroethanol”, Synthesis, 第490-496页。
式III 的化合物
路线图1:
路线图1概述了用于制备式III化合物的一般合成次序。可用碱(包括、但不限于二异丙基氨基锂、六甲基二甲硅烷基氨基化钠、叔丁醇钠或叔丁醇钾)处理商购(例如Sigma-Aldrich Corp., St. Louis, MO) 可得的式1的酯、酰胺或酸,并用商购(例如Sigma-Aldrich Corp., St. Louis, MO)可得的式2的二卤代烷烃烷基化。在Mueller R. 等人, 2004, Journal of Medicinal Chemistry, 47(24), 第6082-6099页中描述的操作,概述了烷基化反应。可以以在Parikka K. 等人, 2009, Beilstein Journal of Organic Chemistry, 5(22), 第1-5页中描述的方式,在有三苯基膦存在下加热式3的烷基化产物,以制备式4的鏻盐。可以以在Le Bigot Y. 等人, 1988, Tetrahedron 44(4), 第1057-1072页中描述的方式,在有商购可得的式5的醛(例如Sigma-Aldrich Corp., St. Louis, MO) 存在下,用碱(包括、但不限于氢氧化钠或氢氧化钾、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠) 处理式4的鏻盐,以产生式6的烯烃,作为顺式和反式异构体的混合物。以在Crispi G. 等人, 1982, Synthesis 9, 第787-788页中描述的方式,通过在低温在醋酸中与无水卤化氢气体(例如溴化氢气体)缩合,可以卤化式6的顺式和反式异构体的混合物,以制备式7的卤代化合物。通过在有碱(包括、但不限于氢氧化钠或氢氧化钾、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠)存在下与商购(例如Acc Corporation, San Diego CA和Ryan Scientific, Mt. Pleasant SC)可得的式8的环状化合物一起加热式7的卤化物(例如7a的溴化物),可以制备式III的终产物。如在文献(例如,Cheng D. 等人, 2008, Chinese Chemical Letters 19(6), 第689-692页)中关于类似化合物所述,可以制备置换产物。
另外,通过在下述文献中描述的方法,可以合成式III化合物:Cheng D. 等人, 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), 第689-692页; Crispi G. 等人, 1982, “Enamine; 42. A simple Synthesis of 3,4-Diaminobiphenyls”, Synthesis (9), 第787-788页; Le Bigot Y. 等人, 1988, “Reactions in a Slightly Hydrated Solid-Liquid Heterogenous培养基: the Wittig Reaction in Alkaline Hydroxide-Aprotic Organic Solvent System”, Tetrahedron 44(4), 第1057-1072页; Mueller R. 等人, 2004, “Long Hydrocarbon Chain Keto Diols and Diacids that Favorably Alter Lipid Disorders in Vivo”, Journal of Medicinal Chemistry, 47(24), 第6082-6099页; Parikka K. 等人, 2009, “An Expedient Synthsis of 5-n-Alkylresorcinols and Novel 5-n-Alkylresorcinols Hapatens”, Beilstein Journal of Organic Chemistry, 5(22) 第1-5页; Cheng D. 等人, 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), 第689-692页; Crispi G. 等人, 1982, “Enamine; 42. A simple Synthesis of 3,4-Diaminobiphenyls”, Synthesis (9), 第787-788页; Le Bigot Y. 等人, 1988, “Reactions in a Slightly Hydrated Solid-Liquid Heterogenous培养基: the Wittig Reaction in Alkaline Hydroxide-Aprotic Organic Solvent System”, Tetrahedron 44(4), 第1057-1072页; Mueller R. 等人, 2004, “Long Hydrocarbon Chain Keto Diols and Diacids that Favorably Alter Lipid Disorders in Vivo”, Journal of Medicinal Chemistry, 47(24), 第6082-6099页; Parikka K. 等人, 2009, “An Expedient Synthsis of 5-n-Alkylresorcinols and Novel 5-n-Alkylresorcinols Hapatens”, Beilstein Journal of Organic Chemistry, 5(22) 第1-5页。
式IV 化合物
路线图1:
路线图1描述了用于制备式IV化合物的合成途径。在某些实施方案中,所述制备包括:在适当的溶剂中搅拌商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO)式1的醛、商购可得的(例如Acc Corporation, San Diego CA和Ryan Scientific, Mt. Pleasant SC) 式2的环状化合物、和无水苯并三唑3,所述溶剂包括、但不限于乙醚、四氢呋喃、二噁烷和甲苯。所述反应可以在加热和不加热反应物下进行,并可以使用干燥的分子筛来除去反应中的水。然后通过研磨、结晶或使用适当载体(例如硅胶或氧化铝)的柱色谱法,可以纯化式4的产物。操作的例子参见:Katritzky A. 等人, 1989, “A General Method for the Preparation of Beta-Amino Esters”, Synthesis (10), 第747-751页。
式 V 的化合物
路线图1:
路线图2:
路线图3:
路线图1概述了用于制备式V化合物的一般合成次序。可用碱(包括、但不限于二异丙基氨基锂、六甲基二甲硅烷基氨基化钠、叔丁醇钠或叔丁醇钾)处理商购(例如Sigma-Aldrich Corp., St. Louis, MO) 可得的式1的酯、酰胺或酸,并用商购(例如Sigma-Aldrich Corp., St. Louis, MO)可得的式2的二卤代烷烃烷基化。在Mueller R. 等人, 2004, Journal of Medicinal Chemistry, 47(24), 第6082-6099页中描述的操作,概述了烷基化反应。可以以在Parikka K. 等人, 2009, Beilstein Journal of Organic Chemistry, 5(22), 第1-5页中描述的方式,在有三苯基膦存在下加热式3的烷基化产物,以制备式4的鏻盐。可以以在Le Bigot Y. 等人, 1988, Tetrahedron 44(4), 第1057-1072页中描述的方式,在有商购可得的式5的醛(例如Sigma-Aldrich Corp., St. Louis, MO) 存在下,用碱(包括、但不限于氢氧化钠或氢氧化钾、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠) 处理式4的鏻盐,以产生式6的烯烃,作为顺式和反式异构体的混合物。
路线图2解释了式6的酯向式9的卤代化合物的转化。通过还原式6化合物中的酯基团,可以制备式7的醇。多种试剂可用于将这样的酯还原为醇,例如, 参见M. Hudlicky , Reductions in Organic Chemistry, 第2版, 1996第212-217页,特此明确地通过引用并入本文。在某些实施方案中,所述还原可以用氢化物型还原剂进行,所述氢化物型还原剂例如氢化铝锂、硼氢化锂、三乙基硼氢化锂、二异丁基氢化铝、三甲氧基氢化铝锂或双(2-甲氧基)氢化铝钠。关于将酯还原为醇的示例性的、但是非限制性的操作,参见Nystrom等人, 1947, J. Am. Chem. Soc. 69:1197;和Moffet等人, 1963, Org. Synth., Collect. 834(4), 氢化铝锂; Brown等人,1965, J. Am. Chem. Soc. 87:5614, 三甲氧基氢化铝锂; Cerny等人, 1969, Collect. Czech. Chem. Commun. 34:1025, 双(2-甲氧基)氢化铝钠; Nystrom等人, 1949, J. Am. Chem. 71:245, 硼氢化锂;和Brown等人, 1980, J. Org. Chem. 45:1, 三乙基硼氢化锂。可以将存在于式7化合物中的醇转化成卤化物,以制备式8的化合物(关于将醇转化成卤化物的不同方法的示例性讨论,参见March, J. Advanced Organic Chemistry; Reactions Mechanisms, and Structure, 第4版, 1992, 第431-433页)。通过用碱水解酯,可以从酯6直接制备式9的化合物,然后使用商购可得的醇、胺和硫醇,进行标准的偶联技术(使用DCC (N,N'-二环己基碳二亚胺)、EDC (1-乙基-3-(3-二甲基氨基丙基)碳二亚胺)、HBTU (O-(苯并三唑-1-基)-N,N,N′,N′-四甲基脲鎓六氟磷酸盐)(例如)。通过用商购可得的烷基化和酰基化试剂烷基化或羰基化,也可以从式7的化合物制备式9的化合物。还可以通过用商购可得的醇、胺和硫醇置换存在于式8化合物中的卤化物,制备式9的化合物。
在路线图3中,以在Crispi G. 等人, 1982, Synthesis 9, 第787-788页中描述的方式,通过在低温在醋酸中与无水卤化氢气体(例如溴化氢气体)缩合,可以卤化式9的顺式和反式异构体的混合物,以制备式10的卤代化合物。可以如在Taillier C. 等人, 2007, Tetrahedron 63(21), 第3589-3592页中所述制备式11化合物(Z2 = N),通过在有碱(包括、但不限于氢氧化钠或氢氧化钾、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠)存在下使其图10的卤化物偶联,形成式12的化合物。如在文献(例如,Cheng D. 等人, 2008, Chinese Chemical Letters 19(6), 第689-692页)中关于类似化合物所述,可以制备置换产物。可以用碱处理式12的酮,并用一系列商购可得的烷基化(或酰基化)试剂烷基化,以形成式14的化合物。通过在Newman M.S. 等人, 1945, Organic Synthesis 25和Mellegaard-Waetzig S. R. 等人, 2006, Tetrahedron, 第7191-7198页中描述的方法,也可以卤化式12的酮,以形成式13化合物。然后可以用商购可得的含有氮、氧或硫亲核体的化合物置换式13的卤代化合物,以制备式14的酮。通过在有碱存在下用商购可得的膦酸酯或鏻盐与式14的酮成烯,可以制备式V的目标化合物。
另外,通过在下述文献中公开的方法,可以合成式V的化合物:Cheng D. 等人, 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), 第689-692页; Crispi G. 等人, 1982, “Enamine; 42. A simple Synthesis of 3,4-Diaminobiphenyls”, Synthesis (9), 第787-788页; M. Hudlicky, Reductions in Organic Chemistry, 第2版, 1996第212-217页; Le Bigot Y. 等人, 1988, “Reactions in a Slightly Hydrated Solid-Liquid Heterogenous培养基: the Wittig Reaction in Alkaline Hydroxide-Aprotic Organic Solvent System”, Tetrahedron 44(4), 第1057-1072页; March, J. Advanced Organic Chemistry; Reactions Mechanisms, and Structure, 第4版, 1992, 第431-433页; Mellegaard-Waetzig S. R. 等人, 2006, “Selenium-Catalyzed Oxidative halogenations”, Tetrahedron, 第7191-7198页; Mueller R. 等人, 2004, “Long Hydrocarbon Chain Keto Diols and Diacids that Favorably Alter Lipid Disorders in Vivo”, Journal of Medicinal Chemistry, 47(24), 第6082-6099页; Newman M.S. 等人, 1945, “2-chlorocyclohexanone”, Organic Synthesis 25; Parikka K. 等人, 2009, “An Expedient Synthsis of 5-n-Alkylresorcinols and Novel 5-n-Alkylresorcinols Hapatens”, Beilstein Journal of Organic Chemistry, 5(22) 第1-5页; Taillier C. 等人, 2007, “Synthesis of 3-Oxooxa and 2-Oxoazacycloak-4-enes by Ring-Closing Metathesis. Application to the Synthesis of an Inhibitor of Cathepsin K”, Tetrahedron 63(21), 第3589-3592页。
式VI的化合物
路线图1:
路线图2:
路线图3:
路线图1描述了用于制备式VI化合物的合成途径。所述制备可以包括:缩合式2和式4的化合物。如在Taillier C. 等人, 2007, Tetrahedron 63(21), 第3589-3592页中所述,用boc保护基可以制备式1的化合物(Z2 = N)。通过在三氟醋酸中搅拌,可以除去保护基,以制备式2的化合物。式4的化合物是商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO),或可以在有阻碍碱(例如二异丙基乙胺) 存在下通过酰化从商购可得的式3化合物制备。可以在极性的非质子溶剂(例如DMF或乙腈)中加热等摩尔量的式2和式4的化合物,以形成式5的置换产物。在文献(例如Cheng D. 等人, 2008, Chinese Chemical Letters 19(6), 第689-692页)中描述了类似的、但是非限制性的置换反应。
路线图2描述了从式5的化合物制备式7的类似物。如在Kall A. 等人, 2010, Synthetic Communications 40(12), 第1730-1735页中所述,通过使含有硫和氮亲核体的商购可得的化合物(例如Sigma-Aldrich Corp., St. Louis, MO)与式5的化合物直接缩合,可以制备式7的类似物。另外,通过在Marx J. 等人, 1983, Tetrahedron 39(9), 第1529-1531页中描述的方法,可以卤化式5的化合物,以形成式6的化合物。可以将式6的化合物与商购可得的含有硫、氮或氧亲核体的化合物(例如Sigma-Aldrich Corp., St. Louis, MO) 一起加热,通过置换形成式7的化合物。
路线图3描述了通过成烯将式7的化合物转化为式VI的目标化合物。可以在有式7的化合物存在下用碱(包括、但不限于氢氧化钠或氢氧化钾、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、二异丙基氨基锂、和氢化钠) 处理商购可得的酰基-鏻盐或酰基-膦酸酯(例如Sigma-Aldrich Corp., St. Louis, MO),以形成式VI的最终化合物。类似操作的非限制性例子参见:Lombardo L. 等人, 1987, Synthetic Communications 8(7), 第463-468页。
另外,通过在下述文献中公开的方法,可以合成式VI的化合物:Cheng D. 等人, 2008, “Synthesis and Activity Evaluation of Some Novel Derivatives of 4,5,6,7-Tetrahydrothiono[3,2-c]-pyridine”, Chinese Chemical Letters 19(6), 第689-692页; Kall A. 等人, 2010, “Microwave-Induced Aza-Michael reaction in Water. A Remakably Simple Procedure”, Synthetic Communications 40(12), 第1730-1735页; Lombardo L. 等人, 1987, “An Improved Procedure for the Conversion of Carbonyl Compounds to Alpha,Beta-Unsaturated Carboxylic Acids”, Synthetic Communications 8(7), 第463-468页; Marx J. 等人, 1983, “A Simple and Convenient Synthesis of Beta-Halo Ketones”, Tetrahedron 39(9), 第1529-1531页; Taillier C. 等人, 2007, “Synthesis of 3-Oxooxa and 2-Oxoazacycloak-4-enes by Ring-Closing Metathesis. Application to the Synthesis of an Inhibitor of Cathepsin K”, Tetrahedron 63(21), 第3589-3592页。
式VII 的化合物
路线图1:
路线图1解释了式VII的化合物的合成。按照在Raines S. 等人, 1976, Journal of Heterocyclic Chemistry 13(4), 第711-716页中描述的操作,在有催化量酸(例如对甲苯磺酸) 存在下,在加热下,可以使商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO)式1的类似物与适当保护的、商购可得的4-哌啶酮(例如Sigma-Aldrich Corp., St. Louis, MO)缩合。可以相应地除去保护基(关于常见保护基的除去,参见Greene T. W. 和Wuts P. G., 1999, Protective Groups in Organic Synthesis第3版, Wiley-Interscience, New York),以形成式4的胺。可以在有额外的碱(包括、但不限于三乙胺、二异丙基乙胺、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、和氢化钠)存在下,使式4的胺与商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO) 式5的酰基氯偶联,以形成式VII的目标酰胺。另外,使用标准的酰胺偶联(使用但不限于DCC (N,N'-二环己基碳二亚胺)、EDC (1-乙基-3-(3-二甲基氨基丙基)碳二亚胺)和HBTU (O-(苯并三唑-1-基)-N,N,N′,N′-四甲基脲鎓六氟磷酸盐)),可以用式4的胺和商业的式5羧酸(例如Sigma-Aldrich Corp., St. Louis, MO) 制备式VII的目标酰胺。
另外,通过在下述文献中公开的方法,可以合成式VII的化合物:Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 第3版, Wiley-Interscience, New York; Raines S. 等人, 1976, “Mannich Reactions. Synthesis of 4,5-Dihydropyrrolo[1,2-a]quinoxalines, 2,3,4,5-Tetrahydro-1H-pyrrolo[1,2-a][1,4]diazapines and 5,6-Dihydropyrrolo[1,2-a][1,4]benzodiazapines”, Journal of Heterocyclic Chemistry 13(4), 第711-716页。
式 VIII 的化合物
路线图1:
路线图1解释了式VIII的酰胺的合成。为了制备式3的中间体,通过在有过量的酸(例如硫酸) 存在下加热,可以使商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO) 式1的胺和苯基肼与2-氯丙烯腈2缩合。在现有文献中存在这类缩合的几个非限制性例子,包括在Ege G. 等人, 1982, Journal of Heterocyclic Chemistry 19(6), 第1265-1266页中描述的操作。以在Quiroga J. 等人, 1998, Journal of Heterocyclic Chemistry 35(2), 第409-412页中描述的方式,可以使式3的中间体与商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO) 麦尔酮酸4和商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO) 式5的醛缩合,以制备式6的酰胺。可以在有碱(包括、但不限于氢氧化钠或氢氧化钾、叔丁醇钾或叔丁醇钠、碳酸钾或碳酸钠、二异丙基氨基锂、和氢化钠)存在下用一系列商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO) 烷化剂(例如烷基卤、酰氯、氯代磺酸酯) 烷基化式6的酰胺,以制备式VIII的目标化合物。
另外,通过在下述文献中公开的方法,可以合成式VIII化合物:Ege G. 等人, 1982, “Aminopyrazoles. III. Novel One-Flask Preparations of 1=Phenylpyrazol-3-amine”, Journal of Heterocyclic Chemistry 19(6), 第1265-1266页。Quiroga J. 等人, 1998, “Reactions of 5-Amino-1-aryl-3-methylpyrazoles with Benzylidene Derivatives of麦尔酮酸: Synthesis and Characterization of Pyrazolo[3,4-b]pyridinones”, Journal of Heterocyclic Chemistry 35(2), 第409-412页。
式 I X 的化合物
路线图1:
路线图2:
路线图1解释了式IX的酰胺的合成。可以使商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO) 图1的胺与任一种烷化剂(例如烷基卤、酰氯、氯代氨基甲酸酯或氯代磺酸酯) 反应,以制备图2的胺。还可以在有还原剂(包括、但不限于硼氢化钠、氰基硼氢化钠和三乙酰氧基硼氢化钠)存在下,通过将图1的胺与商购可得的醛(例如烷基卤、酰氯、氯代磺酸酯)一起搅拌,制备图2的胺。通常使用在Levy D. E. 等人, 2008, Bioorganic & Medicinal Chemistry Letters, 18(7), 第2395-2398页中描述的还原胺化操作。如在Renzi L. 等人, 1956, Gazzetta Chimica Italiana 86, 第1362-1366页中所述,通过在有阻碍碱(例如三乙胺、二异丙基乙胺、二苯乙胺)存在下用商购可得的式3卤化物(例如Sigma-Aldrich Corp., St. Louis, MO) 酰基化式1或式2的结构,可以制备式4的酰胺。以在Demchenko A. M. 等人, 2003, Russian Journal of Organic Chemistry 39(7), 第1025-1028页中描述的方式,通过在有碱存在下用式4的化合物置换式5的胺,可以制备式IX的目标化合物,所述碱包括、但不限于三乙胺、二异丙基乙胺、叔丁醇钾或叔丁醇钠、DMAP、碳酸钾或碳酸钠、或氢化钠。图5的胺可以购自商业来源(例如Sigma-Aldrich Corp., St. Louis, MO),或如路线图2所示制备。
路线图2解释了用于合成式5的胺的一般方法。使用普通文献操作,可以在氮上用合适的保护基(boc、苯甲基或CBZ) 选择性地保护商购可得的(例如Sigma-Aldrich Corp., St. Louis, MO) 式6的氨基-醇。可以用合适的烷化剂(例如烷基卤、酰氯、氯代氨基甲酸酯或氯代磺酸酯)和商购可得的烷化剂(例如Sigma-Aldrich Corp., St. Louis, MO) 烷基化在式7中间体中存在的氧,以形成式8的中间体。然后可以使用标准方法(参见Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 第3版, Wiley-Interscience, New York) 除去保护基,以制备式9的中间体。从前述的商业可得到的试剂,通过烷基化或还原胺化,可以从式9的胺制备式5的胺。
另外,通过在下述文献中公开的方法,可以合成式IX化合物:Demchenko A. M. 等人, 2003, “Synthesis of N5-(Arylcarbonyl)methyl Derivatives of Spinaceamine and 2-Azaspinacaceamine”, Russian Journal of Organic Chemistry 39(7), 第1025-1028页; Greene T. W. and Wuts P. G., 1999, Protective Groups in Organic Synthesis, 第3版, Wiley-Interscience, New York; Levy D. E. 等人, 2008, “Aryl-indolyl Maleimides as Inhibitors of CaMKII-Delta. Part 2.: SAR of the Amine Tether”, Bioorganic & Medicinal Chemistry Letters, 18(7), 第2395-2398页;和Renzi L. 等人, 1956, “The 1,4-Benzodioxan Series V. Some Derivatives of 7-Aminobenzodioxan”, Gazzetta Chimica Italiana 86, 第1362-1366页。
通过参考下述实施例,进一步定义了本发明。
实施例
实施例1. 5-(2-氯苯基)-5-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基戊酸盐酸盐(化合物Ia盐酸盐)
步骤1:5-(2-氯苯基)-2,2-二甲基-戊-4-烯酸乙酯的合成。
将鏻盐(15.0 g, 30.9 mmol)和2-氯苯甲醛(4.3 g, 30.6 mmol) 溶解在二氯甲烷(100 mL)中,并剧烈混合。历时5分钟逐滴加入氢氧化钠溶液(24 g, 50%)。将混合物在室温搅拌2小时。加入水(100 mL),并分离各层。将二氯甲烷级分经硫酸钠干燥、过滤并浓缩。通过在硅胶(150 g)上的柱色谱法,纯化剩余的橙色油(8.3 g),用5%乙酸乙酯在庚烷中的溶液洗脱,得到作为无色油的5-(2-氯苯基)-2,2-二甲基-戊-4-烯酸乙酯(4.02 g, 55%收率),它是顺/反异构体的混合物。1H NMR (300 MHz, CDCl3/TMS):δ= (主要级分; 60:40顺/反异构体混合物) 7.46 (dd, 0.6H, J = 7.5, 1.5 Hz), 7.35 (dd, 0.4H, J = 7.2, 1.5 Hz), 7.32-7.08 (m, 4H), 6.77 (d, 0.6H, J = 15.5 Hz), 6.57 (d, 0.4H, J = 11.7 Hz), 6.13 (dt, 0.6H, J= 15.5, 7.5 Hz), 5.74 (dt, 0.4H, J = 11.7, 7.5 Hz), 4.11 (m, 2H), 2.45 (m, 2H), 1.26-1.15 (m, 9H)。13C NMR (75 MHz, CDCl3/TMS):δ= (主要级分; 60:40顺/反异构体混合物) 177.16, 177.12, 135.55, 133.82, 133.62, 132.60, 130.55, 129.53, 129.38, 129.31, 129.26, 129.15, 128.59, 128.15, 126.82, 126.75, 126.22, 60.52, 44.12, 42.79, 42.40, 38.66, 25.19, 25.08, 14.46, 14.34. MS (GC-EI):C15H19O2Cl (MH) +的计算值:267.1146,实测:267.1143。
步骤2:5-溴-5-(2-氯苯基)-2,2-二甲基戊酸乙酯的合成。
将5-(2-氯苯基)-2,2-二甲基-戊-4-烯酸乙酯(4.0 g, 15.0 mmol)溶解在醋酸(40 mL)中。在冰水浴中冷却烧瓶,在将烧瓶温热至室温的同时,用溴化氢气体在溶液中鼓泡5小时。5小时以后,停止溴化氢气体,并将烧瓶在冰箱中保存过夜(0℃)。在0℃20小时以后,将该溶液倒入冰和水的混合物(200 g)中。用二氯甲烷(2 x 75 mL) 萃取产物。合并二氯甲烷级分,并用饱和的碳酸氢钠溶液(200 mL)和水(100 mL)洗涤。将二氯甲烷经硫酸钠干燥、过滤并浓缩。通过在硅胶(200 g)上的快速柱色谱法纯化剩余的油(5.13 g),用5%乙酸乙酯在庚烷中的溶液洗脱,得到作为无色油的5-溴-5-(2-氯苯基)-2,2-二甲基戊酸乙酯(3.31 g, 63.5%收率)。1H NMR (300 MHz, CDCl3/TMS):δ= 7.57 (dd, 1H, J = 7.5, 1.5 Hz), 7.34-7.16 (m, 3H), 5.39 (t, 1H, J = 7.5 Hz), 4.11 (q, 2H, J = 7.2 Hz), 2.29-2.04 (m, 2H), 1.77 (dt, 1H, J = 12.0, 4.5 Hz), 1.52 (dt, 1H, J = 12.0, 4.2 Hz), 1.23 (t, 3H, J = 7.2 Hz), 1.18 (s, 3H), 1.17 (s, 3H)。13C NMR (75 MHz, CDCl3/TMS):δ= 177.05, 139.06, 132.67, 129.65, 129.31, 128.70, 127.43, 60.49, 50.25, 41.87, 38.78, 34.85, 25.64, 25.08, 14.42. HRMS (CI-TOF):C15H20O2BrCl (MH+)的计算值: 347,实测:267.1146. (在MS中消除)。
步骤3:5-(2-氯苯基)-5-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基戊酸乙酯(化合物Ib)的合成
将4,5,6,7-四氢噻吩并[3,2-c]吡啶(0.65 g, 4.61 mmol)和5-溴-5-(2-氯苯基)-2,2-二甲基戊酸乙酯(1.42 g, 4.08 mmol)溶解在含有少量DMAP晶体的THF (5 mL)和三乙胺(3 mL)中。使用油浴将该溶液加热至85℃过夜。20小时以后,将溶液冷却至室温,并加入碳酸氢钠溶液(20 mL)。混合以后,用二氯甲烷(2x 25 mL)萃取产物。将合并的二氯甲烷级分经硫酸钠干燥、过滤并浓缩。使用硅胶(40 g),通过在Companion色谱站上的MPLC纯化剩余的黄色油,历时20分钟用100% 庚烷至10%乙酸乙酯的梯度洗脱,然后历时40分钟用至100%乙酸乙酯的第二梯度洗脱。收集第二个宽级分,并浓缩,以生成5-(2-氯苯基)-5-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基戊酸乙酯(化合物Ib, 1.11 g, 67.2%收率),为浅黄色油。1H NMR (300 MHz, CDCl3/TMS):δ= 7.46 (dd, 1H, J = 7.80, 1.5 Hz), 7.38(dd, 1H, J = 7.80, 1.5 Hz), 7.30-7.15 (m, 2H), 7.04 (d, 1H, J = 5.1 Hz), 6.70 (d, 1H, J = 5.1 Hz), 4.18 (dd, 1H, J = 8.7, 4.5 Hz), 4.08 (q, 2H, J= 7.2 Hz), 3.79 (d, 1H, J = 14.4 Hz), 3.45 (d, 1H, J = 14.4 Hz), 2.90-2.62 (m, 4H), 2.0-1.85 (m, 1H), 1.82-1.68 (m, 1H), 1.49 (dt, 1H, J = 13.5, 4.2 Hz), 1.30 (dt, 1H, J = 13.5, 4.5 Hz), 1.20 (t, 3H, J= 7.2 Hz), 1.10 (s, 6H)。13C NMR (75 MHz, CDCl3/TMS):δ= 177.59, 138,99, 134.95, 134.25, 133.59, 129.64, 129.11, 128.07, 126.73, 125.43, 122.61, 63.81, 60.38, 50.63, 47.95, 42.22, 36.43, 28.22, 26.17, 25.52, 25.38, 14.47. MS (MMI-TOF):C22H28NO2ClS (MH+)的计算值:406.1602;实测:406.1576。
步骤4: 5-(2-氯苯基)-5-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基戊酸盐酸盐。
将5-(2-氯苯基)-5-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基戊酸乙酯(1.0 g, 2.46 mmol) 溶解在含有氢氧化钾(1 g)的水(5 mL)和乙醇(5 mL) 中。将混合物加热至回流保持6小时。冷却至室温以后,在减压下除去乙醇。加入水(25 mL),并用浓盐酸将溶液酸化至pH=5-6。用二氯甲烷(2x25 mL)萃取产物。将合并的二氯甲烷萃取物经硫酸钠干燥、过滤并浓缩。通过在硅胶(20 g)上的柱色谱法纯化粗产物(1.06 g),用庚烷/乙酸乙酯(1:1) 洗脱。合并含有产物的级分,浓缩,并溶解于二氯甲烷(25 mL)中。用盐酸(2N)在乙醚(6 mL)中的溶液酸化二氯甲烷溶液。在减压下除去溶剂。将剩余的固体在室温在高真空下干燥至恒重,得到5-(2-氯苯基)-5-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基戊酸盐酸盐(化合物Ia盐酸盐, 0.71 g, 69.6%收率),为灰白色固体。1H NMR (300 MHz, DMS-d6/TMS):δ= 12.20 (br s, 1H), 11.83 (br s, 0.5H), 11.61 (br s, 0.5H), 8.10 (m, 1H), 7.77-7.41 (m, 4H), 6.97 (d, 1H, J = 4.5 Hz), 6.81 (d, 1H, J = 4.5 Hz), 4.90-4.70 (m, 1.5H), 4.42-4.37 (m, 0.5H), 4.11-4.01 (m, 1H), 3.90-3.80 (m, 0.5H), 3.51 (m, 0.5H), 3.20-2.84 (m, 2H), 2.37 (m, 1H), 2.14 (m, 1H), 1.28 (m, 1H), 1.04 (s, 3H), 1.02 (s, 3H), 0.90 (m, 1H)。(旋转异构体的混合物)。13C NMR (75 MHz, DMS-d6/TMS):δ= 177.64, 134.50, 131.23, 130.89, 129.72, 129.38, 128.02, 127.33, 125.06, 124.68, 64.62, 63.52, 54.63, 49.73, 48.54, 47.93, 47.20, 40.72, 35.21, 25.90 (d), 24.78, 24.37, 21.79, 21.31 (d), 14.99. MS (MMI-TOF):C20H25NO2Cl2S (MH+)的计算值:378.1289,实测:378.1271. CHN分析:计算:57.97 C, 6.08 H, 3.38 N, 17.11 Cl,实测:56.07 C, 6.25 H, 3.12 N, 18.55 Cl,CHN数据的最佳拟合:C20H25Cl2NO2S + 0.65 H2O + 0.05 HCl。
实施例2. 6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸盐酸盐(化合物Ic盐酸盐)
步骤1:(4-乙氧基羰基-4-甲基)三苯基溴化鏻的合成.
将三苯基膦(6.6 g, 25.3 mmoL) 加入5-溴-2,2-二甲基戊酸乙酯(6 g, 25.3 mmoL)在甲苯(55 mL)中的溶液中。将该溶液加热至回流(油浴122℃)保持24 h。蒸发甲苯,并用庚烷(20 mL)和乙醚(20 mL)洗涤残余物。在高真空下干燥剩余的固体,得到(4-乙氧基羰基-4-甲基)三苯基溴化鏻(11 g, 87.3%),为灰白色粉末(熔点245-250℃)。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.94-7.63 (m, 15H), 3.99 (q, 2H, J = 6.9 Hz), 3.75 (m, 2H), 1.91 (m, 2H), 1.61 (m, 2H), 1.10 (m, 9H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):176.79, 134.81, 133.29 (d, J = 10 Hz), 130.22 (d, J = 12 Hz), 117.79 (d,J = 85 Hz), 60.14, 41.94, 40.60 (d, J = 16 Hz), 24.95, 22.98 (d, J = 50 Hz), 18.37, 14.09. MS (FIA-ESI-TOFM):C27H32BrOP (MH)+的计算值:419.2134;实测:419.2138。
步骤2:6-(2-氯苯基)-2,2-二甲基己-5-烯酸乙酯的合成.
将(4-乙氧基羰基-4-甲基)三苯基溴化鏻(7 g, 14 mmol)和2-氯苯甲醛(1.96 g, 14 mmol)在CH2Cl2 (21 mL)中尽可能剧烈地搅拌,并逐滴加入50% NaOH溶液(8 mL)。该过程以后,继续搅拌1.5 h。将该混合物转移至分离器,并加入二氯甲烷(70 mL)和水(70 mL)。在混合以后,除去水层,并用二氯甲烷(3×70 mL)萃取。用盐水(50 mL)洗涤合并的有机萃取物,经Na2SO4干燥,浓缩,并通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/10至1/6)纯化,得到6-(2-氯苯基)-2,2-二甲基己-5-烯酸乙酯(2.81 g, 72.2%),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.35-7.08 (m, 4H), 6.46 (d, 1H, J = 11.7 Hz), 5.76-5.68 (m, 1H), 4.04 (q, 2H, J = 7.2 Hz), 2.23-2.08 (m, 2H), 1.74-1.61 (m, 2H), 1.20 (t, 3H, J = 7.2 Hz), 1.13 (s, 6H)。(顺式和反式异构体的混合物,列出了主要的/顺式异构体)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.34, 135.54, 133.49, 130.34, 129.30, 128.03, 126.40, 126.14, 60.32, 42.13, 40.53, 29.06, 25.33, 14.30. (顺式和反式异构体的混合物,列出了主要的/顺式异构体)。MS (GC-CI):C16H21ClO2 (MH)+的计算值:281.1303;实测:282.1324
步骤3:6-溴-6-(2-氯苯基)-2,2-二甲基己酸乙酯的合成.
将6-(2-氯苯基)-2,2-二甲基己-5-烯酸乙酯(15 g, 53.6 mmol) 溶解在冰醋酸(150 mL)中。在使干燥的溴化氢在溶液中鼓泡8 h的同时,将溶液在冰浴(约15℃)中冷却。将所述反应混合物倒入冰水(250 mL)中,并用乙酸乙酯(3×100 mL)萃取。用饱和的NaHCO3溶液(100 mL)洗涤合并的有机萃取物,经Na2SO4干燥,并在减压下浓缩。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/20至1/10)纯化粗产物(18 g),得到6-溴-6-(2-氯苯基)-2,2-二甲基己酸乙酯(15.5 g, 80.14%),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.58 (d, 1H, J = 7.8 Hz), 7.34 (t, 1H, J = 7.8 Hz), 7.27 (d, 1H, J = 7.2 Hz), 7.21 (t, 1H, J = 7.2 Hz), 5.45 (t, 1H, J = 8.1 Hz), 4.09 (q, 2H, J = 6.9 Hz), 2.19 (m, 2H), 1.55 (m, 2H), 1.24 (t, 3H, J = 6.9 Hz), 1.14 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.45, 139.25, 132.64, 129.64, 129.27, 128.75, 127.43, 60.36, 49.97, 42.17, 39.84, 39.51, 25.41, 25.20, 23.67, 14.43. MS (GC-CI):C16H22BrClO2 (MH)+的计算值:361.0564;实测:361.0547。
步骤4:6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸乙酯(化合物Ie)的合成
将4,5,6,7-四氢-噻吩并[3,2-c ]吡啶盐酸盐(2.5 g, 8.8 mmol) 与氢氧化钠(2.7 g)在水(80 mL)中混合。用二氯甲烷(3×20 mL)萃取对应的游离胺,将其经Na2SO4干燥、过滤并浓缩,得到4,5,6,7-四氢-噻吩并[3,2-c ]吡啶(1.22 g)。将4,5,6,7-四氢-噻吩并[3,2-c ]吡啶(1.22 g, 8.8 mmol)和6-溴-6-(2-氯苯基)-2,2-二甲基己酸乙酯(2.5 g, 8.8 mmol) 在DMF (90 mL)中混合,并加入碳酸钾(1.82 g, 13.2 mmol)。将混合物加热至70℃保持1 h,然后在60℃过夜。将混合物冷却至室温,并加入水(100 mL)。用乙醚(3×150 mL)萃取产物。用水(3×80 mL)洗涤合并的醚萃取物,经Na2SO4干燥,过滤,并在减压下浓缩。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/10)纯化粗产物,得到6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸乙酯(化合物Ie, 1.26 g, 42.04%),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.49 (d, 1H, J = 7.8 Hz), 7.37 (d, 1H, J = 8.1 Hz), 7.26-7.14 (m, 2H), 7.05 (d, 1H, J = 5.4 Hz), 6.70 (d, 1H, J = 5.4 Hz), 4.19 (dd, 1H, J= 8.7, 4.5 Hz), 4.02 (q, 2H, J = 7.2 Hz), 3.81 (d, 1H, J = 14.4 Hz), 3.47 (d, 1H, J = 14.4 Hz), 2.88-2.66 (m, 4H), 1.95-1.74 (m, 2H), 1.52-1.46 (m, 2H), 1.13 (t, 3H, J = 7.2 Hz), 1.15-1.08 (m, 2H), 1.07 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.63, 139.16, 134.91, 134.21, 133.55, 129.53, 129.03, 128.03, 126.81, 124.46, 122.65, 63.47, 60.33, 50.73, 48.04, 42.31, 41.04, 33.48, 26.14, 25.58, 25.12, 21.07, 14.44. MS (GC-CI):C23H30ClNO2S (MH)+的计算值:420.1758;实测:420.1725。
步骤5:6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸盐酸盐的合成.
将6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸乙酯(2.5 g, 7.33 mmol) 加入乙醇(100 mL)和氢氧化钠(1.8 g, 43.8 mmol)在水(32 mL)中的溶液中。将混合物加热至回流保持6.5小时。在减压下蒸发溶液,并将水(100 mL) 加入残余物中。用乙酸乙酯/庚烷= 1/10 (100 mL) 萃取水溶液。抛弃有机层,并用10 N盐酸溶液将水溶液调至pH=6。用二氯甲烷(3×100 mL)萃取产物,将其经Na2SO4干燥,过滤,并在减压下浓缩,得到6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸(1.6 g, 68.7%),为黄色油。将6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸(3.97 g, 10.2 mmol)溶解在36% HCl (0.95 mL)和水(59 mL)中。搅拌10分钟以后,用乙酸乙酯/庚烷(20/80, 110 mL)萃取水溶液。抛弃有机萃取物,将水性级分冷冻干燥,得到6-(2-氯苯基)-6-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基己酸盐酸盐(化合物Ic盐酸盐,3.48 g,79.8%收率,通过HPLC确定为99.7% 纯度),为白色固体,在136-138℃熔化。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.69 (s, 1H), 7.47-7.38 (m, 3H), 7.21 (d, 1H, J = 3.9 Hz), 6.71 (d, 1H, J = 3. 9 Hz), 4.92 (m, 1H), 4.31-4.15 (m, 2H), 3.15-3.05 (m, 4H), 2.24-2.14 (m, 2H), 1.47-1.36 (m, 2H), 1.08-1.06 (m, 2H), 0.99 (s, 6H)。(在NMR中,旋转异构体的混合物)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):181.10, 137.13, 132.80, 132.77, 132.04, 131.79, 130.48, 129.73, 128.73, 126.62, 126.14, 66.14, 51.52, 42.87, 40.92, 32.04, 25.88, 25.47, 23.45, 22.50. MS (FIA-ESI-TOF):C21H26ClNO2S (MH)+的计算值:392.1446;实测:392.1453. CHN分析:计算:58.87 C, 6.35 H, 3.27 N, 16.55 Cl, 7.48 S,实测:57.80 C, 6.44 H, 3.15 N, 16.14 Cl, 7.15 S,CHN数据的最佳拟合:C21H27Cl2NO2S + 0.55 H2O
实施例3. 1 2-(2-氯苯基)-2-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5-基)乙醇(化合物Is)
向氯吡格雷硫酸氢盐(10 g, 23.8 mmol)在去离子水(300 mL) 中的溶液中,分成小份加入碳酸氢钠(4 g, 47.6 mmol)。混合以后,加入叔丁基甲基醚(200 mL),并将溶液搅拌1小时。分离各层,并再用叔丁基甲基醚(2×100 mL)萃取水层。用盐水(100 mL)洗涤合并的有机萃取物,经硫酸钠干燥,过滤,并在减压下浓缩。将剩余的褐色油在高真空下干燥至恒重,得到氯吡格雷游离碱(8.24 g, 106%收率,其包括微量的MTBE)。在氮气氛下,将氢化铝锂(0.46 g, 12 mmol)在无水乙醚(8 mL)中的溶液逐滴加入氯吡格雷游离碱(3.22 g, 10 mmol)在无水乙醚(4 mL)中的溶液中。以保持乙醚轻轻回流的速率,加入氢化铝锂。当加入结束时,将混合物在回流温度搅拌另外30分钟。30分钟以后,在冰/水浴中将溶液冷却至0℃。用乙酸乙酯(2 mL,缓慢地加入)分解过量的LiAlH4,随后加入6N HCl溶液(20 mL,在剧烈搅拌下缓慢地加入)。将该混合物转移至分液漏斗。分离水性级分,并用乙醚(30 mL,随后抛弃)萃取。用6 N NaOH溶液(20 mL)将水性级分调至pH =7,并用乙醚(4×15 mL)萃取。用盐水(20 mL)洗涤合并的乙醚萃取物,经MgSO4干燥,并在真空下浓缩,得到2-(2-氯苯基)-2-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5-基)乙醇(化合物Is, 2.12 g, 75.4%收率),为白色固体。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.45-7.40 (m, 2H), 7.23 (m, 2H), 7.04 (d, 1H, J = 5.1 Hz), 6.70 (d, 1H, J = 5.1 Hz), 4.47 (dd, IH, J= 6.9, 5.8 Hz), 3.97-3.91 (m, 1H), 3.81-3.74 (m, 2H), 3.62 (d, 1H, J = 14.4 Hz), 3.01-2.95 (m, 1H), 2.84 (m, 2H) 2.76-2.68 (m, 1H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):135.31, 134.92, 133.81, 133.26, 130.03, 129.35, 128.85, 126.72, 125.34, 122.82, 64.53, 61.55, 50.20, 47.67, 26.15. MS (HR, DIP-CI):C15H17ClNOS (MH)+的计算值:294.0714,实测:294.0715
实施例4. 4-(2-氯苯基)-4-(6,7-二氢-4H - 噻吩并[3,2-c ]吡啶-5-基)丁-1-醇(化合物Id)
步骤1. 5-[2-氯-1-(2-氯苯基)乙基]-4,5,6,7-四氢噻吩并[3,2-c ]吡啶的合成.
向2-(2-氯苯基)-2-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5-基)乙醇(9.7 g, 33.1 mmol)在THF (280 mL)中的溶液中,逐滴加入亚硫酰氯(7.9 g, 66.2 mmol)。将该溶液在室温搅拌1.5 h (通过TLC监测)。在减压下蒸发过量的亚硫酰氯。将残余物溶解在CH2Cl2 (200 mL)中,用饱和的NaHCO3溶液洗涤(至pH = 7),经Na2SO4干燥,并在真空下浓缩,得到5-[2-氯-1-(2-氯苯基)乙基]-4,5,6,7-四氢噻吩并[3,2-c ]吡啶(10.1 g, 98.1%),为浅黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.63 (d, J = 6.3 Hz, 1H), 7.36-7.22 (m, 3H), 7.05 (d, J = 5.1 Hz, 1H), 6.70 (d, J = 5.1 Hz, 1H), 5.62 (t, J = 6.3, 1H), 3.71 (s, 2H), 3.10-3.07 (m, 2H), 2.95-2.91 (m, 2H), 2.86-2.84 (m, 2H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):137.72, 133.43, 132.79, 129.56, 129.47, 129.00, 127.43, 125.31,122.70, 64.37, 56.60, 53.24, 50.98, 25.24. MS (HR, GC-CI):C15H15Cl2NS (MH)+的计算值:312.0375,实测:312.0349。
步骤2. 2-[2-氯苯基)-2-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5-基)乙基]丙二酸二乙酯的合成。
在氮气氛下,向乙醇(38 mL)中加入分成小块的钠(1.03 g, 44.9 mmol)。冷却得到的乙醇钠溶液,并逐滴加入丙二酸二乙酯(7.4 g, 46.2 mmol)。将反应混合物在室温搅拌5分钟,然后逐滴加入5-[2-氯-1-(2-氯苯基)乙基]-4,5,6,7-四氢噻吩并[3,2-c ]吡啶(14 g, 44.9 mmol)在乙醇(30 mL)中的溶液。将混合物加热至回流保持2小时。2小时以后,在减压下除去溶剂,并加入水(100 mL)。用CH2Cl2 (3×200 mL)萃取产物,并将合并的萃取物经Na2SO4干燥,过滤,并在减压下浓缩,得到粗制的2-[2-氯苯基)-2-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5-基)乙基]丙二酸二乙酯(17 g, 86.7%),为褐色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.36 (d, J = 7.8 Hz, 1H), 7.15 (m, 3H), 7.02 (d, J = 5.1 Hz, 1H), 6.69 (d, J = 5.1 Hz, 1H), 4.45-4.37 (m, 1H), 3.96-3.74 (m, 5H), 3.6 (d, J = 14.4 Hz, 1H), 3.48 (d, J = 14.4 Hz, 1H), 3.00-2.95 (m, 1H), 2.81-2.60 (m, 4H), 1.05 (t, J = 7.2 Hz, 3H), 0.94 (t, J = 7.2 Hz, 3H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):168.01, 167.77, 138.09, 134.28, 133.92, 133.34, 129.81, 127.98, 127.76, 126.88, 125.05, 122.38, 61.57, 61.50, 61.23, 56.56, 53.25, 50.72, 40.02, 25.21, 13.88, 13.81. MS (HR, GC-CI):C22H27ClNO4S (MH)+的计算值:436.1344,实测:436.1318
步骤3. 4-(2-氯苯基)-4-(6,7-二氢-4H - 噻吩并[3,2-c ]吡啶-5-基)丁酸乙酯(化合物If)的合成
在搅拌下,将2-[2-氯苯基)-2-(6,7-二氢-4H - 噻吩并[3,2-c ]吡啶-5-基)乙基]丙二酸二乙酯(17.5 g, 40.2 mmol)和氯化锂(6.8 g, 161.9 mmol)在DMSO (230 mL)和水(3.9 mL)中的混合物加热至回流(油浴,约190℃) 保持4小时。将所述反应混合物冷却至室温,用水(150 mL)稀释,并用乙酸乙酯(3×150 mL)萃取。用水(3×80 mL)、盐水(80 mL)洗涤合并的有机萃取物,经MgSO4干燥。过滤产物,在减压下浓缩,并在高真空下干燥。所述操作产生粗产物(15.4 g)。通过柱色谱法(硅胶,乙酸乙酯/庚烷1:3)纯化粗产物,得到4-(2-氯苯基)-4-(6,7-二氢-4H - 噻吩并[3,2-c ]吡啶-5-基)丁酸乙酯(化合物If, 11.1 g, 76.1%收率),为浅黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.34 (d, J= 7.8 Hz, 1H), 7.24-7.10 (m, 3H), 7.03 (d, J = 5.1 Hz, 1H), 6.71 (d, J = 5.1 Hz, 1H), 4.15-4.02 (m, 1H), 3.92 (q, J = 7.2 Hz, 2H), 3.67 (d, J = 14.4 Hz, 1H), 3.51 (d, J = 14.4 Hz, 1H), 2.97-2.80 (m, 4H), 2.75-2.53 (m, 4H), 1.06 (t, J = 7.2 Hz, 3H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):172.25, 139.97, 134.09, 133.99, 133.46, 129.77, 127.76, 127.60, 127.01, 125.22, 122.46, 62.68, 60.32, 53.43, 51.14, 38.41, 36.49, 25.59, 14.20. MS (HR, GC-CI):Calcd. C19H23ClNO2S (MH)+ 364.1133,实测:364.1152。
步骤4. 4-(2-氯苯基)-4-(6,7-二氢-4H - 噻吩并[3,2-c ]吡啶-5-基)丁-1-醇的合成.
在氮气氛下,将4-(2-氯苯基)-4-(6,7-二氢-4H - 噻吩并[3,2-c ]吡啶-5-基)丁酸乙酯(1.0 g, 2.75 mmol)在无水乙醚(3 mL)中的溶液逐滴加入氢化铝锂(0.13 g, 3.31 mmol)在无水乙醚(7 mL)中的溶液中。以保持乙醚轻轻回流的速率,加入氢化铝锂。当加入结束时,将混合物回流搅拌另外30分钟。用冰水浴将反应物冷却至0℃。用乙酸乙酯(1 mL,缓慢地加入)分解过量的LiAlH4。随后在剧烈搅拌下缓慢地加入6 N HCl溶液(10 mL)。分离水,并用乙醚(30 mL,随后抛弃)萃取。用6 N NaOH溶液(15 mL,至pH = 7)中和水性部分,并用乙醚(3×20 mL)萃取。用盐水(15 mL)洗涤合并的有机层,经MgSO4干燥,并在减压下浓缩,得到4-(2-氯苯基)-4-(6,7-二氢-4H - 噻吩并[3,2-c ]吡啶-5-基)丁-1-醇(化合物Id, 0.77 g, 87.2%收率),为黄色粘稠的油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.36 (d, 1H, J = 7.5 Hz), 7.25-7.12 (m, 3H), 7.07 (d, 1H, J = 5.1 Hz), 6.72 (d, 1H, J = 5.1 Hz), 6.55 (br s, 1H), 3.82-3.54 (m, 5H), 3.2-3.13 (m, 1H), 2.95 (br t, 2H, J = 4.8 Hz), 2.82-2.75 (m, 1H), 2.70-2.60 (m, 2H), 2.06-1.90 (m, 2H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):142.33, 133.14, 133.06, 132.63, 129.70, 127.75, 127.68, 127.28, 125.16, 123.11, 63.80, 62.19, 53.41, 51.01, 39.78, 39.43, 24.88. MS (HR, DIP-CI):C17H21ClNOS (MH)+的计算值:322.1027,实测:322.1006。
实施例5. 1-[(2-氯苯基)-(6,7-二氢-4H - 噻吩并[3,2-c]吡啶-5-基)-甲基]-1H -苯并三唑(化合物IVa)
将4,5,6,7-四氢-噻吩并[3,2-c]吡啶(0.5 g, 3.59 mmol)、苯并三唑(428 mg, 3.59 mmol)和2-氯苯甲醛(504 mg, 3.59 mmol) 溶解在乙醚中,并在有分子筛(4Å)存在下在氩气氛下在室温搅拌3天。3天后,过滤乙醚,并用饱和的碳酸氢钠溶液(20 mL)洗涤。将乙醚经硫酸钠干燥、过滤、并浓缩。将剩余的无色泡沫溶解在庚烷/乙醚(5 mL/1mL)中,并在冰箱(-10℃)中保存过夜。在-10℃保存20小时以后,倾析出庚烷,并将剩余的油在高真空下在室温干燥至恒重,得到作为无色泡沫的1-[(2-氯苯基)-(6,7-二氢-4H - 噻吩并[3,2-c]吡啶-5-基)-甲基]-1H -苯并三唑(化合物IVa, 0.84 g, 61.7%收率),通过NMR表明为80:20的异构体混合物。1H NMR (300 MHz, CDCl3/TMS):δ= 8.08 (d, 1H, J = 8.1 Hz), 7.91-7.79 (m, 2H), 7.55 (d, 1H, J = 8.4 Hz), 7.53-7.24 (m, 4H), 7.11 (s, 1H), 7.04 (d, 1H, J = 5.1 Hz), 6.61 (d, 1H, J = 5.1 Hz), 3.90 (d, 1 H, J = 14.4 Hz), 3.69 (d, 1H, J = 14.4 Hz), 3.21-2.95 (m, 2H), 2.88 (m, 2H)。(主要异构体)。13C NMR (75 MHz, CDCl3/TMS):δ= 145.48, 133.78, 133.45, 132.94, 132.65, 130.10, 129.40, 127.50, 126.98, 126.56, 124.97, 124.00, 122.79, 119.80, 110.39, 84.85, 49.59, 47.28, 25.50. (主要异构体)。MS (HR, DIP-CI):(MH+)的计算值:379.0779,实测:379.0752。
实施例6. 7-(2-氯苯基)-7-(6,7-二氢-4H - 噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸盐酸盐(化合物Ig盐酸盐)
步骤1. (5-乙氧基羰基-5-甲基己基)-三苯基溴化鏻的合成。
将6-溴-2,2-二甲基己酸乙酯(38.0 g, 0.15 mol)和三苯基膦(40.0 g, 0.15 mol) 溶解在甲苯(100 mL)中,并在氩气氛下加热至回流保持24小时。加热24小时以后,将烧瓶冷却至室温,并在室温搅拌另外24小时。48小时以后,在减压下除去甲苯。将剩余物溶解在二氯甲烷(100 mL)中,并逐滴加入叔丁基甲基醚(600 mL)中。从沉淀物中倾析出醚,将沉淀物在高真空下干燥至恒重,得到(5-乙氧基羰基-5-甲基己基)-三苯基溴化鏻(71.5 g, 93.1%收率),为浅黄色泡沫。1H NMR (300 MHz, CDCl3/TMS):δ= 7.87-7.57 (m, 15H), 4.05 (1, 2H, J = 7.2 Hz), 3.67 (m, 2H), 1.78-1.45 (m, 6H), 1.20 (t, 3H, J = 7.2 Hz), 1.12 (s, 6H)。13C NMR (75 MHz, CDCl3/TMS):δ= 177.02, 134.56 (d, 3C, J= 2.4 Hz), 132.94 (d, 6C, J = 10.1 Hz), 129.99 (d, 6C, J = 12.6 Hz), 117.44 (d, 3C, J = 85.4 Hz), 59.78, 41.47, 39.12, 25.44 (d, 1C, J = 15.5 Hz), 24.62, 22.59 (d, 1C, J = 4.1 Hz), 21.96, 13.83. HRMS (ESI-TOF):(M+)的计算值:433.2291,实测:433.2330。
步骤2. 7-(2-氯苯基)-2,2-二甲基庚-6-烯酸乙酯的合成。
将(5-乙氧基羰基-5-甲基己基)-三苯基溴化鏻(35.0 g, 0.0619 mol) 溶解在含有2-氯苯甲醛(8.70 g, 0.0619 mol)的二氯甲烷(100 mL)中。历时10分钟,分份加入氢氧化钠(10 g, 0.25 mol)在水(10 mL)中的溶液。将混合物在室温搅拌2小时。2小时以后,分离各层,并用水(2×150 mL)洗涤二氯甲烷级分,经硫酸钠干燥,过滤,并在减压下浓缩。穿过硅胶(200 g)过滤剩余的褐色油(18.99 g),用乙酸乙酯/庚烷(1:9)洗脱。合并含有产物的级分,并在减压下浓缩。剩余的黄色油(10.5 g)被2-氯苯甲醛(30-40%)污染。通过溶解在含有额外鏻盐(25 g, 0.048 mol)的新鲜二氯甲烷(100 mL) 中,再处理该物质。分份加入氢氧化钠(10 g, 0.25 mol)在水(10 mL)中的溶液,将混合物在室温搅拌4小时。分离各层,并用水(2×100 mL)洗涤二氯甲烷级分,经硫酸钠干燥,过滤,并在减压下浓缩。通过在硅胶(200 g)上的柱色谱法纯化剩余的褐色油,用乙酸乙酯/庚烷(1:9)洗脱。合并含有产物的级分,并在减压下浓缩,得到7-(2-氯苯基)-2,2-二甲基庚-6-烯酸乙酯(12.0 g, 60%收率),为浅黄色液体。1H NMR (300 MHz, CDCl3/TMS):δ= 7.47 (d, 0.4 H, J = 7.5 Hz), 7.36-7.07 (m, 3.6 H), 6.74 (d, 0.4 H, J = 15.6 Hz), 6.50 (d, 0.6 H, J = 11.4 Hz), 6.16 (dt, 0.4 H, J = 15.6, 7.2 Hz), 5.75 (dt, 0.6 H, J = 11.4, 7.2 Hz), 4.10 (m, 2H), 2.26-2.12 (m, 2H), 1.61-1.34 (m, 4H), 1.29-1.17 (m, 3H), 1.14 (s, 6H)。(顺/反异构体的混合物)。13C NMR (75 MHz, CDCl3/TMS):δ= 177.59, 135.63 (2峰), 133.67, 133.45, 133.31, 132.38, 130.31, 129.41, 129.19, 127.84, 127.73, 126.57, 126.47, 126.24, 126.00, 60.16, 42.12, 42.07, 40.22, 33.56, 28.83, 25.21, 25.16, 24.69, 14.30. (顺/反异构体的混合物)。HRMS (MMI-TOF):C22H29Cl2NO2S (MH+)的计算值:406.1202,实测:406.1594。
步骤3. 7-溴-7-(2-氯苯基)-2,2-二甲基庚酸乙酯的合成。
在氩气氛下,将7-(2-氯苯基)-2,2-二甲基庚-6-烯酸乙酯(11.75 g, 0.040 mole)溶解在醋酸(80 mL) 中。在冰水浴中冷却烧瓶。在将溶液缓慢地温热至室温的同时,用溴化氢气体在溶液中缓慢地鼓泡3小时。加入冰(250 g),并在混合以后,用乙酸乙酯(2×100 mL) 萃取产物。合并乙酸乙酯萃取物,并用饱和的碳酸氢钠溶液(3×75 mL)和水(100 mL)洗涤。该溶液经硫酸钠干燥,过滤,并在减压下浓缩。通过在硅胶(250 g)上的柱色谱法纯化剩余的油(14.06 g),用3:1 (随后2:1) 庚烷/乙酸乙酯洗脱。合并含有产物的级分,并在减压下浓缩,得到7-溴-7-(2-氯苯基)-2,2-二甲基庚酸乙酯(12.9 g, 86%收率),为澄清的油。1H NMR (300 MHz, CDCl3/TMS):δ= 7.57 (d, 1H, J = 7.50 Hz), 7.33-7.15 (m, 3H), 5.43 (t, 1H, J = 6.9 Hz), 4.08 (q, 2H, J = 7.2 Hz), 2.30-2.21 (m, 2H), 1.55-1.42 (m, 3H), 1.34-1.19 (m, 5H), 1.13 (s, 6H)。13C NMR (75 MHz, CDCl3/TMS):δ= 177.72, 139.41, 132.69, 129.67, 129.28, 128.92, 127.48, 60.36, 50.17, 42.26, 40.57, 39.19, 28.59, 25.38, 25.34, 24.45, 14.50. MS (HR, DIP-CI):(MH+)的计算值:375.0726,实测:375.0726。
步骤4. 7-(2-氯苯基)-7-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸乙酯(化合物Ih)的合成
将4,5,6,7-四氢-噻吩并[3,2-c]吡啶盐酸盐(3.2 g, 18.9 mmol) 溶解在水(120 mL)中,并加入50% 氢氧化钠溶液(10 mL)。用二氯甲烷(2×25 mL)萃取对应的游离碱,将其经硫酸钠干燥、过滤并浓缩。在氩气氛下,将剩余的油(2.58 g, 18.9 mmol)溶解在DMF (10 mL)中。加入碳酸钾(5 g)和7-溴-7-(2-氯苯基)-2,2-二甲基庚酸乙酯(7.0 g, 18.63 mmol),并将混合物在75℃搅拌44小时。将烧瓶冷却至室温,并加入水(50 mL)。用二氯甲烷(2×75 mL) 萃取产物,将其经硫酸钠干燥、过滤并浓缩。通过在硅胶(140 g)上的柱色谱法纯化剩余的褐色油(8.94 g),用庚烷-乙酸乙酯(4:1至2:1) 洗脱。合并含有产物的级分,浓缩,并在高真空下在室温干燥至恒重,得到7-(2-氯苯基)-7-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸乙酯(化合物Ih, 3.85 g, 49%收率),为澄清的油。1H NMR (300 MHz, CDCl3/TMS):δ= 7.53 (dd, 1H, J = 7.5,1.2 Hz), 7.40 (dd, 1H, J = 7.8,1.2 Hz), 7.31-7.18 (m, 2H), 7.08 (d, 1H, J = 5.1 Hz), 6.74 (d, 1H, J = 5.1 Hz), 4.22 (dd, 1H, J = 8.7, 4.5 Hz), 4.12 (q, 2H, J = 7.2 Hz), 3.83 (d, 1H, J= 14.1 Hz), 3.49 (d, 1H, J = 14.1 Hz), 2.92-2.68 (m, 4H), 2.08-1.73 (m, 2H), 1.46 (m, 2H), 1.30-1.19 (m, 7H), 1.15 (s, 6H)。13C NMR (75 MHz, CDCl3/TMS):δ= 178.00, 139.41, 134.99, 134.26, 133.62, 129.53, 128.13, 128.03, 126.87, 125.51, 122.67, 63.69, 60.37, 50.79, 48.15, 42.33, 40.84, 33.11, 26.19, 25.40, 14.55. HRMS (DIP-CI):(M+H+)的计算值:434.1921,实测:434.1871。
步骤5. 7-(2-氯苯基)-7-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸盐酸盐的合成。
将7-(2-氯苯基)-7-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸乙酯(2.80 g, 7.23 mmol)溶解在含有氢氧化钾(10 g, 250 mmol)的乙醇(40 mL)和水(25 mL)中。将混合物加热至回流保持24小时。24小时以后,将烧瓶冷却至室温,并浓缩。加入水(50 mL),并用10%乙酸乙酯在庚烷中的溶液萃取。用浓盐酸酸化水性级分(至pH= 6),并用二氯甲烷(2 x 100 mL) 萃取产物。合并二氯甲烷萃取物,经硫酸钠干燥、过滤、并浓缩。通过在硅胶(100 g)上的快速柱色谱法纯化剩余的黄褐色油(3.0 g),用庚烷-乙酸乙酯(2:1) 洗脱,得到7-(2-氯苯基)-7-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸,为白色固体(2.1 g, 71.6%收率)。将产物溶解在含有盐酸(1%)的水(ASTM 1, 200 mL)和丙酮(25 mL)中。在减压下除去大部分丙酮和多余的盐酸。将剩余的水(200 mL) 冷冻干燥,得到7-(2-氯苯基)-7-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸盐酸盐(化合物Ig盐酸盐, 2.2 g,熔点97-100℃, 通过HPLC确定为99.47% 纯度),为白色固体。1H NMR (300 MHz, DMSO-d6):δ= 12.25 (br s, 0.5H), 12.12 (br s, 0.5H), 8.24 (br s, 1H), 7.60-7.38 (m, 4H), 6.94 (d, 1H, J = 4.5 Hz), 6.77 (d, 1H, J = 4.5 Hz), 4.90-4.68 (m, 1.5H), 4.38 (dd, 0.5H, J = 14.4, 5.4 Hz), 4.01 (m, 1H), 3.80 (dd, 0.5H, J = 14.4, 5.1 Hz), 3.60-3.30 (m, 1.5H), 3.20-2.85 (m, 2H), 2.42 (m, 1H), 2.23 (m, 1H), 1.40-1.0 (m, 5H), 1.01 (s, 6H), 0.79 (m, 1H)。(旋转异构体的混合物)。13C NMR (75 MHz, DMSO-d6):δ= 178.41, 134.70, 134.60, 131.83, 131.49, 131.23, 131.05, 129.93, 129.75, 128.38, 128.35, 128.10, 125.43, 125.26, 124.87, 124.86, 64.35, 63.17, 49.87, 48.70, 48.00, 47.13, 41.16, 30.78, 30.30, 30.12, 25.77, 25.02, 24.14, 21.78, 21.37。(旋转异构体的混合物)。HRMS (MMI-TOF):C22H29Cl2NO2S (MH+)的计算值:406.1202,实测:406.1594。
实施例7. 8-(2-氯苯基)-8-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基辛酸盐酸盐(化合物Io盐酸盐)
步骤1. 2-(2-硝基乙烯基)-呋喃的合成。
将2-糠醛(18.2 g, 190 mmol)和乙酸铵(13 g, 169 mmol) 加入硝基甲烷(169 mL)中,并将混合物加热至回流保持45 min。浓缩溶液,并加入二氯甲烷(250 mL)。用水(2×250 mL)洗涤二氯甲烷溶液,经硫酸钠干燥,并过滤,得到粗产物。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/10至2/3)纯化,得到纯的2-(2-硝基乙烯基)-呋喃(14 g, 53.0%),为黄色粉末(熔点71-74℃)。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.78 (d, 1H, J = 13.2 Hz), 7.59 (m, 1H), 7.52 (d, 1H, J = 13.5 Hz), 6.90 (d, 1H, J = 3.6 Hz), 6.58 (m, 1H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):146.90, 146.67, 134.93, 125.51, 120.12, 113.46。
步骤2:2-呋喃-2-基-乙胺的合成。
在氩气氛下在0℃,向氢化铝锂(13.4 g, 353 mmol)在无水THF (400 ml)中的混合物中,历时30分钟逐滴加入2-(2-硝基乙烯基)-呋喃(14 g, 101 mmol)在THF (100 ml)中的溶液。停止冷却,并将烧瓶回流加热另外3.5小时。将所述反应混合物冷却至室温,在剧烈搅拌下缓慢地加入水(13 mL)。加入水以后,加入15% 氢氧化钠溶液(13 mL),随后再次加入水(36 mL)。剧烈搅拌10 min以后,过滤固体,并用乙醚(3×150 mL)洗涤。合并的滤液经Na2SO4干燥,过滤,并在减压下浓缩,得到2-呋喃-2-基-乙胺(11 g, 85.6%收率),为褐色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.31 (m, 1H), 6.29 (d, 1H, J = 2.1 Hz), 6.05 (d, 1H, J = 2.1 Hz), 2.96 (t, 2H, J = 6.6 Hz,), 2.76 (t, 2H, J= 6.6 Hz), 1.84 (br, 2H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):153.82, 141.21, 110.15, 106.00, 40.79, 32.35。
步骤3:6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-甲酸叔丁酯的合成。
将福尔马林(37%甲醛水溶液, 4 g, 49.2 mmol) 逐滴加入2-呋喃-2-基-乙胺(5.5 g, 43.3 mmol,纯净的)中,并将混合物在室温搅拌30分钟。用乙醚(3×80 mL)萃取粗制的中间体1。合并乙醚萃取物,经硫酸钠干燥、过滤并浓缩。通过使氯化氢在溶液中鼓泡1小时,用氯化氢气体饱和DMF (35 mL)。将剩余的油溶解在DMF (10 mL)中,并加入到DMF/HCl溶液中。在室温混合3小时以后,在高真空下除去DMF。加入甲基叔丁基醚,并通过用水(100 mL)萃取,除去微量的DMF,所述水已经用饱和的碳酸氢钠溶液调至pH = 11。所述醚溶液经硫酸钠干燥、过滤并浓缩。在室温,将剩余的粗制的中间体4,5,6,7-四氢呋喃并[3,2-c]吡啶(0.87 g, 7.1 mmol)在二氯甲烷(50 mL) 中的溶液逐滴加入含二碳酸二叔丁酯(1.6 g, 7.4 mmol)的CH2Cl2 (50 mL)中。将所述反应混合物在相同温度搅拌1.5小时,通过TLC监测。减压蒸发溶剂,并通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/9)纯化粗产物,得到纯的6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-甲酸叔丁酯(0.56 g, 35.4%),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.19 (s, 1H), 6.13 (d, 1H, J = 1.5 Hz), 4.25 (s, 2H), 3.63 (m, 2H), 2.59 (d, m), 1.44 (s, 9H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):154.78, 146.85, 141.23, 108.19, 85.03, 79.82, 41.58, 40.05, 28.52, 23.90。
步骤4:4,5,6,7-四氢呋喃并[3,2-c]吡啶盐酸盐的合成。
向含6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-甲酸叔丁酯(0.56 g, 2.51 mmol)的甲醇(50 mL)中,加入浓盐酸溶液(37%, 1.4 mL)。将混合物在室温搅拌5.5小时,通过TLC监测。在减压下蒸发甲醇,得到4,5,6,7-四氢呋喃并[3,2-c]吡啶盐酸盐(0.5 g, 100%收率),为黄色粉末。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):7.47 (s, 1H), 6.42 (s, 1H), 4.20 (s, 2H), 3.57 (m, 2H), 3.03 (m, 2H)。13C NMR (场: 75 MHz, 溶剂:CDCl3/TMS) δ(ppm):147.23, 143.89, 112.23, 109.50, 43.10, 42.37, 21.70。
步骤5: 8-(2-氯苯基)-8-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基辛酸乙酯(化合物Ip)
在氩气氛下在室温,在DMF (36 mL)中混合4,5,6,7-四氢呋喃并[3,2-c]吡啶(0.47 g, 3.82 mmol)、8-溴-8-(2-氯苯基)-2,2-二甲基辛酸乙酯(1.47 g, 3.78 mmol)和碳酸钾(0.77 g, 5.58 mmol)。在氩气氛下,将混合物加热至60℃保持3天,通过TLC监测。在减压下蒸发DMF,并将残余物溶解在乙醚(120 mL)中,用水(3×30 mL)、盐水(30 mL)洗涤,并经Na2SO4干燥。通过柱色谱法(硅胶,乙酸乙酯/庚烷1/9) 纯化粗制的油,得到纯的8-(2-氯苯基)-8-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基辛酸乙酯(化合物Ip, 0.50 g, 30.3%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):7.48 (d, 1H, J = 7.5 Hz), 7.36 (d, 1H, J = 7.8 Hz), 7.32-7.15 (m, 3H), 6.17 (s, 1H), 4.20 (dd, 1H, J = 9.0, 4.8 Hz), 4.08 (q, 2H, J = 7.2 Hz), 3.62 (d, 1H, J = 13.5 Hz), 3.11 (d, 1H, J = 13.5 Hz), 2.88-2.58 (m, 4H), 1.95-1.70 (m, 2H), 1.45-1.40 (m, 2H), 1.30-1.0 (m, 15H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):178.01, 149.01, 140.94, 139.50, 135.00, 129.57, 129.04, 128.03, 126.85, 115.92, 108.90, 63.43, 60.36, 47.65, 47.54, 42.36, 40.91, 33.27, 30.57, 25.67, 25.41, 25.06, 24.73, 14.55。HRMS (HR, DART-TOF):(M+H)+的计算值:432.2300,实测:432.2316。
步骤6:8-(2-氯苯基)-8-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基辛酸盐酸盐的合成
将8-(2-氯苯基)-8-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基辛酸乙酯(0.5 g, 1.16 mmol) 加入乙醇(20 mL)和含氢氧化钠(0.32 g, 8.0 mmol)的水(6.6 mL)的混合物中。将混合物加热至回流保持6.5小时。在减压下蒸发溶剂,并将水(20 mL)加入残余物中。用乙酸乙酯/庚烷1/10的混合物(10 mL)洗涤水溶液,并抛弃萃取物。用浓盐酸酸化水性级分至pH=6,并用二氯甲烷(3×20 mL)萃取产物。将合并的二氯甲烷萃取物经硫酸钠干燥,过滤,并在减压下浓缩,得到8-(2-氯苯基)-8-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基辛酸(0.25 g, 46.9%),为黄色油。将该物质溶解在乙醚(5 mL)中,并加入盐酸溶液(2N HCl在乙醚中, 0.34 mL)中。加入水(12 mL),并在混合以后,分离水性部分,并冷冻干燥,得到8-(2-氯苯基)-8-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基辛酸盐酸盐(化合物Io盐酸盐, 0.16 g, 浅黄色粉末, 59.3%收率)。1H NMR (场: 300 MHz, 溶剂: CD3OD /TMS) δ(ppm):7.77 (br s, 1H), 7.63-7.48 (m, 5H), 6.37 (br s, 1H), 5.05 (m, 1H), 4.18 (m, 1H), 3.62 (m, 1H), 3.02 (m, 2H), 2.36-2.24 (m, 2H), 1.42-1.40 (m, 2H), 1.32 (m, 2H), 1.20-1.18 (m, 4H), 1.11 (s, 6H), 1.08-0.92 (m, 1H)。13C NMR (场: 75 MHz, 溶剂: CD3OD/TMS) δ(ppm):181.70, 147.11, 144.55, 137.40, 132.15, 131.89, 130.08, 128.21, 129.87, 112.37, 109.71, 66.34, 52.10, 50.05, 43.07, 41.60, 35.10, 31.67, 30.56, 26.64, 25.85, 22.31。HRMS (HR, DIP-CI):C23H30ClNO3 (M+H)+的计算值:404.1987,实测:404.2009. CHN分析:计算:62.73 C, 7.09 H, 3.18 N, 16.10 Cl,实测:59.13 C, 7.04 H, 3.03 N, 13.58 Cl。
实施例8。8-(2-氯苯基)-2,2-二甲基-8-(1,4,6,7-四氢吡咯并[3,2-c ]吡啶-5-基)-辛酸盐酸盐(化合物Iq盐酸盐)
步骤1:吡咯并[3,2-c]吡啶-1-甲酸叔丁酯的合成。
在室温,将含二甲基氨基吡啶(DMAP) (2.08 g, 16.9 mmol)的乙腈(20 mL) 逐滴加入含5-氮杂吲哚(2.0 g, 16.9 mmol)的乙腈(70 mL) 中。搅拌2小时以后,在相同温度分份加入二碳酸二叔丁酯(3.68 g, 16.9 mmol)。在2.5小时以后,在减压下蒸发溶剂,并通过柱色谱法(硅胶,乙酸乙酯/庚烷1/10至1/2)纯化残余物(5.4 g),得到吡咯并[3,2-c]吡啶-1-甲酸叔丁酯(2.72 g, 92.4%收率),为亮黄色油。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):8.87 (s, 1H), 8.48 (d, 1H, J = 5.7 Hz), 7.98 (d, 1H, J = 5.1 Hz), 7.60 (d, 1H, J = 3.3 Hz), 6.63 (d, 1H, J = 3.0 Hz), 1.68 (s, 9H)。13C NMR (场:75 MHz, 溶剂: CDCl3/TMS) δ(ppm):148.82, 143.75, 143.51, 139.50, 126.69, 109.83, 105.46, 84.60, 28.05。
步骤2:1-叔-丁氧基羰基-5-[1-(2-氯苯基)-7-甲基辛基]-1H -吡咯并[3,2-c]吡啶-5-鎓溴化物) 的合成。
将吡咯并[3,2-c]吡啶-1-甲酸叔丁酯(0.81 g, 3.73 mmol)、8-溴-8-(2-氯苯基)-2,2-二甲基辛酸乙酯(1.44 g, 3.73 mmol)、和乙腈(25 mL)的混合物在45℃搅拌52小时。在减压下蒸发溶剂,并用乙醚(3×20 mL)洗涤残余物,得到1-叔-丁氧基羰基-5-[1-(2-氯苯基)-7-甲基辛基]-1H -吡咯并[3,2-c]吡啶-5-鎓溴化物(1.37 g, 60.6%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):10.52 (s, 1H), 8.84 (d, 1H, J = 7.2 Hz), 8.23 (d, 1H, J = 7.8 Hz), 7.91 (t, 1H, J = 3.6 Hz), 7.54-7.31 (m, 4H), 7.39 (s, 1H), 6.95 (s, 1H), 6.42 (t, 1H, J = 7.2 Hz), 4.09 (q, 2H, J= 7.2 Hz), 2.67 (m, 1H), 1.70 (s, 9H), 1.42-1.19 (m, 8H), 1.21 (t, 3H, J = 6.9 Hz), 1.10 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):178.00, 147.44, 140.17, 136.60, 133.19, 131.97, 131.45, 130.42, 130.03, 128.64, 113.10, 108.61, 88.15, 71.55, 60.34, 42.23, 40.54, 34.54, 29.69, 28.13, 26.42, 25.28, 24.92, 14.46。HRMS (HR, DIP-CI):C30H40ClN2O2 (M+H)+的计算值:未发现(在分析时分解)。
步骤3:5-[1-(2-氯苯基)-7-乙氧基羰基-7-甲基辛基]-4,5,6,7-四氢吡咯并[3,2-c ]吡啶-1-甲酸叔丁酯的合成。
在室温,历时30分钟,将硼氢化钠部分(0.16 g, 4.28 mmol) 加入1-叔-丁氧基羰基-5-[1-(2-氯苯基)-7-甲基辛基]-1H -吡咯并[3,2-c]吡啶-5-鎓溴化物(1.3 g, 2.14 mmol)在70% EtOH (30 mL) 中的溶液中。加入结束以后,将混合物搅拌0.5小时。在减压下蒸发溶剂,并加入水(60 mL)。用二氯甲烷(3×80 mL)萃取产物。合并的有机萃取物经硫酸钠干燥、过滤并浓缩。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/10至1/3) 纯化得到的叔胺,得到5-[1-(2-氯苯基)-7-乙氧基羰基-7-甲基辛基]-4,5,6,7-四氢吡咯并[3,2-c ]吡啶-1-甲酸叔丁酯(0.53 g, 46.9%),为黄色油。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):7.52 (d, 1H, J = 7.8 Hz), 7.37 (d, 1H, J = 7.8 Hz), 7.27 (t, 1H, J = 6.6 Hz), 7.19 (t, 1H, J = 7.5 Hz), 7.12 (d, 1H, J = 3.3 Hz), 5.95 (d, 1H, J = 3.3 Hz), 4.16-4.13 (m, 1H), 4.09 (q, 2H, J = 7.2 Hz), 3.31 (d, 1H, J = 13.8 Hz), 2.85-2.59 (m, 4H), 1.95-1.76 (m, 2H), 1.55 (s, 9H), 1.45-1.40 (m, 2H), 1.28-1.14 (m, 6H), 1.21 (t, 3H, J = 7.2 Hz), 1.11 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):178.00, 149.47, 139.70, 134.90, 129.45, 129.15, 127.89, 127.60, 126.80, 120.94, 119.76, 109.18, 83.24, 63.80, 60.30, 48.78, 42.32, 40.88, 33.20, 30.56, 28.31, 26.45, 25.44, 25.38, 25.03, 14.52. HRMS (HR):C30H43ClN2O2 (M+H)+的计算值:531.2990,实测:531.2933。
步骤4:8-(2-氯苯基)-2,2二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c ]吡啶-5-基)-辛酸乙酯(化合物Ir)的合成
在氩气氛下,在搅拌下,将5-[1-(2-氯苯基)-7-乙氧基羰基-7-甲基辛基]-4,5,6,7-四氢吡咯并[3,2-c ]吡啶-1-甲酸叔丁酯(0.12 g, 0.23 mmol) 溶解在二氯甲烷(5 mL) 中。将TFA (0.48 mL, 4.75 mmol) 加入该溶液中。1小时以后,TLC表明原料消失。将混合物倒入冰冷的水(50 mL)中,并用二氯甲烷(3×20 mL)萃取。合并的有机层经硫酸钠干燥,过滤,并在减压下浓缩。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/9至1/3) 纯化粗产物,得到8-(2-氯苯基)-2,2二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c ]吡啶-5-基)-辛酸乙酯(化合物Ir, 65 mg, 65.9%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):7.80 (s, 1H), 7.54 (d, 1H, J = 7.5 Hz), 7.37 (d, 1H, J = 7.8 Hz), 7.26-7.16 (m, 2H), 6.60 (s, 1H), 5.94 (s, 1H), 4.17 (m, 1H), 4.08 (q, 2H, J = 7.2 Hz), 3.75 (d, 1H, J = 13.2 Hz), 3.35 (d, 1H, J = 13.2 Hz), 2.83-2.57 (m, 4H), 2.01-1.56 (m, 4H), 1.45-1.40 (m, 2H), 1.21 (t, 3H, J = 7.2 Hz), 1.24-1.11 (m, 4H), 1.11 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):178.14, 140.07, 134.95, 129.43, 129.25, 127.82, 126.79, 125.12, 116.43, 115.93, 105.80, 63.89, 60.37, 48.79, 48.32, 42.37, 40.94, 33.34, 30.64, 25.55, 25.41, 25.08, 24.07, 14.56. HRMS (HR, DART-TOF):C25H35ClN2O2 (M+H)+的计算值:431.2460,实测:431.2476。
步骤5:8-(2-氯苯基)-2,2二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-辛酸(化合物Iq) 的合成.
将8-(2-氯苯基)-2,2二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c ]吡啶-5-基)-辛酸乙酯(0.11 g, 0.25 mmol) 加入乙醇(15 mL)、水(1.35 mL)和氢氧化钠(0.07 g, 1.75 mmol)的混合物中。将该混合物加热至回流(95-98℃油浴) 保持11小时。冷却至室温以后,在减压下蒸发溶剂。将水(10 mL) 加入残余物中,并用10 N盐酸调至pH = 6。用二氯甲烷(3×10 mL)萃取产物。用水(10 mL)、盐水(10 mL)洗涤合并的有机层,经Na2SO4干燥。过滤以后,将二氯甲烷溶液在减压下浓缩,得到粗产物。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 3/7)纯化粗产物,得到8-(2-氯苯基)-2,2二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-辛酸(化合物Iq, 0.06 g, 59.8%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂:CD3OD/TMS) δ(ppm):7.60 (d, 1H, J = 7.5 Hz), 7.48 (d, 1H, J = 7.8 Hz), 7.41-7.30 (m, 2H), 6.58 (d, 1H, J = 2.7 Hz), 5.89 (d, 1H, J = 2.7 Hz), 4.56-4.52 (m, 1H), 3.96 (d, 1H, J = 13.5 Hz), 3.65 (d, 1H, J = 13.5 Hz), 2.97-2.65 (m, 4H), 2.00-1.87 (m, 2H), 1.43-1.39 (m, 2H), 1.28-1.18 (m, 6H), 1.11 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CD3OD /TMS) δ(ppm) 183.09, 136.97, 136.77, 131.03, 130.05, 130.31, 128.80, 124.29, 118.35, 113.24, 105.71, 65.49, 50.79, 49.81, 43.48, 42.01, 33.13, 30.39, 26.74, 26.19, 25.98, 23.34。
步骤6:8-(2-氯苯基)-2,2-二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c ]吡啶-5-基)-辛酸盐酸盐的合成.
将8-(2-氯苯基)-2,2二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-辛酸(0.09 g, 0.22 mmol) 溶解在乙醚(5mL)中,并加入HCl溶液(2N HCl在乙醚中)中。加入水(5 mL),在混合以后,将有机层抛弃。将水溶液冷冻干燥,得到8-(2-氯苯基)-2,2二甲基-8-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-辛酸盐酸盐(化合物Iq盐酸盐, 0.09 g, 86.5%收率),为浅黄色粉末。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):10.28 (br, 1H), 7.61 (br s, 1H), 7.48-7.40 (m, 3H), 6.57 (d, 1H, J = 10.8 Hz), 5.86-5.70 (2 s, 1H), 4.79-4.52 (m, 3H), 4.19-3.86 (m, 2H), 3.45-2.77 (m, 4H), 2.22-2.06 (m, 2H), 1.26-0.65 (m, 8H), 0.97 (s, 6H)。(在NMR中,构象异构体的混合物)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm) 181.66, 137.12, 132.64, 132.39, 129.68, 122.62, 119.72, 109.61, 105.95, 105.76, 66.09, 51.79, 50.73, 43.06, 41.57, 37.95, 31.80, 30.52, 26.59, 25.78, 21.67. HRMS (HR, DIP-CI):C23H31ClN2O2 (M+H)+的计算值:403.2147,实测:403.2158. CHN分析:计算:62.87 C, 7.34 H, 6.37 N, 16.14 Cl,实测:60.04 C, 7.05 H, 5.92 N, 15.88 Cl。
实施例9. (3-羧甲烯-5-巯基哌啶-1-基)-(2-氯苯基)-乙酸甲酯盐酸盐(化合物VIa盐酸盐)
步骤1:烯丙基-(1-羟基烯丙基)-氨基甲酸叔丁酯的合成。
在15℃冷却的同时,将丁二烯一氧化物(1, 5.0 g, 71.3 mmol) 加入烯丙胺(2, 16 mL)和水(1 mL) 中。将混合物加热至回流(100℃)保持6小时。冷却至室温以后,在减压下在室温除去挥发物。将剩余的含有烯丙基胺的油溶解在二噁烷(100 mL)和水(20 mL)中。在水浴中冷却烧瓶,并加入1M氢氧化钠溶液(80 mL)。加入二碳酸二叔丁酯(17.95 g, 82.2 mmol),并将溶液在室温搅拌过夜。18小时以后,在减压下除去大部分二噁烷。用乙醚(2×100 mL)萃取剩余的水/二噁烷溶液(40 mL)。将醚经硫酸钠干燥,过滤,并在减压下浓缩。通过在硅胶(250 g)上的柱色谱法纯化剩余的油(19.0 g),用庚烷/乙酸乙酯(4:1至1:1)洗脱,得到烯丙基-(1-羟基烯丙基)-氨基甲酸叔丁酯(10.5 g, 65%收率),为澄清的油。1H NMR (300 MHz, CDCl3/TMS):δ= 5.82-5.67 (m, 2H), 5.28-5.02 (m, 4H), 4.25 (m, 1H), 3.80-3.65 (m, 2H), 3.22 (m, 1H), 1.39 (s, 9H)。13C NMR (75 MHz, CDCl3/TMS):δ= 138.52, 135.38, 133.88, 117.42, 116.34, 115.58, 80.30, 72.53, 63.42, 61.04, 53.41, 51.77, 28.50。
步骤2:3-羟基-3,6-二氢-2H-吡啶-1-甲酸叔丁酯的合成。
用氩气在含烯丙基-(1-羟基烯丙基)-氨基甲酸叔丁酯(5.0 g, 21.9 mmol)的二氯甲烷(350 mL)中鼓泡5分钟。将烧瓶置于氩气氛下,并加入Grubb催化剂I (0.48 g)。将混合物在氩下在室温搅拌过夜。18小时以后,浓缩溶液,并通过在硅胶(150 g)上的柱色谱法纯化剩余的油,用庚烷/乙酸乙酯(4:1至1:1)洗脱。合并含有产物的级分,在减压下浓缩,并在高真空下在室温干燥至恒重,得到3-羟基-3,6-二氢-2H-吡啶-1-甲酸叔丁酯(4.20 g, 96%收率),为浓的褐色油。1H NMR (300 MHz, CDCl3/TMS):δ= 5.85-5.65 (m, 2H), 4.13 (m, 1H), 3.80 (s, 2H), 3.66 (m, 1H), 3.26 (m, 1H), 1.39 (s, 9H)。13C NMR (75 MHz, CDCl3/TMS):δ= 155.06, 128.81, 126.79, 80.11, 67.53, 66.83, 63.60, 47.38, 43.36, 28.56。
步骤3:3-氧代-3,6-二氢-2H-吡啶-1-甲酸叔丁酯的合成。
在氩气氛下,将3-羟基-3,6-二氢-2H-吡啶-1-甲酸叔丁酯(3.1 g, 17.3 mmol) 溶解在二氯甲烷(30 mL)中。历时1小时分份加入氯铬酸吡啶鎓(5 g, 23 mmol)。穿过硅胶(100 g)过滤二氯甲烷溶液,用二氯甲烷洗脱。通过在硅胶(silica)筒(40 g)上的MPLC (Companion),纯化粗制的褐色固体(2.84 g),用100% 庚烷洗脱,随后用乙酸乙酯/庚烷(0至60%)梯度洗脱。合并含有产物的级分,浓缩,并在高真空下在室温干燥1小时,得到3-氧代-3,6-二氢-2H-吡啶-1-甲酸叔丁酯(2.1 g, 61%收率),为澄清的油,其在低温静置固化。1H NMR (300 MHz, CDCl3/TMS):δ= 6.95 (m, 1H), 6.08 (d, 1H, J= 10.5 Hz), 4.15 (m, 2H), 4.02 (br s, 2H), 1.39 (s, 9H)。13C NMR (75 MHz, CDCl3/TMS):δ= 193.11, 154.02, 147.17, 127.40, 80.89, 51.97, 42.69, 28.41。
步骤4:1,6-二氢-2H-吡啶-3-酮三氟醋酸盐的合成。
在氩气氛下,将3-氧代-3,6-二氢-2H-吡啶-1-甲酸叔丁酯(0.42 g, 2.13 mmol) 溶解在二氯甲烷(5 mL)中。加入三氟醋酸(1.5 mL),并将混合物在室温搅拌4小时。在减压下浓缩TFA/二氯甲烷溶液,并在高真空下在室温干燥1小时。将剩余的含有1,6-二氢-2H-吡啶-3-酮三氟醋酸盐的褐色油(0.44 g, 97%收率) 立即用于下一步。1H NMR (300 MHz, CDCl3-CD3OD/TMS):δ= 7.12 (d, 1H, J= 10.5 Hz), 6.8 (br, 2H), 6.32 (d, 1H, J = 10.5 Hz), 4.06 (m, 2H), 3.90 (br s, 2H)。13C NMR (75 MHz, CDCl3-CD3OD /TMS):δ= 187.85, 160.79 (q, J = 38 Hz), 132.63, 128.31, 115.71 (q, J = 284 Hz), 49.32, 41.11。
步骤5:溴-(2-氯苯基)-乙酸甲酯的合成。
在氩气氛下在室温,将甲醇(1 mL) 加入甲苯(10 mL)中。在水浴中冷却烧瓶,并加入2M (三甲基甲硅烷基)重氮甲烷(5 mL, 10 mmol),随后历时5分钟逐份加入α-溴-2-氯苯基醋酸(2.2 g, 8.81 mmol)。在另外10分钟以后,在减压下除去甲苯/甲醇。通过在硅胶(silica)筒(40 g)上的MPLC (companion),用乙酸乙酯在庚烷中的梯度(10% 至50%)历时20分钟,纯化粗制的油。合并含有产物的级分,浓缩,并在高真空下在室温干燥1小时,得到溴-(2-氯苯基)-乙酸甲酯(2.0 g, 86%收率),为澄清的液体,其在低温(-10℃)固化。1H NMR (300 MHz, CDCl3/TMS):δ= 7.75 (d, 1H, J = 6.9 Hz), 7.39-7.26 (m, 3H), 5.91 (s, 1H), 3.80 (s, 3H)。13C NMR (75 MHz, CDCl3/TMS):δ= 168.29, 133.82, 133.30, 130.93, 130.47, 129.83, 127.66, 53,80, 43.08。
步骤6:(2-氯苯基)-(3-氧代-3,6-二氢-2H -吡啶-1-基)-乙酸甲酯的合成。
在氩气氛下在室温,将1,6-二氢-2H-吡啶-3-酮三氟醋酸盐(1.27 g, 6.0 mmol)和溴-(2-氯苯基)-乙酸甲酯(1.50 g, 6.15 mmol) 溶解在DMF (5 mL) 中。加入碳酸钾(2 g, 14.5 mmol),并将混合物在室温搅拌3小时。加入水(25 mL),并用二氯甲烷(2×25 mL) 萃取产物。合并二氯甲烷萃取物,经硫酸钠干燥、过滤并浓缩。在硅胶(30 g)上纯化剩余的油,用庚烷-乙酸乙酯(3:1) 洗脱。合并含有产物的级分,在减压下浓缩,并在高真空下在室温干燥2小时,得到(2-氯苯基)-(3-氧代-3,6-二氢-2H -吡啶-1-基)-乙酸甲酯(0.50 g, 29.7%收率),为浅黄色油。1H NMR (300 MHz, CDCl3/TMS):δ= 7.50-7.39 (m, 2H), 7.30-7.26 (m, 2H), 7.02-6.97 (m, 1H), 6.09 (d, 1H, J = 10.5 Hz), 4.94 (s, 1H), 3.70 (s, 3H), 3.49 (m, 2H), 3.39 (d, 1H, J = 15.9 Hz), 3.31 (d, 1H, J = 15.9 Hz)。13C NMR (75 MHz, CDCl3/TMS):δ= 194.81, 170.41, 148.28, 134.74, 132.10, 129.95, 129.73, 129.69, 127.44, 126.99, 66.70, 57.89, 52.14, 49.16. HRMS (GC-CI):(M+H+)的计算值:280.0740,实测:280.0727。
步骤7:(2-氯苯基)-(3-巯基-5-氧代哌啶-1-基)-乙酸甲酯的合成。
在氩气氛下,将(2-氯苯基)-(3-氧代-3,6-二氢-2H -吡啶-1-基)-乙酸甲酯(0.45 g, 1.6 mmol) 溶解在甲醇(100 mL) 中。加入几滴三乙胺,并用氩气在溶液中鼓泡5分钟。停止氩,用硫化氢在溶液中鼓泡45分钟。停止硫化氢,将溶液置于氩气氛保持另外30分钟。通过用氩气在溶液中鼓泡10分钟,除去过量的硫化氢。在减压下除去甲醇,并将粗制的(2-氯苯基)-(3-巯基-5-氧代哌啶-1-基)-乙酸甲酯(0.48 g, 96%收率, 黄色玻璃) 直接用于下一步。1H NMR (300 MHz, CDCl3/TMS):δ= 7.40-7.20 (m, 4H), 4.88 (3×s, 1H), 3.70 (s, 3H), 3.40-3.00 (m, 4H), 2.90-2.60 (m, 2H), 2.45-2.15 (m, 1H)。13C NMR (75 MHz, CDCl3/TMS):δ= 203.04, 170.75, 135.07, 132.49, 130.30, 129.96, 129.85, 127.04, 67.06, 60.38 (4峰), 57.19 (2峰), 52.29, 45.88 (3峰), 39.06 (4峰)。HRMS (GC-CI):(M+H+)的计算值:314.0618,实测:314.0588。
步骤8:3-叔-丁氧基羰基甲烯基-5-巯基哌啶-1-基)-(2-氯苯基)-乙酸甲酯(化合物VIb) 的合成
在室温在氩气氛下,将粗制的(2-氯苯基)-(3-巯基-5-氧代哌啶-1-基)-乙酸甲酯(0.36 g, 1.14 mmol)溶解在THF (5 mL, 经过氩鼓泡)中,并加入P,P-二甲基膦酰基乙酸叔丁酯(0.50 g, 2.23 mmol)和60% 氢化钠(0.075 g, 1.87 mmol)在THF (4 mL, 经过氩鼓泡)中的混合物中。20分钟以后,加入二氯甲烷(25 mL, 经过氩鼓泡),并用水(25 mL, 经过氩鼓泡)洗涤混合物。该二氯甲烷溶液经硫酸钠干燥,过滤,并在减压下浓缩。通过在硅胶(15 g)上的柱色谱法纯化剩余的黄色油,用庚烷-乙酸乙酯(4:1, 经过氩鼓泡)洗脱,得到3-叔-丁氧基羰基甲烯基-5-巯基哌啶-1-基)-(2-氯苯基)-乙酸甲酯(0.28 g, 59%收率),为澄清的凝胶,将其在氩气氛下保持在低温,以防止氧化为二硫化物。1H NMR (300 MHz, CDCl3/TMS):δ= 7.60-7.15 (m, 4H), 5.55 (m, 1H), 4.77 (m, 1H), 3.67 (s, 3H), 3.20-2.00 (m, 7H), 1.41 (m, 9H)。13C NMR (75 MHz, CDCl3/TMS):δ= 170.63 (2×), 165.26 (2×), 151.09 (3×), 134.64, 133.11 (4×), 129.83 (5×), 127.21, 118.35 (2×), 80.32 (2×), 67.57 (5×), 57.28 (6×), 52.28, 42.49-40.14 (8×), 34.54, 28.38 (2×)。HRMS (DIP-CI):(M+H+)的计算值:412.1349,实测:412.1312。
步骤9:(3-羧甲烯-5-巯基哌啶-1-基)-(2-氯苯基)-乙酸甲酯盐酸盐的合成.
将3-叔-丁氧基羰基甲烯基-5-巯基哌啶-1-基)-(2-氯苯基)-乙酸甲酯(0.18 g, 0.43 mmol)溶解在二氯甲烷和三氟醋酸的1:1混合物(6 mL,经过氩鼓泡)中,所述混合物已经用氩鼓泡。将该溶液在氩气氛下在室温搅拌3小时。加入二氯甲烷(25 mL),然后用5% 碳酸氢钠溶液(2×25 mL,经过氩鼓泡)洗涤。将二氯甲烷经硫酸钠干燥,过滤,并在减压下浓缩。加入二氯甲烷(5 mL,经过氩鼓泡),并通过加入2N盐酸在乙醚中的溶液(2 mL,经过氩鼓泡),形成盐。加入额外的乙醚(25 mL,经过氩鼓泡),过滤固体沉淀物,并在高真空下在室温干燥3小时,得到(3-羧甲烯-5-巯基哌啶-1-基)-(2-氯苯基)-乙酸甲酯盐酸盐(化合物VIa盐酸盐, 0.12 g, 70%收率),为浅粉红色固体。1H NMR (300 MHz, DMSO/TMS):δ= 7.65-7.25 (m, 4H), 5.71 (m, 1H), 4.91 (m, 1H), 3.66 (s, 3H), 3.40-2.00 (m, 7H)。(8种异构体的混合物)。13C NMR (75 MHz, DMSO/TMS):δ= 169.60-165.99 (6峰), 194.52 (4峰), 133.46-127.01 (13峰), 119.50-117.50 (6峰), 66.94-65.45 (7峰), 56.74-49.17 (12峰), 33.31 (br)。HRMS (DIP-CI):(M+H+)的计算值:356.0723,实测:356.0712. CHN分析:计算:48.99 C, 4.88 H, 3.51 N,实测:49.25 C, 5.00 H, 3.57 N。
实施例10. 8-(2-氯苯基)-8-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基辛酸(化合物Im)
步骤1:6-乙氧基羰基-6-甲基庚基) 三苯基溴化鏻的合成。
将三苯基膦(14.8 g, 56.6 mmoL) 加入7-溴-2,2-二甲基庚酸乙酯(15.0 g, 56.6 mmoL)在甲苯(110 mL)中的溶液中。将该溶液加热至回流(油浴122℃)保持24 h。蒸发甲苯,并用庚烷(2×60 mL)、乙醚(2×60 mL)洗涤残余物,并在高真空中干燥至恒重,得到6-乙氧基羰基-6-甲基庚基) 三苯基溴化鏻(24.39 g, 81.8%收率),为灰白色粉末(熔点165-170℃)。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.88-7.82 (m, 9H), 7.75-7.72 (m, 6H), 4.06 (q, 2H, J = 6.9 Hz), 3.76 (m, 2H), 1.64 (m, 4H), 1.46-1.41 (m, 2H), 1.20 (t, 5H, J = 6.9 Hz), 1.10 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.82, 134.99, 133.69 (d, J = 10 Hz), 130.57 (d, J = 12 Hz), 117.69 (d, J = 85 Hz), 60.29, 42.12, 40.09, 30.94 (d, J = 16 Hz), 25.26 (d, J = 41 Hz), 23.18 (d, J =41 Hz), 14.40. HRMS (FIA-ESI-TOFM)):C29H36BrO2P (M+H)+的计算值:447.2447,实测:447.2446。
步骤2:8-(2-氯苯基)-2,2-二甲基-辛-7-烯酸乙酯的合成.
将6-乙氧基羰基-6-甲基庚基) 三苯基溴化鏻(24 g, 45.5 mmol)和2-氯苯甲醛(6.38 g, 45.5 mmol)在CH2Cl2 (60 mL) 中尽可能剧烈地搅拌,并逐滴加入50% NaOH溶液(24 mL)。加入结束以后,将混合物继续搅拌3小时。将该混合物转移至分离器,并用二氯甲烷(200 mL)和水(200 mL)洗涤。用二氯甲烷(3×150 mL)萃取水性部分。用盐水(150 mL)洗涤合并的二氯甲烷萃取物,经Na2SO4干燥,浓缩,并通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/10至1/6)纯化,得到8-(2-氯苯基)-2,2-二甲基-辛-7-烯酸乙酯(10 g, 71.6%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.46 (dd, 1H, J = 7.5, 1.5 Hz), 7.36-7.06 (m, 3H), 6.49 (d, 1H, J =11.4 Hz), 5.74 (m, 1H), 4.09 (m, 2H), 2.20 (m, 2H), 1.58-1.36 (m, 4H), 1.21 (m, 5H), 1.13 (s, 6H)。(主要异构体)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.76, 135.78, 134.00, 133.69, 130.48, 129.52, 127.94, 126.69, 126.27, 60.25, 42.26, 40.68, 33.14, 30.27, 28.46, 25.28, 24.73, 14.32. (主要异构体)。HRMS (FIA-ESI-TOFM):C18H25ClO2 (M + Na)的计算值:331.1435,实测:331.1446。
步骤3:8-溴-8-(2-氯苯基)2,2-二甲基辛酸乙酯的合成。
将8-(2-氯苯基)-2,2-二甲基-辛-7-烯酸乙酯(7 g, 22.8 mmol) 溶解在冰醋酸(60 mL)中。在用干燥的溴化氢在溶液中鼓泡8 h的同时,在冰浴(约15℃)中冷却溶液。将所述反应混合物倒入冰水(130 mL)中,并用乙酸乙酯(3×50 mL)萃取。用饱和的NaHCO3溶液(100 mL)洗涤合并的有机层,经Na2SO4干燥,并在减压下浓缩。通过柱色谱法(200 g硅胶,用乙酸乙酯/庚烷= 1/20至1/10洗脱)纯化粗产物(10 g),得到8-溴-8-(2-氯苯基)2,2-二甲基辛酸乙酯(7.46 g, 81.8%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.59 (d, 1H, J = 7.8 Hz), 7.34 (t, 1H, J = 7.5 Hz), 7.26 (d, 1H, J = 6.9 Hz), 7.19 (t, 1H, J = 7.2 Hz), 5.45 (t, 1H, J = 7.5 Hz), 4.09 (q, 2H, J = 7.2 Hz), 2.26-2.09 (m, 4H), 1.52-1.46 (m, 2H), 1.41-1.31 (m, 2H), 1.28-1.18 (m, 2H), 1.20 (t, 3H, J = 7.2 Hz), 1.14 (s, 6H)。13C NMR (场: 75 MHz, 溶剂:CDCl3/TMS) δ(ppm):177.80, 139.43, 132.65, 129.64, 129.24, 128.88, 127.45, 60.28, 50.30, 42.25, 39.21, 29.48, 27.98, 25.36, 24.88, 14.49. HRMS (GC-Cl):C18H26BrClO2 (M+H)+的计算值:389.0883,实测:389.0867。
步骤4:8-(2-氯苯基)-8-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基辛酸乙酯(化合物In) 的合成
将4,5,6,7-四氢噻吩并[3,2-c ]吡啶盐酸盐(1.30 g, 7.19 mmol) 加入含氢氧化钠(1.38 g)的水(86 mL)中,并用二氯甲烷(3×20 mL)萃取。将二氯甲烷经硫酸钠干燥、过滤并浓缩,制备游离碱(1.0 g)。将4,5,6,7-四氢噻吩并[3,2-c ]吡啶游离碱(1.0 g, 7.19 mmol)和8-溴-8-(2-氯苯基)2,2-二甲基辛酸乙酯(2.8 g, 7.19 mmol) 溶解在含碳酸钾(1.49 g, 10.79 mmol)的DMF (75 mL) 中。将该混合物加热至65-70℃过夜。在18小时以后,将混合物冷却至室温,并加入水(50 mL)。用乙醚(3×50 mL)萃取产物。用水(3×50 mL)洗涤合并的有机萃取物,经硫酸钠干燥,并在减压下浓缩。通过在硅胶上的柱色谱法纯化粗产物,用乙酸乙酯/庚烷(1/10)洗脱,得到8-(2-氯苯基)-8-(6,7-二氢-4H -噻吩并[3,2-c]吡啶-5yl)-2,2-二甲基辛酸乙酯(化合物In, 1.4 g, 43.5%),为黄色油。1H NMR (场:300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.49 (dd, 1H, J = 7.5, 1.5 Hz), 7.35 (dd, 1H, J = 8.1, 1.2 Hz), 7.24 (dt, 1H, J = 7.5, 1.0 Hz), 7.16 (dt, 1H, J = 7.8, 1.5 Hz), 7.02 (d, 1H, J = 5.0 Hz), 6.68 (d, 1H, J = 5.0 Hz), 4.18 (dd, 1H, J= 8.7, 4.2 Hz), 4.08 (q, 2H, J = 6.9 Hz), 3.82 (d, 1H, J = 14.1 Hz), 3.48 (d, 1H, J = 14.1 Hz), 2.88-2.66 (m, 4H), 1.96-1.76 (m, 2H), 1.46-1.41 (m, 2H), 1.26-1.18 (m, 6H), 1.21 (t, 3H, J = 6.9 Hz), 1.11 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.92, 134.92, 134.21, 133.53, 129.48, 129.06, 127.94, 126.77, 125.44, 122.60, 63.67, 60.27, 50.73, 48.08, 42.29, 40.85, 33.02, 30.52, 26.13, 25.39, 25.01, 14.51. HRMS (FIA-ESI-TOFM):C25H34ClNO2S (M+H)+的计算值:448.2072,实测:448.2067。
步骤5:8-(2-氯苯基)-8-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基辛酸(化合物Im) 的合成.
将8-(2-氯苯基)-8-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基辛酸乙酯(0.86 g, 1.92 mmol) 加入乙醇(32 mL)和氢氧化钠(0.53 g, 13.4 mmol)在水(10.4 mL)中的溶液中。将混合物加热至回流保持6.5小时。在减压下浓缩溶液,并将水(43 mL) 加入残余物中。用乙酸乙酯/庚烷(1/10, 43 mL)萃取任何原料。抛弃庚烷萃取物。用10 N盐酸溶液将剩余的水溶液调至pH = 6。用CH2Cl2 (3×40 mL)萃取产物,经Na2SO4干燥,浓缩,并通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/10至1/3)进行纯化,得到8-(2-氯苯基)-8-(6,7-二氢-4H -噻吩并[3,2-c ]吡啶-5yl)-2,2-二甲基辛酸(化合物Im,0.35 g,43.8%收率,通过HPLC确定为99.6% 纯度),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):10.43 (br, 1H), 7.52 (d, 1H, J = 7.50 Hz), 7.39 (d, 1H, J = 8.10 Hz), 7.28-7.15 (m, 2H), 7.03 (d, 1H, J = 5.0 Hz), 6.69 (d, 1H, J = 5.0 Hz), 4.24 dd, 1H, J = 9.3, 4.2 Hz), 3.83 (d, 1H, J = 14.40 Hz), 3.51 (d, 1H, J = 14.40 Hz), 2.92-2.73 (m, 4H), 1.99-1.81 (m, 2H), 1.46-1.41 (m, 2H), 1.28-1.15 (m, 6H), 1.13 (s, 6H)。(t, 3H, J = 7.2 Hz), 1.15-1.08 (m, 2H), 1.07 (s, 6H)。13C NMR (场:75 MHz, 溶剂: CDCl3/TMS) δ(ppm):184.02, 138.85, 135.16, 133.87, 133.42, 129.60, 129.17, 128.17, 126.92, 125.52, 122.74, 63.53, 50.54, 47.90, 42.29, 40.70, 32.95, 30.52, 25.75, 25.64, 25.26, 24.96. HRMS (FIA-ESI):C23H30ClNO2S (M+H)+的计算值:420.1759,实测:420.1779. CHN分析:计算:65.77 C, 7.20 H, 3.33 N,实测:60.77 C, 6.68 H, 3.11 N。CHN的最佳拟合: C23H30ClNO2S + HCl
实施例11. 7-(2-氯苯基)-7-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸盐酸盐(化合物Ii盐酸盐)
步骤1:7-(2-氯苯基)-7-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸乙酯(化合物Ij) 的合成
在氩气氛下在室温,在DMF (13 mL)中混合4,5,6,7-四氢呋喃并[3,2-c]吡啶(0.16 g, 1.3 mmol)、7-溴-7-(2-氯苯基)-2,2-二甲基庚酸乙酯(0.49 g, 1.3 mmol)和碳酸钾(0.27 g, 1.95 mmol)。在氩气氛下,将该混合物加热至65℃过夜。继续在65℃反应48小时。冷却至室温并在减压下浓缩以后,通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/20至1/5)纯化粗产物(0.28 g),得到7-(2-氯苯基)-7-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸乙酯(化合物Ij, 0.26 g, 48.1%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂:CDCl3/TMS) δ(ppm):7.42 (d, 1H, J = 7.5 Hz), 7.29 (d, 1H, J = 7.2 Hz), 7.17 (t, 1H, J = 8.7 Hz), 7.14 (s, 1H), 7.09 (t, 1H, J = 8.1 Hz), 6.08 (s, 1H), 4.12 (t, 1H, J = 4.2 Hz), 4.01 (q, 2H,J = 7.2 Hz), 3.98 (d, 1H, J = 14.4 Hz), 3.26 (d, 1H, J = 13.5 Hz), 2.79-2.53 (m, 4H), 1.90-1.73 (m, 2H), 1.35-1.32 (m, 2H), 1.20-1.06 (m, 7 H), 1.04 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.91, 148.97, 140.89, 139.39, 134.93, 129.50, 129.00, 128.02, 126.84, 115.82, 108.84, 63.33, 60.54, 47.61, 47.51, 42.28, 40.78, 33.21, 26.23, 25.35, 24.65, 14.50。
步骤2:7-(2-氯苯基)-7-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸盐酸盐的合成.
将7-(2-氯苯基)-7-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸乙酯(0.26 g, 0.62 mmol) 加入乙醇(10 mL)和氢氧化钠(0.17 g, 4.4 mmol)在水(3.3 mL)中的混合物溶液中,将混合物加热至回流保持6.5小时,此时TLC表明原料消失。在减压下除去乙醇,并将额外的水(15 mL) 加入残余物中。用乙酸乙酯/庚烷1/10的混合物(10 mL)洗涤水性部分,随后抛弃所述混合物。用浓盐酸调节水性级分至pH=6。用二氯甲烷(3×15 mL) 萃取产物。合并的二氯甲烷层经硫酸钠干燥,过滤,并在减压下浓缩。得到产物酸(0.17 g,70.1%收率,通过HPLC确定为97.23% 纯度),为黄色油。将一部分物质(0.15 g, 0.38 mmol) 溶解在乙醚(5 mL)中,并加入2N HCl在醚中的溶液(0.21 mL)中。用水(10 mL)萃取固体沉淀物,并冷冻干燥,得到7-(2-氯苯基)-7-(6,7-二氢-4H -呋喃并[3,2-c]吡啶-5-基)-2,2-二甲基庚酸(化合物Ii盐酸盐,0.11 g,68.8%收率,通过HPLC确定为99.08% 纯度),为浅黄色粉末。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):7.80 (br s, 1H), 7.61-7.48 (m, 4H), 6.43 (s, 0.5H), 6.29 (s, 0.5H), 5.07 (m, 1H), 4.68 (d, 0.5H, J= 13.8 Hz), 4.30 (d, 0.5H, J= 13.8 Hz), 4.25-3.95 (m, 1H), 3.64-3.40 (m, 1H), 3.09-2.95 (m, 2H), 2.40-2.27 (m, 2H), 1.46-0.87 (m, 6H), 1.11 (s, 6H), 1.09 (br s, 1H)。(旋转异构体的混合物)。13C NMR (场: 75 MHz, 溶剂: CD3OD/TMS) δ(ppm):181.47, 147.11, 141.52, 137.12, 132.84, 132.09 131.86, 130.50, 129.75, 112.35, 109.69, 66.20 (br), 43.03, 41.40, 33.10, 31.71, 30.18, 27.36, 25.92, 25.70, 25.63, 23.80, 22.26, 14.50。(旋转异构体的混合物)。HRMS (DIP-CI):C22H28ClNO3 (M+H)+的计算值:390.1830,实测:390.1792. CHN分析:C22H29NCl2O3S: 的计算值61.97 C, 6.86 H, 3.28 N, 16.61 Cl;实测:60.28 C, 6.88 H, 3.13 N, 16.24 Cl。
实施例12. 7-(2-氯苯基)-2,2-二甲基-7-(1,4,6,7-四氢吡咯并[3,2-c]吡啶-5-基)-庚酸盐酸盐(化合物Ik盐酸盐)
步骤1:1-叔-丁氧基羰基-5-[1-(2-氯苯基)-6-甲基庚基]-1 H -吡咯并[3,2-c]吡啶-5-鎓溴化物的合成。
将1H-吡咯并[3,2-c]吡啶-1-甲酸叔丁酯(0.81 g, 3.73 mmol)、7-溴-7-(2-氯苯基)-2,2-二甲基辛酸乙酯(1.40 g, 3.73 mmol)在乙腈(25 mL)中的混合物搅拌并温热至45℃。52小时以后,结束反应。在减压下蒸发溶剂,用乙醚(3×20 mL)洗涤残余物,得到1-叔-丁氧基羰基-5-[1-(2-氯苯基)-6-甲基庚基]-1 H -吡咯并[3,2-c]吡啶-5-鎓溴化物(1.47 g, 66.5%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):10.54 (s, 1H), 8.84 (d, 1H, J = 6.9 Hz), 8.23 (d, 1H, J = 7.8 Hz), 7.72 (s, 1H), 7.53 (t, 1H, J = 4.2 Hz), 7.45 (t, 1H, J = 7.2 Hz), 7.39 (d, 1H, J = 3.6 Hz), 7.31 (s, 1H), 6.96 (s, 1H), 6.47 (t, 1H, J = 7.2 Hz), 4.10 (q, 2H, J = 5.7 Hz), 2.72 (d, 2H, J = 6.6 Hz), 2.40 (m, 1H), 1.69 (s. 9H), 1.50-1.32 (m, 4H), 1.21 (t, 3H, J = 3.3 Hz), 1.12 (s, 6H)。(粗制的混合物. HRMS (DIP-CI):失败,在分析时分解。
步骤2:5-[1-(2-氯苯基)-6-乙氧基羰基-6-甲基庚基]-4,5,6,7-四氢-吡咯并[3,2-c]吡啶-1-甲酸叔丁酯的合成.
向1-叔-丁氧基羰基-5-[1-(2-氯苯基)-6-甲基庚基]-1 H -吡咯并[3,2-c]吡啶-5-鎓溴化物(2.4 g, 4.0 mmol)在70% EtOH (60 mL) 中的溶液中,加入(在室温,在剧烈搅拌下,逐份) 硼氢化钠(0.30 g, 8.0 mmol)。当硼氢化钠完全加入时,停止搅拌,并将混合物搅拌0.5小时。在减压下蒸发溶剂,并加入水(60 mL)。用二氯甲烷(3×150 mL) 萃取产物,合并的有机萃取物经硫酸钠干燥,过滤,并在减压下浓缩。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/10至1/3)纯化粗制的残余物,得到5-[1-(2-氯苯基)-6-乙氧基羰基-6-甲基庚基]-4,5,6,7-四氢-吡咯并[3,2-c]吡啶-1-甲酸叔丁酯(1.45 g, 60.9%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CD3OD/TMS) δ(ppm):7.50 (dd, 1H, J = 7.5, 1.2 Hz), 7.37 (d, 1H, J = 8.1, 1.5 Hz), 7.27-7.10 (m, 3H), 5.94 (d, 1H, J = 3.3 Hz), 4.16-4.13 (m, 1H), 4.09 (q, 2H, J = 6.9 Hz), 3.62 (d, 1H, J = 14.1 Hz), 3.28 (d, 1H, J = 13.5 Hz), 2.85-2.60 (m, 4H), 1.98-1.76 (m, 2H), 1.55 (s, 9H), 1.42 (t, 2H, J = 6.9 Hz), 1.28-1.16 (m, 4H), 1.21 (t, 3H, J = 7.2 Hz), 1.11 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):177.98, 149.49, 139.66, 134.90, 129.44, 129.15, 127.93, 127.60, 126.84, 120.92, 119.77, 109.20, 83.26, 63.76, 60.33, 48.81, 48.19, 42.30, 40.80, 33.23, 32.13, 28.31, 26.07, 25.35, 22.96, 14.52。HRMS (DART-TOF):C29H41ClN2O2 (M+H)+的计算值:517.2828,实测:517.2833。
步骤3:7-(2-氯苯基)-2,2二甲基-7-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-庚酸乙酯(化合物Il)的合成
在氩气氛下,将5-[1-(2-氯苯基)-6-乙氧基羰基-6-甲基庚基]-4,5,6,7-四氢-吡咯并[3,2-c]吡啶-1-甲酸叔丁酯(1.0 g, 1.94 mmol) 溶解在二氯甲烷(40 mL)中。将三氟乙酸(4.0 mL, 40.7 mmol) 加入该溶液中。在2小时以后,将该混合物倒入冰/水(90 mL)中,并用二氯甲烷(3×20 mL)萃取。合并的有机层经硫酸钠干燥,过滤,并在减压下浓缩。通过柱色谱法(硅胶,乙酸乙酯/庚烷= 1/9至1/3)纯化粗产物,得到7-(2-氯苯基)-2,2二甲基-7-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-庚酸乙酯(化合物Il, 330 mg, 41.2%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: CDCl3/TMS) δ(ppm):7.93 (br, 1H), 7.53 (d, 1H, J = 7.5 Hz), 7.35 (d, 1H, J = 7.8 Hz), 7.26-7.13 (m, 2H), 6.58 (s, 1H), 5.93 (s, 1H), 4.16 (dd, 1H, J= 9.3, 3.9 Hz), 4.10 (q, 2H, J = 7.2 Hz), 3.75 (d, 1H, J = 13.5 Hz), 3.39 (d, 1H, J = 13.2 Hz), 2.80-2.56 (m, 4H), 2.04-1.78 (m, 2H), 1.44-1.39 (m, 2H), 1.21 (t, 3H, J = 6.9 Hz), 1.26-1.16 (m, 4H), 1.11 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm):178.01, 139.87, 134.94, 129.40, 129.22, 127.86, 126.81, 125.01, 116.45, 115.70, 105.69, 63.80, 60.37, 48.78, 48.29, 42.31, 40.81, 33.29, 26.15, 25.36, 23.95, 14.52. HRMS (DART-TOF):C24H33ClN2O2 (M+H)+的计算值:717.2303,实测417.2295。
步骤4:7-(2-氯苯基)-2,2二甲基-7-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-庚酸(化合物Ik)的合成
将7-(2-氯苯基)-2,2二甲基-7-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-庚酸乙酯(0.21 g, 0.51 mmol) 加入乙醇(10 mL)、水(2.70 mL)和氢氧化钠(0.14 g, 3.54 mmol)的混合物中。将该混合物加热至回流(95-98℃油浴) 保持16小时。26小时以后,将烧瓶冷却至室温,并在减压下蒸发溶剂。将水(15 mL)加入残余物中,并用乙醚(15 mL)萃取产物。抛弃萃取物,并用10 N盐酸将水性部分调至pH = 6。用二氯甲烷(3×15 mL) 萃取产物,并用水(20 mL)和盐水(20 mL)洗涤合并的二氯甲烷层。二氯甲烷溶液经硫酸钠干燥,过滤,并在减压下浓缩。通过柱色谱法(硅胶,MeOH/CH2Cl2 = 1/9) 纯化粗产物,得到7-(2-氯苯基)-2,2二甲基-7-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-庚酸(化合物Ik, 0.11 g, 57.9%收率),为黄色油。1H NMR (场: 300 MHz, 溶剂: DMSO/TMS) δ(ppm):12.10 (br, 1H), 10.30 (br, 1H), 7.53 (m, 1H), 7.43 (d, 1H, J = 7.2 Hz), 7.36-7.25 (m, 2H), 6.50 (s, 1H), 5.71 (s, 1H), 4.10 (m, 1H), 3.39 (m, 5H), 2.75 (m, 1H), 1.14 (m, 2H), 1.34 (m, 2H), 1.23-1.07 (m, 4H), 1.03 (s, 6H)。13C NMR (场: 75 MHz, 溶剂: DMSO /TMS) δ(ppm) 178.45, 178.97, 133.83, 129.07, 128.23, 126.93, 123.97, 115.83, 113.99, 104.19, 63.11, 48.27, 47.60, 41.19, 40.06, 31.59, 25.84, 25.03, 24.96, 24.55, 23.34。
步骤5:7-(2-氯苯基)-2,2-二甲基-7-(1,4,6,7-四氢吡咯并[3,2-c]吡啶-5-基)-庚酸盐酸盐的合成
将7-(2-氯苯基)-2,2二甲基-7-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-庚酸(0.26 g, 0.67 mmol) 溶解在乙醚(15 mL)中,并加入氯化氢溶液(2N HCl在乙醚中)。用水(15 mL)萃取醚溶液,并抛弃有机层。将水溶液冷冻干燥,得到7-(2-氯苯基)-2,2二甲基-7-(1,4,6,7-四氢-吡咯并[3,2-c]吡啶-5-基)-庚酸盐酸盐(化合物Ik盐酸盐, 0.19 g, 61.9%收率),为浅黄色粉末(通过HPLC确定为纯度97.33%,熔点168-172℃)。1H NMR (场:300 MHz, 溶剂: CD3OD/TMS) δ(ppm):10.26 (br s, 1H), 7.65 (br s, 1H), 7.45-7.38 (m, 4H), 6.54-6.50 (2 s, 1H), 5.84 (s, 0.5H), 5.66 (s, 0.5H), 4.79 (m, 1H), 4.51 (m, 1H), 4.14-3.82 (m, 2H), 3.43-2.76 (m, 4H), 2.43-2.09 (m, 2H), 1.25-1.14 (m, 4H), 1.39-0.65 (m, 2H), 0.96 (s, 6H)。(构象异构体的混合物)。13C NMR (场: 75 MHz, 溶剂: CDCl3/TMS) δ(ppm) 181.52, 137.17, 132.64, 132.32, 131.75, 130.09, 129.79, 122.85, 119.88, 109.70, 106.00, 65.79, 51.79, 51.10, 42.98, 41.43, 31.84, 27.28, 25.096, 25.76, 21.78. MS (HR, DIP-CI):C22H30Cl2N2O2 (M+H)+的计算值:389.1994,实测:389.1996. CHN分析:计算:62.12 C, 7.11 H, 6.59 N, 16.67Cl,实测:59.23 C, 7.04 H, 6.26 N, 16.93 Cl。
实施例13. 6-[1-(2-二甲基氨基嘧啶-5-基甲基)-哌啶-4-基]-2-吗啉-4-基-嘧啶-4-醇, 化合物IIa
步骤1:4-(2-乙氧基羰基乙酰基)-哌啶-1-甲酸叔丁酯的合成
将含有哌啶-1,4-二甲酸1-叔丁酯4-乙酯(0.50 g, 1.94 mmol)的DMF (5 mL) 与乙酸乙酯(0.38 mL, 3.88 mmol)和叔丁醇钾(0.33 g, 2.92 mmol)混合。将该混合物加热至50℃保持20小时。冷却至室温以后,加入水(50 mL),并用乙醚萃取产物。所述醚萃取物经硫酸钠干燥、过滤、并浓缩。通过在硅胶上的柱色谱法纯化粗产物,用庚烷-乙酸乙酯洗脱,得到4-(2-乙氧基羰基乙酰基)-哌啶-1-甲酸叔丁酯(0.27 g, 47%收率)。1H NMR (300 MHz, CDCl3/TMS):δ= 4.20 (q, 2H, J = 7.2 Hz), 4.18-4.05 (m, 2H), 3.50 (s, 2H), 2.86-2.70 (m, 2H), 2.68-2.55 (m, 1H), 1.90-1.78 (m, 2H), 1.60-1.50 (m, 2H), 1.45 (s, 9H), 1.28 (t, 3H, J = 7.2 Hz)。13C NMR (75 MHz, CDCl3/TMS):δ= 204.21, 167.20, 154.67, 79.91, 61.70, 48.93, 47.56, 43.41, 28.73, 27.56, 14.46。
步骤2:4-(6-羟基-2-吗啉-4-基嘧啶-4-基)-哌啶-1-甲酸叔丁酯的合成
将吗啉-4-甲脒氢溴酸盐(0.267 g)和4-(2-乙氧基羰基乙酰基)-哌啶-1-甲酸叔丁酯(0.38 g, 1.27 mmol)在乙醇(10 mL)中搅拌,并加入重氮基双环十一烷(diazobicycloundecane)(285 μl)。将反应物在室温搅拌18小时。在减压下除去乙醇,并加入水(25 mL)。用醋酸酸化溶液(至pH = 4)。用二氯甲烷(4×30 mL)萃取产物。除去二氯甲烷,并在硅胶(Combiflash)上纯化粗产物2次,用10% 甲醇在二氯甲烷中的溶液洗脱,得到4-(6-羟基-2-吗啉-4-基嘧啶-4-基)-哌啶-1-甲酸叔丁酯(0.15 g, 40.5%收率),为白色固体。1H NMR (300 MHz, CDCl3/TMS):δ= 5.63 (s, 1H), 4.25-4.05 (m, 2H), 3.75 (m, 8H), 2.75-2.65 (m, 2H), 2.47-2.35 (m, 1H), 1.00-1.75 (m, 2H), 1.70-1.50 (m, 2H), 1.46 (s, 9H)。13C NMR (75 MHz, CDCl3/TMS):δ= 173.35, 166.87, 154.96, 154.06, 98.88, 79.70, 66.72, 45.13, 44.24, 30.64, 28.82, 27.58。
步骤3:2-吗啉-4-基-6-哌啶-4-基-嘧啶-4-醇盐酸盐的合成
将4-(6-羟基-2-吗啉-4-基嘧啶-4-基)-哌啶-1-甲酸叔丁酯(0.19 g, 0.52 mmol) 加入2 N氯化氢在乙醚中的溶液中,并在室温搅拌18小时。通过过滤,分离得到的产物,并用醚洗涤,得到2-吗啉-4-基-6-哌啶-4-基-嘧啶-4-醇盐酸盐(0.16 g, 91%收率),为白色固体。1H NMR (300 MHz, CD3OD/TMS):δ= 6.17 (s, 1H), 3.90-3.78 (m, 8H), 3.60-3.50 (m, 2H), 3.26-3.1 (m, 3H), 2.32-2.20 (m, 2H), 2.00-1.80 (m, 2H)。13C NMR (75 MHz, CD3OD/TMS):δ= 171.60, 164.86, 155.10, 97.63, 66.93, 45.13, 46.92, 44.81, 38.29, 28.41。
步骤4:6-[1-(2-二甲基氨基嘧啶-5-基甲基)-哌啶-4-基]-2-吗啉-4-基-嘧啶-4-醇的合成
将2-吗啉-4-基-6-哌啶-4-基-嘧啶-4-醇盐酸盐(0.16 g, 0.47 mmol)、2-二甲基氨基-嘧啶-5-醛(86 mg, 0.57 mmol)、和一滴醋酸在无水二氯甲烷(10 mL) 中在室温混合3小时。加入氰基硼氢化钠(88 mg, 1.42 mmol),并将混合物搅拌3天。将该混合物倒入饱和的碳酸氢钠溶液中,并用二氯甲烷(3×50 mL)萃取。在硅胶上纯化该物质,用10% 甲醇在二氯甲烷中的溶液洗脱,得到6-[1-(2-二甲基氨基嘧啶-5-基甲基)-哌啶-4-基]-2-吗啉-4-基-嘧啶-4-醇(化合物IIa, 0.11 g, 69%收率),为白色固体. CHN分析:C20H29N7O2的计算值: 60.13 C, 7.32 H, 24.54 N;实测:57.31 C, 7.11 H, 23.68 H。最佳拟合是C20H29N7O2+ 1H2O; 57.54 C, 7.48 H, 23.48 N。1H NMR (300 MHz, CDCl3/CD3OD/TMS):δ= 8.26 (s, 2H), 5.65 (s, 1H), 3.85-3.60 (m, 8H), 3.42 (m, 2H), 3.19 (s, 6H), 3.15-2.85 (m, 3H), 2.40-2.00 (m, 2H), 2.18-1.60 (m, 4H)。13C NMR (75 MHz, CDCl3/CD3OD/TMS):δ= 173.66, 161.63, 158.91, 153.90, 116.63, 98.76, 66.56, 57.62, 53.12, 44.94, 43.65, 37.48, 30.26。
实施例14. 制备N-(2,3-二氢苯并[1,4]二氧杂环己烯-6-基)-2-(2-甲氧基-乙基氨基)-乙酰胺, 化合物XIa
将2-氯-N-(2,3-二氢-1,4-苯并二氧杂环己烯-6-基)乙酰胺(2 mmol)、碳酸钾(10 mmol)、2-甲氧基乙胺(2 mmol)和无水DMF (4 mL)的混合物在100℃搅拌2小时(通过TLC监测)。将该混合物冷却至室温,用冷水(30 mL)处理。通过过滤,收集沉淀物,用乙醚洗涤,并干燥,得到N-(2,3-二氢苯并[1,4]二氧杂环己烯-6-基)-2-(2-甲氧基-乙基氨基)-乙酰胺(化合物XIa, 96 mg, 36%)。1H NMR (300 MHz, CDCl3/ TMS):δ= 9.29 (s, 1H), 7.24 (d, 1H, J = 2.4 Hz), 6.97 (dd, 1H, J = 8.7, 2.4 Hz), 6.78 (d, 1H, J = 8.7 Hz), 4.32-4.15 (m, 4H), 3.46 (t, 2H, J = 4.8 Hz), 3.36 (s, 3H), 3.34 (s, 2H), 2.82 (t, 2H, J = 4.8 Hz), 2.17 (s, 1H)。13C NMR (75 MHz, CDCl3/CD3OD/TMS):δ= 169.48, 143.16, 139.87, 131.43, 116.84, 112.74, 108.89, 71.41, 64.28, 64.12, 58.68, 52.57, 49.35。
实施例15. 制备6-[1-(3-羟基-4-甲氧基苯甲基)-哌啶-4-基]-2-哌啶-1-基-嘧啶-4-醇, 化合物IIb
步骤1:4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯的合成
将含哌啶-1,4-二甲酸1-叔丁酯4-乙酯(0.50 g, 1.94 mmol)的DMF (5 mL) 与乙酸乙酯(0.38 mL, 3.88 mmol)和叔丁醇钾(0.33 g, 2.92 mmol)混合。将该混合物加热至50℃保持20小时。冷却至室温以后,加入水(50 mL),并用乙醚萃取产物。将醚萃取物经硫酸钠干燥、过滤、并浓缩。通过在硅胶上的柱色谱法纯化粗产物,用庚烷-乙酸乙酯洗脱,得到4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯(0.27 g, 47%收率)。
步骤2:4-(6-羟基-2-哌啶-1-基-嘧啶-4-基)-哌啶-1-甲酸叔丁酯的合成
将钠(1 mol) 溶解在无水乙醇(400 mL)中。向得到的溶液中,小心地分份加入哌啶-1-甲脒(0.5 mol)。然后,逐滴加入4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯(0.5 mol),并将混合物回流搅拌4-6 h (通过TLC监测),冷却至室温,浓缩,用水(300 mL)稀释,并用醋酸酸化至pH-4。通过过滤,收集形成的沉淀物,用水洗涤,并干燥,得到4-(6-羟基-2-哌啶-1-基-嘧啶-4-基)-哌啶-1-甲酸叔丁酯(89 g, 49%)。
步骤3:6-哌啶-4-基-2-哌啶-1-基-嘧啶-4-醇盐酸盐的合成
将4-(6-羟基-2-哌啶-1-基-嘧啶-4-基)-哌啶-1-甲酸叔丁酯(0.05 mol)在15% HCl的二噁烷溶液(100 mL)中的混悬液回流搅拌2小时。反应结束以后,冷却混合物,过滤沉淀物,用无水乙醚洗涤,并干燥,得到6-哌啶-4-基-2-哌啶-1-基-嘧啶-4-醇盐酸盐(12 g, 81%)。
步骤4:6-[1-(3-羟基-4-甲氧基-苯甲基)-哌啶-4-基]-2-哌啶-1-基-嘧啶-4-醇的合成
将6-哌啶-4-基-2-哌啶-1-基-嘧啶-4-醇盐酸盐(2.0 mmol)、3-羟基-4-甲氧基-苯甲醛(2.6 mmol)、三乙胺(4.0 mmol)和3滴醋酸在无水二氯甲烷(20 mL) 中的混合物在室温搅拌3小时。然后,分份加入三乙酰氧基硼氢化钠(6.0 mmol),并继续搅拌48小时(通过TLC监测)。用饱和碳酸氢钠水溶液(20 mL)淬灭混合物,并用二氯甲烷(2x10 mL)萃取产物。用盐水洗涤萃取物,并经硫酸钠干燥。在真空中蒸发溶剂,得到粗产物。通过柱色谱法(硅胶,乙酸乙酯/己烷) 进行纯化,得到6-[1-(3-羟基-4-甲氧基-苯甲基)-哌啶-4-基]-2-哌啶-1-基-嘧啶-4-醇(化合物IIb, 542 mg, 68%)。
实施例16. 制备6-(1-(4-(甲基氨基)苯甲基)哌啶-4-基)-2-(哌啶-1-基)嘧啶-4-醇, 化合物IIc
步骤1:4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯的合成
将含哌啶-1,4-二甲酸1-叔丁酯4-乙酯(0.50 g, 1.94 mmol)的DMF (5 mL) 与乙酸乙酯(0.38 mL, 3.88 mmol)和叔丁醇钾(0.33 g, 2.92 mmol)混合。将该混合物加热至50℃保持20小时。冷却至室温以后,加入水(50 mL),并用乙醚萃取产物。将醚萃取物经硫酸钠干燥、过滤、并浓缩。通过在硅胶上的柱色谱法纯化粗产物,用庚烷-乙酸乙酯洗脱,得到4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯(0.27 g, 47%收率)。
步骤2:4-(6-羟基-2-(哌啶-1-基)嘧啶-4-基)哌啶-1-甲酸叔丁酯的合成
将钠(1 mol) 溶解在无水乙醇(400 mL)中。向该得到的溶液中,小心地分份加入哌啶-1-甲脒(0.5 mol)。然后,逐滴加入4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯(0.5 mol),并将混合物回流搅拌4-6 h (通过TLC监测),冷却至室温,浓缩,用水(300 mL)稀释,并用醋酸酸化至pH-4。通过过滤,收集形成的沉淀物,用水洗涤,并干燥,得到4-(6-羟基-2-哌啶-1-基-嘧啶-4-基)-哌啶-1-甲酸叔丁酯(80 g, 44%)。
步骤3:2-(哌啶-1-基)-6-(哌啶-4-基)嘧啶-4-醇盐酸盐的合成
将4-(6-羟基-2-哌啶-1-基-嘧啶-4-基)-哌啶-1-甲酸叔丁酯(0.05 mol)在15% HCl的二噁烷溶液(100 mL)中的混悬液回流搅拌2小时。反应结束以后,冷却混合物,过滤沉淀物,用无水乙醚洗涤,并干燥,得到2-吗啉-4-基-6-哌啶-4-基-2-哌啶-1-基-嘧啶-4-醇盐酸盐(12 g, 82%)。
步骤4:6-(1-(4-(甲基氨基)苯甲基)哌啶-4-基)-2-(哌啶-1-基)嘧啶-4-醇的合成
将6-哌啶-4-基-2-哌啶-1-基-嘧啶-4-醇盐酸盐(2.0 mmol)、2-甲基氨基嘧啶-5-甲醛(2.6 mmol)、三乙胺(4.0 mmol)和3滴醋酸在无水二氯甲烷(20 mL)中的混合物在室温搅拌3小时。然后,分份加入三乙酰氧基硼氢化钠(6.0 mmol),并继续搅拌48小时(通过TLC监测)。用饱和碳酸氢钠水溶液(20 mL)淬灭混合物,并用二氯甲烷(2x10 mL)萃取产物。用盐水洗涤萃取物,并经硫酸钠干燥。在真空中蒸发溶剂,得到粗产物。通过柱色谱法(硅胶,乙酸乙酯/己烷)纯化,得到6-[1-(2-甲基氨基嘧啶-5-基甲基)-哌啶-4-基]-2-哌啶-1-基-嘧啶-4-醇(化合物IIc, 637 mg, 83%)。
实施例17. 制备异色满-1-基(5'H-螺[哌啶-4,4'-吡咯并[1,2-a]喹喔啉]-1-基)甲酮, 化合物VIIa
步骤1:4,5-二氢-吡咯并[1,2-a]喹喔啉-螺-4-哌啶-1-甲酸叔丁酯的合成
将2-吡咯-1-基-苯胺(0.05 mol)和N-Boc-哌啶酮(0.05 mol) 溶解在乙醇(50 mL)中,加入对甲苯磺酸一水合物作为催化剂,在氩下将搅拌的反应混合物加热至回流2-3 h。通过TLC监测反应。过滤形成的产物,用冷乙醇洗涤,并干燥,得到4,5-二氢-吡咯并[1,2-a]喹喔啉-螺-4-哌啶-1-甲酸叔丁酯(7 g, 44%)。1H NMR (300 MHz, CDCl3/TMS) δ7.30 (d, J = 7.5 Hz,1H), 7.18-7.12 (m, 1H), 6.97 (t, J = 7.4 Hz,1H), 6.84 (t, J = 7.6 Hz,1H), 6.78 (d, J = 8.1 Hz,1H), 6.30 (t, J = 3.0 Hz,1H), 6.07-6.02 (m, 1H), 4.21 (s, 1H), 3.95-3.77 (m, 2H), 3.30-3.17 (m, 2H), 2.15-1.90 (m, 2H), 1.90-1.78 (m, 2H), 1.47 (s, 9H)。13C NMR (75 MHz, CDCl3/TMS) δ154.55, 133.87, 133.03, 125.53, 124.59, 119.45, 116.06, 114.49, 114.26, 109.83, 102.62, 79.69, 51.10, 39.58, 35.50, 28.38。
步骤2:游离胺的合成
将4,5-二氢-吡咯并[1,2-a]喹喔啉-螺-4-哌啶-1-甲酸叔丁酯(0.1 mol) 溶解在异丙醇(100 mL)中,并加热至回流。向剧烈搅拌的混合物中,逐滴加入30-40 mL HCl (14-16%)在二噁烷中的溶液。放出气体。产物盐酸盐形成为白色沉淀物。将混合物加热至回流保持30-40分钟,以完成反应。过滤,得到4,5-二氢吡咯并[1,2-a]喹喔啉-螺-4-哌啶盐酸盐。将4,5-二氢吡咯并[1,2-a]喹喔啉-螺-4-哌啶盐酸盐溶解在水(50 mL)中,并用固体碳酸钾淬灭至pH 7-8。通过过滤,收集沉淀物,得到4,5-二氢-吡咯并[1,2-a]喹喔啉螺-4-哌啶(22 g 94%),为游离碱。1H NMR (300 MHz, CDCl3/TMS) δ7.28 (d, J = 7.8 Hz,1H), 7.15-7.11 (m, 1H), 6.96 (t, J = 7.5 Hz,1H), 6.86-6.76 (m, 2H), 6.29 (t, J = 3.0 Hz,1H), 6.08-6.04 (m, 1H), 4.39 (s, 1H), 3.69 (s, 1H), 3.07-2.85 (m, 4H), 2.03-1.90 (m, 2H), 1.90-1.79 (m, 2H)。13C NMR (75 MHz, CDCl3/TMS) δ134.13, 134.01, 125.45, 124.49, 119.11, 115.88, 114.41, 114.02, 109.77, 102.53, 66.96, 51.26, 42.07, 36.54。
步骤3:异色满-1-基(5'H-螺[哌啶-4,4'-吡咯并[1,2-a]喹喔啉]-1-基)甲酮的合成
将4,5-二氢吡咯并[1,2-a]喹喔啉-螺-4-哌啶(2.0 mmol)、三乙胺(2 mmol)、异色满-1-甲酸(2 mmol)和苯并三唑-1-基-氧基-三-(二甲基氨基)-鏻六氟磷酸盐(2 mmol)在二氯甲烷(10 mL)中的混合物在室温搅拌16小时。用饱和碳酸氢钠水溶液(10 mL) 淬灭混合物,搅拌2小时,并用二氯甲烷(2x10 mL)萃取产物。合并的有机层经硫酸钠干燥,并减压浓缩,得到粗产物。通过柱色谱法(硅胶,乙酸乙酯/己烷)纯化,得到异色满-1-基(5'H-螺[哌啶-4,4'-吡咯并[1,2-a]喹喔啉]-1-基)甲酮。1H NMR (300 MHz, CDCl3/TMS) δ7.31-7.05 (m,6H), 6.96 (dd, J = 13.6, 6.1 Hz,1H), 6.87-6.71 (m, 2H), 6.30 (t, J = 3.1 Hz,0.5H), 6.25 (t, J = 3.1 Hz,0.5H), 6.07 (d, J = 2.1 Hz,0.5H), 5.88 (d, J = 2.1 Hz,0.5H), 5.52 (s, 1H), 4.28-4.03 (m, 3H), 3.91-3.68 (m, 2H), 3.48-3.12 (m, 2H), 3.10-2.96 (m, 1H), 2.12-1.78 (m, 2.5H), 1.68-1.54 (m, 2H), 1.44-1.36 (m, 0.5H)。13C NMR (75 MHz, CDCl3/TMS) δ168.30, 133.82, 132.75, 132.61, 132.31, 132.18, 129.01, 127.24, 126.29, 125.68, 124.63, 119.64, 116.30, 114.50, 114.36, 109.91, 102.92, 102.61, 79.59, 79.39, 64.55, 51.36, 41.63, 41.50, 38.67, 36.30, 35.69, 35.52, 28.00。
在60℃,向(2-乙基-苯基)-肼1 (35 mmol)和乙二胺四乙酸二钠(20 mg)在甲醇(60 ml)中的溶液中,逐滴加入2-氯丙烯腈2 (105 mmol),并将所述混合物在回流下搅拌8小时。然后,加入浓硫酸(94 mmol),并将混合物进一步加热6小时。冷却至室温以后,用无水碳酸钠(105 mmol)淬灭混合物,并在减压下除去溶剂。用水(100 mL)处理残余物,并用乙酸乙酯(3*100 mL)萃取产物。合并的有机萃取物经无水硫酸镁干燥,并在减压下除去溶剂。通过柱色谱法纯化残余物,得到(28 mmol) 2-(2-乙基-苯基)-2H-吡唑-3-基胺3。1H NMR (300 MHz, CDCl3/TMS) δ7.44-7.35 (m, 3H), 7.30-7.25 (m, 2H), 5.57 (d, J = 2.1 Hz,1H), 3.57 (br s, 2H), 2.49 (q, J = 7.6 Hz,2H), 1.10 (t, J = 7.6 Hz,3H),. 13C NMR (75 MHz, CDCl3/TMS) δ145.20, 142.60, 139.62, 135.93, 129.47, 129.41, 127.88, 126.55, 88.70, 24.04, 14.34。
向含2-(2-乙基-苯基)-2H-吡唑-3-基胺3 (25.9 mmol)的EtOH (150 mL) 中加入3-乙氧基-4-羟基-苯甲醛5 (28.5 mmol),随后加入麦尔酮酸4 (28.5 mmol)。将所述反应混合物加热至75℃,然后在2小时以后,将所述反应混合物冷却至室温,并在减压下浓缩。用水(100 mL)处理残余物,并用二氯甲烷(200 mL) 萃取产物。将有机萃取物经无水硫酸镁干燥,并减压除去溶剂。通过柱色谱法纯化残余物,得到(11 mmol) 4-(3-乙氧基-4-羟基-苯基)-1-(2-乙基-苯基)-1,4,5,7-四氢-吡唑并[3,4-b]吡啶-6-酮6。1H NMR (300 MHz, CDCl3/TMS) δ7.44-7.35 (m, 3H), 7.30-7.25 (m, 2H), 5.57 (d, J = 2.1 Hz,1H), 3.57 (br s, 2H), 2.49 (q, J = 7.6 Hz,2H), 1.10 (t, J = 7.6 Hz,3H);13C NMR (75 MHz, CDCl3/TMS) δ145.20, 142.60, 139.62, 135.93, 129.47, 129.41, 127.88, 126.55, 88.70, 24.04, 14.34。
实施例17. 制备6-[1-(2-氨基-嘧啶-5-基甲基)-哌啶-4-基]-2-哌啶-1-基-嘧啶-4-醇, 化合物IId
在0 C,向N-Boc-哌啶-4-甲酸乙酯(0.5 mol)和乙酸乙酯(3 mol)的混合物中,分份加入t-BuOK(1.5 mol)。将所述混合物在室温搅拌3h (通过TLC监测),浓缩至一半体积,用水(200 mL)稀释,并用乙醚萃取。有机层经硫酸钠干燥,并在真空中蒸发。通过柱色谱法(硅胶,乙酸乙酯/己烷) 纯化,得到4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯(78 g, 52%)
将钠(1 mol) 溶解在无水乙醇(400 mL)中。向该得到的溶液中,小心地分份加入哌啶-1-甲脒(0.5 mol)。然后,逐滴加入4-(2-乙氧基羰基-乙酰基)-哌啶-1-甲酸叔丁酯(0.5 mol),并将混合物回流搅拌4-6 h (通过TLC监测),冷却至室温,浓缩,用水(300 mL)稀释,并用醋酸酸化至pH-4。通过过滤,收集形成的沉淀物,用水洗涤,并干燥,得到4-(6-羟基-2-哌啶-1-基-嘧啶-4-基)-哌啶-1-甲酸叔丁酯(82 g, 45%)。
将4-(6-羟基-2-哌啶-1-基-嘧啶-4-基)-哌啶-1-甲酸叔丁酯(0.05 mol)在15% HCl的二噁烷溶液(100 mL)中的混悬液回流搅拌2小时。反应结束以后,冷却混合物,过滤沉淀物,用无水乙醚洗涤,并干燥,得到6-哌啶-4-基-2-哌啶-1-基-嘧啶-4-醇盐酸盐(12 g, 82%)。
将6-哌啶-4-基-2-哌啶-1-基-嘧啶-4-醇(2.0 mmol)、2-氨基-嘧啶-5-甲醛(2.6 mmol)、三乙胺(4.0 mmol)和3滴醋酸在无水二氯甲烷(20 mL) 中的混合物在室温搅拌3小时。然后,分份加入三乙酰氧基硼氢化钠(6.0 mmol),并继续搅拌48小时(通过TLC监测)。用饱和碳酸氢钠水溶液(20 mL)淬灭混合物,并用二氯甲烷(2x10 mL)萃取产物。用盐水洗涤萃取物,并经硫酸钠干燥。在真空中蒸发溶剂,得到粗产物。通过柱色谱法(硅胶,乙酸乙酯/己烷) 纯化,得到6-[1-(2-氨基-嘧啶-5-基甲基)-哌啶-4-基]-2-哌啶-1-基-嘧啶-4-醇(606 mg, 82%)
实施例18. 制备异色满-1-基(5'H-螺[哌啶-4,4'-吡咯并[1,2-a]喹喔啉]-1-基)甲酮, 化合物VIIa
步骤1:4,5-二氢-吡咯并[1,2-a]喹喔啉-螺-4-哌啶-1-甲酸叔丁酯的合成
将2-吡咯-1-基-苯胺(0.05 mol)和N-Boc-哌啶酮(0.05 mol) 溶解在乙醇(50 mL)中,加入对甲苯磺酸一水合物作为催化剂,在氩下将搅拌的反应混合物加热至回流2-3 h。通过TLC监测反应。过滤形成的产物,用冷乙醇洗涤,并干燥,得到4,5-二氢-吡咯并[1,2-a]喹喔啉-螺-4-哌啶-1-甲酸叔丁酯(7 g, 44%)。
步骤2:游离胺的合成
将4,5-二氢-吡咯并[1,2-a]喹喔啉-螺-4-哌啶-1-甲酸叔丁酯(0.1 mol)溶解在异丙醇(100 mL)中,并加热至回流。向剧烈搅拌的混合物中,逐滴加入30-40 mL HCl (14-16%)在二噁烷中的溶液。放出气体。产物盐酸盐形成为白色沉淀物。将混合物加热至回流保持30-40分钟,以完成反应。过滤,得到4,5-二氢吡咯并[1,2-a]喹喔啉-螺-4-哌啶盐酸盐。将4,5-二氢吡咯并[1,2-a]喹喔啉-螺-4-哌啶盐酸盐溶解在水(50 mL)中,并用固体碳酸钾淬灭至pH 7-8。通过过滤,收集沉淀物,得到4,5-二氢-吡咯并[1,2-a]喹喔啉螺-4-哌啶(22 g 94%),为游离碱。
步骤3:异色满-1-基(5'H-螺[哌啶-4,4'-吡咯并[1,2-a]喹喔啉]-1-基)甲酮的合成
将4,5-二氢吡咯并[1,2-a]喹喔啉-螺-4-哌啶(2.0 mmol)、三乙胺(2 mmol)、异色满-1-甲酸(2 mmol)和苯并三唑-1-基-氧基-三-(二甲基氨基)-鏻六氟磷酸盐(2 mmol)在二氯甲烷(10 mL)中的混合物在室温搅拌16小时。用饱和碳酸氢钠水溶液(10 mL)淬灭混合物,搅拌2小时,并用二氯甲烷(2x10 mL) 萃取产物。合并的有机层经硫酸钠干燥,并减压浓缩,得到粗产物。通过柱色谱法(硅胶,乙酸乙酯/己烷)纯化,得到异色满-1-基(5'H-螺[哌啶-4,4'-吡咯并[1,2-a]喹喔啉]-1-基)甲酮(化合物VIIa, 527 mg, 66%)。
生物学测定
实施例19. 使用P2Y13受体转染的细胞系,本发明的示例性化合物的激动剂活性
使用Ca++流量测定(与荧光染料检测结合),以参照化合物作为阳性参照,测定化合物对P2Y13 GPCR转染的细胞的激动剂活性。在该测定中使用2种细胞系:稳定表达人P2Y13受体的1321N1-P2Y13,和作为对照的1321N1亲本细胞系。以300万细胞/烧瓶,分别将1321N1亲本和1321N1 P2Y13接种进T25烧瓶中。在5% CO2中,在37摄氏度温育细胞过夜。次日,使用Fugene转染试剂(Roche’s Fugene Reagent),用Gqi5蛋白转染细胞。
转染24小时以后,从烧瓶中收集细胞,并以8000细胞/孔接种进384-孔平板中。在Gqi5转染以后48小时,进行测定。
方法:根据生产商的方案(Molecular Devices FLIPR Calcium 4 Assay试剂盒(R8142)),进行Ca++流量测定。简而言之,从孔中抽吸细胞培养基,并用25µL汉克氏缓冲液或PBS缓冲液替换。将25µL Ca++染料加入每个测定孔中。将平板在5% CO2中在37℃温育1小时。温育以后,将平板转移至FlexStation III (FlexStation III (Molecular Devices))。将化合物自动地注射进每个孔中。
结果:得到本发明的示例性化合物的EC50值。结果显示在图1和表3中。还将所述EC50值与参照化合物的EC50值进行对比。
测定了本发明的其它示例性化合物对P2Y13受体转染的细胞的激动剂活性,并显示在图2中,并将EC50总结在表4中。
表3:从图1的数据测得的代表性化合物对P2Y13受体活性的EC50
表4:从图2的数据测得的对P2Y13受体活性的EC50值
参照化合物的化学名是二氯-(((((2R,3S,4R,5R)-3,4-二羟基-5-(6-(2-(甲硫基)乙基氨基)-2-(3,3,3-三氟丙基硫基)-9H - 嘌呤-9-基)四氢呋喃-2-基)甲氧基)(羟基) 磷酰基氧基)(羟基)磷酰基)-甲基膦酸。所述参照化合物也称作坎格雷洛®。它的化学结构如下所示:
在人肝细胞(HepG2) 细胞中的体外HDL内化
通过对HepG2细胞的体外测定,测量了本发明的示例性化合物对HDL内化的影响(图3)。
在有本发明的示例性化合物(终浓度1 μM)存在下,将3H胆固醇胆甾醇油酸酯[胆甾醇基-1,2-3H(N)] 标记的HDL ] (可从PerkinElmer http://www.perkinelmer.com/Catalog/Product/ID/NET746L001MC得到) 加给HepG2细胞。以前报道,所述参照化合物会在体外增加HepG2细胞中的HDL摄入(Jacquet S, Malaval C, Martinez LO, Sak K, Rolland C, Perez C, Nauze M, Champagne E, TercéF, Gachet C, Perret B, Collet X, Boeynaems JM, Barbaras R. The nucleotide receptor P2Y13 is a key regulator of hepatic high-density lipoprotein (HDL) endocytosis. Cell Mol Life Sci. 2005 Nov;62(21):2508-15),并用作HepG2细胞的HDL摄入的阳性对照。经确定,化合物XIa、IIc、IIa和Ih (R-异构体)对HepG2细胞的HDL摄入具有强烈影响;化合物IIb、VIIa和VIIIa对HepG2细胞的HDL摄入具有不大的影响。
在静脉内注射选择的分子以后对胆汁酸生理学的影响
胆汁酸源自胆固醇,它们的合成是胆固醇的分解代谢的主要途径。肝脏的HDL摄入与胆汁酸分泌和胆汁胆固醇清除直接关联。为了证实本发明的示例性化合物对胆汁生理学的影响,将所述化合物注射进C57Bl6小鼠的尾巴中,并在4小时以后,处死小鼠,分析胆汁含量(图5)。观察到胆汁酸浓度以及胆汁体积的增加。所有化合物与对照化合物(参照化合物)同样好地或甚至更好地产生响应。
选择的分子对胆汁生理学的剂量响应
在经口管饲法以后,确定本发明的示例性化合物的剂量响应。选择的剂量是0.003、0.03和0.3 mg/kg。通过经口管饲法施用选择的本发明的化合物,并在6小时以后,处死小鼠,分析胆汁含量(图6-11)。对于所有化合物的所有浓度,观察到胆汁酸、胆汁胆固醇和胆汁磷脂的剂量依赖性的增加。化合物IIa、VIIIa、IIc和XIa在0.003 mg/kg表现出作用;化合物IIb和VIIa在0.3 mg/kg表现出作用。
每天用化合物IIa治疗持续1周以后对胆汁酸生理学的影响
在以前的实验中,施用本发明的示例性化合物1次,并在6小时以后测量对胆汁生理学的影响。通过经口管饲法,每天1次地施用化合物IIa,持续1周,以允许观察更长时间的治疗对胆汁生理学的影响。在处死当天,所述条件与剂量响应实验类似。观察化合物IIa对胆汁分泌的净影响。与媒介物治疗相比,单一胆汁的体积也实现显著增加。
在Hepa 1-6细胞中的P2Y13抑制
为了将特异性的P2Y13 HDL摄入与本发明的示例性化合物相关联,研究了小鼠肝细胞系(Hepa 1-6)中的受抑制的P2Y13。使用编码小鼠P2Y13 shRNA的慢病毒颗粒(得自Open Biosystem的pTRIPZ载体),其可通过将多西环素加给细胞进行诱导。在多西环素存在下,观察到P2Y13 mRNA的减少(图11)。在有和没有化合物IIa和多西环素存在下,测量了该细胞系的HDL摄入(图12)。当用多西环素处理细胞时,观察到HDL摄入的显著减少(8.5%) (媒介物组-p<0.05)。在化合物IIa存在下,也观察到该减少(17.5%-p<0.005),因而表明化合物IIa的靶标是P2Y13。
材料和方法
胆汁酸试剂盒购自Diazyme;磷脂和总胆固醇试剂盒购自Biolabo;小鼠C57Bl6购自Janvier。小鼠pGIPZ慢病毒P2Y13 shRNAmir (RMM4431-98723221) 购自Openbiosystem,并亚克隆进pTRIZ (多西环素可诱导的载体)中。由Vectalys (法国) 制备1 mL慢病毒产物,其具有1.2E8转导单位/mL。用于实时PCR测定的小鼠P2Y13引物(Mm 00546978-m1)、GAPDH引物(Mm03302249-g1)和核糖体18S引物(Mm02601777-g1) 购自Applied。HepG2细胞(HB-8065)和Hepa 1-6 (CRL-1830) 得自ATCC。
脂蛋白制备
通过在KBr梯度上的连续超速离心,从小鼠血浆中纯化出脂蛋白。在密度d=1.21,得到HDL。在PBS中透析纯化的HDL,并用于摄入实验。
HDL标记
干燥3H胆固醇胆甾醇油酸酯[胆甾醇基-1,2-3H(N)] (Perkin Elmer)溶液(250 μL),并再悬浮于丙酮(250 μL)中。将该溶液与脂蛋白脱脂血清(20 mL,40mg/mL)和DTNB (0.4 μM终浓度)混合。加入HDL (7 mg),并在轻轻旋转下在4℃温育过夜。如以前所述,在KBr梯度上纯化HDL。
HepG2和shP2Y13转导的Hepa 1-6 HDL摄入
以300,000细胞/孔,将转导的Hepa 1-6细胞铺板在6x-孔平板中。每天用多西环素(10µM)处理这些细胞持续3天(对于P2Y13熄灭),或者不用多西环素处理(质粒“渗漏”对照)。摄入当天,用DMEM洗涤细胞1次,并在DMEM中在37℃温育1小时。将75 μg放射标记的HDL (6500 dpm/ μg的HDL3.)加入培养基中,并在37℃温育10分钟。用DMEM (2 mL)洗涤细胞1次,并在DMEM中在4℃温育1.5小时,然后发生解离。然后,用冷的DMEM (2 ml)再洗涤细胞1次,并通过加入己烷:异丙醇(3:2)溶液(1mL),从HepG2细胞中回收掺入的胆固醇醚。
HDL摄入
以300,000细胞/孔,将肝细胞铺板在6x-孔平板中。2天以后,用DMEM洗涤细胞1次,并在DMEM中在37℃温育1小时。将75 μg放射标记的HDL (6500 dpm/ μg) 加入培养基中,并在37℃温育10分钟。用DMEM (2mL)洗涤细胞1次,并在DMEM中在4℃温育1.5小时,然后发生解离。然后,用冷的DMEM (2 ml)再洗涤细胞1次,并通过加入己烷:异丙醇(3:2)溶液(1mL),从HepG2细胞中回收掺入的胆固醇醚。
胆固醇流出
如以前所述(Wang等人, 2007),在第0天给药之前,和在化合物IIa施用4周以后,在收集的兔血浆的血液样品中定量胆固醇流出能力。简而言之,将用[3H]-胆固醇(2µCi/ml; Perkin-Elmer)标记的氧化的LDL (25µg/ml)加给在2.5% 血清培养基中培养24h的J774巨噬细胞。在用2.5% (v/v) 兔血浆或无ß-脂蛋白的血浆温育6h以后,测量[3H]-胆固醇释放(PTA试剂盒–Biolabo, 法国)。一式三份地进行所有测定。在每个平板上包括兔血浆(校准物),并通过将血浆样品除以该校准物血浆,将所述值标准化,以确定样品的标准化胆固醇流出能力。
动物方案
动物居住和护理符合指导性86/609/EEC的推荐,且方案得到常设伦理学委员会的批准。
ApoE-/-小鼠“流动停止模型”: 结扎apoE-/-小鼠的左颈动脉,并饲喂西餐。每天1次地给这些小鼠施用100µg/kg (0.5% CMC, 0.2% 吐温80)的化合物IIa,持续2周。
ApoE-/-“高胆固醇饮食模型”: 给小鼠饲喂西餐(0.2% 胆固醇, 21% 酪乳) 2个月,然后每天1次地施用100µg/kg (0.5% CMC, 0.2% 吐温80) 的化合物IIa,持续另外4周。处死小鼠,收集主动脉用于生化和免疫组织学表征。在心脏基底处分离一组主动脉(n = 10),并放入含有3 ml氯仿-甲醇2:1 (v/v)的玻璃管中,并用豆甾醇作为内部标准品。然后,如上所述进行分析。首先用PBS充分洗涤另一组主动脉(n = 10),然后用4% 低聚甲醛(500µl)洗涤,最后用组织tek OCT (500µl)洗涤。接着,将主动脉埋入OCT中,并在-20℃冷冻。加工彼此间隔10 μm的3阶冷冻切片,并用Oil Red O (ORO)、Sirius red和苏木精/曙红染色。使用F4/80第一大鼠单克隆抗体(Abcam)检测巨噬细胞,并用大鼠抗小鼠CD106 (AbDSerotec)检测VCAM。
新西兰兔。 在高胆固醇饮食饲喂(0.5% 胆固醇)2个月和2周清除期以后,每天1次地用30、100和300µg/kg的化合物IIa处理动物(通过经口管饲法)持续4周。在第0天给药之前,和在化合物IIa施用4周以后,收集血液样品。在最后一次抽血以后,对肝脏、胆囊、心脏、从心脏(左心室) 至髂动脉的胸和腹主动脉取样。
巨噬细胞和小鼠 apoE-/-颈动脉的Oil Red O染色。将在福尔马林中固定的左颈动脉埋入OCT中,并在-20℃冷冻。加工彼此间隔50 μm的4阶冷冻切片,并用Oil Red O (ORO)染色。使用第一大鼠单克隆抗CD-68抗体(Abcam)检测巨噬细胞。
兔主动脉染色.在中性福尔马林缓冲液中固定兔主动脉,并埋入石蜡中。用苏木精-曙红-Saffron将纵向3µm切片染色,用于组织学分析。将所述组织切片脱石蜡,并在无蛋白的封闭液中温育,以抑制用于兔单核细胞和巨噬细胞的免疫染色的第一单克隆抗体(得自Dako Corp.的RAM11,1:100稀释)和平滑肌细胞-特异性的肌动蛋白(得自Dako Corp. 的HHF35,1:100稀释)的非特异性结合。以1:300稀释度使用第二生物素化的抗-山羊抗体(Dako)。
Hepa 1-6转导
从Vectalys得到在HEK 293T中的慢病毒生产和慢病毒生产授权。在转导前一天,将Hepa 1-6细胞以75,000细胞铺板在24-孔板中。转导后24小时,计数细胞的数目。除去其它孔的培养基,并用1 ml DMEM 10% 血清(含有8µg/ml聚凝胺)替换。通过加入40转导单位的病毒载体(1.2E8TU/ml) /细胞,转导细胞。转导后17小时,更换培养基,并建立转导的Hepa细胞库。
QPCR
就定量PCR实验而言,用RiboPure™Kit (AM1924 Ambion)从培养的细胞中纯化RNA。用High Capacity RNA-to-cDNA试剂盒(4387406 Applied BioSystem),将RNA逆转录为单链cDNA。根据生产商的操作(Applied Biosystems),使用Taqman技术进行QPCR。
分析
胆汁酸. 在PBS (1:5000)中稀释胆汁酸,并根据稍微改进的生产商的方案,测量浓度。标准曲线中的胆汁酸浓度在50-200 mM范围内,并进行酶反应20分钟。将4 μl 1:5000胆汁稀释液用于浓度测定。
胆汁磷脂. 根据生产商的方案,测量胆汁磷脂。简而言之,标准曲线在0-20 μg/ml之间,并在测定中测量4 μl胆汁。在37℃进行反应5分钟。
胆汁胆固醇. 根据生产商的方案,测量胆汁胆固醇。 简而言之,标准曲线在0-60 μg/ml之间,并在测定中测量10 μl胆汁(1:10稀释)。在37℃进行反应5分钟。
实施例20. 化合物IIa对P2Y13r途径的影响
通过HDL-P2Y13r途径的刺激,化合物IIa在小鼠中表现出对动脉粥样硬化进展的抑制的显著作用。还使用携带P2Y13r-shRNA的腺病毒,在被感染的apoE-/-小鼠中在体内证实了P2Y13r途径的特异性。此外,化合物IIa会诱发在用胆固醇食物饲喂的兔的主动脉中预先建立的粥样硬化斑块的消退。这些结果证实,P2Y13r在HDL代谢和动脉粥样硬化中起非常关键的作用,并且P2Y13r是研究动脉粥样硬化生物学的有用治疗靶标。
使用了设计为口服活性的P2Y13受体激动剂的杂环化合物。基于它们在表达P2Y13受体的1321N1细胞中的特异性结合和活性(未显示),选择这些化合物(Kim, Y. C.等人.Synthesis of pyridoxal phosphate derivatives with antagonist activity at the P2Y13 receptor. Biochem Pharmacol 70, 266-274 (2005))。进一步试验了所述化合物在体外在小鼠肝细胞(Hepa)和人肝细胞瘤细胞(HepG2)中刺激[3H]-胆固醇-标记的-HDL摄入的倾向。还使用P2Y13 siRNA探针,证实了摄入的特异性(未显示)。假定P2Y13r激动剂在动物中的施用会增加胆固醇逆向转运(即,肝脏的HDL摄入),并随后增强胆汁酸和胆汁胆固醇分泌。首先,当给C57Bl/6J小鼠静脉内地施用化合物IIa (10 nmol/kg)时,在治疗4 h以后,观察到胆汁酸(BA)和游离胆固醇向胆汁中的分泌的增加,该效应稍微高于坎格雷洛(The Medicines Company),后者是一种已知的P2Y13r激动剂(图16 A和B) (Jacquet, S.等人. The nucleotide receptor P2Y13 is a key regulator of hepatic high-density lipoprotein (HDL) endocytosis. Cell Mol Life Sci 62, 2508-2515 (2005))。通过经口管饲法以低至30µg/kg的剂量施用P2Y13r激动剂,证实了动物胆汁中的胆汁酸和胆固醇分泌的刺激(图16 C和D)。用P2Y13r激动剂观察到的这种胆汁酸分泌增加,可以是BA从肝内总汇分泌至胆管的结果(Schwartz, C. C., Halloran, L. G., Vlahcevic, Z. R., Gregory, D. H. & Swell, L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science 200, 62-64 (1978)),或者是在HDL摄入刺激以后肝脏通过P2Y13r途径从头合成BA的结果。清楚地证实了在30µg/kg P2Y13r激动剂时肝脏胆汁酸含量的增加,这与肝脏的HDL摄入的刺激相一致,后者又导致BA合成的增加(图16 E)。值得注意的是,在治疗4h以后,血浆胆固醇和HDL没有受到显著影响(图16 F),但是未酯化胆固醇例外,后者代表已经描述的特异性的HDL胆固醇肝脏摄入(Schwartz, C. C., Halloran, L. G., Vlahcevic, Z. R., Gregory, D. H. & Swell, L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science 200, 62-64 (1978))。为了进一步描绘在用化合物IIa治疗小鼠以后HDL在胆汁分泌的增加中的涉入,在体内进行了[3H]-胆固醇标记的小鼠HDL (图16G)、[3H]-胆固醇酯标记的小鼠HDL (图16H)和[3H]-胆固醇标记的小鼠LDL (图16I) 的肝吸收分析。用10 nmol/kg的P2Y13r激动剂对动物的静脉内治疗表现出HDL与LDL相比的吸收的特异性刺激,这支持了HDL介导的胆固醇逆向转运的刺激的假设。
研究了激动剂的P2Y13r途径刺激对apoE-/-小鼠中的粥样硬化斑块病变的影响,所述apoE-/-小鼠在短期西餐(即高胆固醇) 饮食以后非常易于形成动脉粥样硬化。为了模仿人病理学,使用在“流动停止模型”中描述的方法在apoE-/-小鼠中形成动脉粥样硬化病变(Godin, D., Ivan, E., Johnson, C., Magid, R. & Galis, Z. S. Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation 102, 2861-2866 (2000); Ivan, E.等人. Expansive arterial remodeling is associated with increased neointimal macrophage foam cell content: the murine model of macrophage-rich carotid artery lesions. Circulation 105, 2686-2691 (2002); Lessner, S. M., Martinson, D. E. & Galis, Z. S. Compensatory vascular remodeling during atherosclerotic lesion growth depends on matrix metalloproteinase-9 activity. Arterioscler Thromb Vasc Biol 24, 2123-2129 (2004))。简而言之,结扎小鼠的左颈动脉以造成局部炎症,在西餐饲喂2周以后产生界限清楚的动脉粥样硬化病变。与高胆固醇饮食一起伴随施用P2Y13r激动剂2周,基于病变胆固醇含量,表明与对照动物相比动脉粥样硬化病变的进展的剂量依赖性抑制(Riedmüller K, Metz S, Bonaterra GA, Kelber O, Weiser D, Metz J, Kinscherf R. Cholesterol diet and effect of long-term withdrawal on plaque development and composition in the thoracic aorta of New Zealand White rabbits. Atherosclerosis 210 407-13(2010)) (图17A)。通过组织病理学分析对用苏木精-伊红(图17B、E、H)、oil-red O染色(图17C、F、I)和用CD68抗体(对巨噬细胞特异性的)(图17D、G、J)治疗的颈动脉的斑块组分的定量进一步证实了该观察结果。所述染色清楚地表明治疗的动物的颈动脉的脂质含量的下降。在“流动停止模型”apoE-/-小鼠的非炎症性的右颈动脉中,也观察到P2Y13r激动剂对胆固醇沉积的抑制(例如,对于媒介物和100µg/kg的化合物IIa,分别为18.2±3.6和7.5±3.4 nmol/mg组织)。
为了证实P2Y13r特异性,在颈动脉结扎和西餐之前,用腺病毒感染小鼠,所述腺病毒携带模拟shRNA或靶向P2Y13r的特异性shRNA,其中观察到P2Y13r的强抑制(图18 A)。与高胆固醇饮食一起伴随施用P2Y13r激动剂(在0.1 mg/kg/天)持续2周,表明模仿腺病毒对结扎的颈动脉的胆固醇含量的显著下降(下降60%),从而表明与对照动物相比动脉粥样硬化病变进展的显著抑制(图18 B)。通过组织病理学分析,进一步证实了用苏木精-曙红和oil-red O染色处理过的颈动脉的该观察结果(未显示)。在用靶向P2Y13r的shRNA治疗过的小鼠中,消除了P2Y13r激动剂对颈动脉的胆固醇含量的影响,这强化了当P2Y13r表达受到抑制时P2Y13r在该模型中的特异性(图18 B)。还观察到P2Y13r对血浆胆固醇含量(图18 C)和更具体地对LDL和VLDL胆固醇(在该特定动物模型中的主要脂蛋白类型)(分别为图18 E和D)的这种特异性作用,而在模仿腺病毒P2Y13r激动剂治疗的小鼠中观察到下降,在P2Y13r shRNA激动剂治疗的小鼠中消除。这些数据代表第一手证据:P2Y13r的刺激对动脉粥样硬化进展具有积极影响。
在胆固醇饮食饲喂的apoE-/-小鼠中进一步评价了主动脉中的斑块的进展的预防(图19)。在8周饮食和随后用100µg/kg/天的化合物IIa治疗4周以后,治疗的动物具有显著减少的斑块面积(图4B、4G和4J)、胆固醇含量(图19A)、VCAM1表达(使用特异性的抗体CD106)(图4F、4I和4L)、主动脉壁和斑块的下降的巨噬细胞含量(F4/80抗体-图19C)、以及下降的脂质染色(图19D、H和K)。壁的胶原含量提高(图19E)。
还研究了下述问题:P2Y13r途径的刺激是否会影响高胆固醇饮食饲喂的兔中预诱导的动脉粥样硬化的消退。在2个月高胆固醇饮食(在饮食中含0.5%胆固醇)和随后的3周清除期以后,这些动物形成在主动脉中的动脉粥样硬化病变。另外,它们表达胆固醇酯转移蛋白(CETP,在小鼠中不表达),且也呈现与人类类似的脂蛋白模式。在病变诱导和清除期以后,用口服施用的化合物II(在最高达0.3 mg/kg的剂量)治疗这些动物,持续4周。通过整个动脉的胆固醇含量测得,激动剂治疗的动物的主动脉中的粥样硬化斑块明显消退(减少了30%) (图20 A)。此外,使用苏木精-曙红、HHF35抗体(对平滑肌细胞特异性的)和RAM11抗体(对巨噬细胞特异性的) 对主动脉的不同部分(从近端至远端)的免疫组织学分析,证实了斑块的消退(未显示)。通过HPLC分析的脂蛋白分布表明了HDL胆固醇(图20 D)以及兔apoA-I(通过SELDI-TOF分析测得)的增加趋势,而VLDL和LDL稍微下降(分别为图20 B和C)。在治疗的兔的肝脏中没有观察到LDL受体和SR-BI的mRNA表达的变化(通过QPCR测量)(未显示)。这些观察结果表明,P2Y13r的活化对ß-脂蛋白代谢没有直接影响,也得到血浆VLDL、LDL apoB和甘油三酯的适当调节的证实。如以前在小鼠中观察到的(图16),在用P2Y13r激动剂治疗的兔子中,肝脏中的胆汁酸含量增加(图20G),这强化了P2Y13r途径在兔子观察结果中的作用。对血浆HDL水平的这种相对不大的影响,可能是由于新HDL颗粒的新合成,后者可以补偿由于化合物IIa效应引起的肝脏对成熟HDL的吸收。作为结果,观察到的与肝脏中的apoA-I mRNA表达的增加(图21B)相关联的血浆apoA-I的增加(图21A)有利于新HDL颗粒在循环中的产生。
另外,由于P2Y13r介导的HDL有效周转和它们从巨噬细胞流出胆固醇的强能力,新生的HDL颗粒可以有效地促进粥样硬化斑块的消退。
为了评价该假设,使用Lipoprint®技术,分析了治疗的动物中的HDL颗粒的大小。治疗的动物的HDL含量表现出非常一致的模式:与对照动物相比,大HDL的明确减少和“中间”大小的HDL颗粒的增加(图21 C和D)。就通过促进胆固醇从存在于病变处的巨噬细胞的流出而从粥样硬化斑块除去胆固醇而言,认为这些中间HDL颗粒是更有效的颗粒(Khera, A. V.等人. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364, 127-135 (2011); deGoma, E. M., deGoma, R. L. & Rader, D. J. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol 51, 2199-2211 (2008))。最后,使用得自治疗的动物的血浆(图21E)或apoB-颗粒消除的血浆(图21F)测定[3H]-胆固醇加载的J774巨噬细胞中的胆固醇流出,表明在用P2Y13r激动剂治疗以后胆固醇的剂量依赖性的流出,这指示得自治疗的动物的血浆HDL会更有效地从巨噬细胞流出胆固醇(Khera, A. V.等人.Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364, 127-135 (2011))。
综上所述,首次在体内证实,P2Y13受体是HDL代谢(即,在胆固醇逆向转运过程中,和在它们的最终重要生理学功能即动脉粥样硬化保护中)的关键配偶体。这些数据支持了下述机理:通过P2Y13r途径刺激肝脏的HDL摄入或胞吞,会促进肝脏的胆固醇分解代谢和在胆汁中的分泌。肝脏对大HDL颗粒的吸收还可能刺激新生的HDL颗粒的从头合成,并由此提高血清的流出能力。换而言之,在体内证实了对HDL的功能的积极直接干预,而不是仅仅在HDL水平起作用,可以对动脉粥样硬化病理学具有突出影响。这些数据也支持,就动脉粥样硬化疾病或它的并发症的治疗而言,P2Y13r激动剂(诸如化合物IIa和本发明的其它化合物)是非常有用的药理学治疗剂。
- 还没有人留言评论。精彩留言会获得点赞!