一种用于检测羧酸酯酶的荧光探针及其制备方法与应用与流程
本发明属于分析检测技术领域,具体涉及一种荧光探针及其制备方法与在羧酸酯酶检测中的应用。
背景技术:
羧酸酯酶是丝氨酸酯酶超家族中的一种酶,能够催化水解许多外源性基底物,如催化脂肪酸酯水解成酸、醇和水。由于羧酸酯酶能催化许多基底物,被广泛用于有机合成、工业生产,也是重要药物靶标和前药活化剂。此外羧酸酯酶还在有机磷化合物的解毒作用、药物的修饰或清除中发挥了重要的作用。开发一种能够应用于羧酸酯酶的检测及监控的新方法是药物合成、解毒及工业生产等各个领域的研究热点。
据报道,迄今为止,已有许多用于检测羧酸酯酶的分析方法,主要包括高效液相色谱法、化学发光法和荧光法等。
高效液相色谱法用于检测羧酸酯酶,用C18反相色谱柱,先用缓冲液作溶剂,设定流速和柱温。淋洗一段时间后,改变线性梯度,淋洗几分钟。之后柱子用淋洗液冲洗,冲洗完后,继续用开始条件淋洗。在通过UV探测器后,柱子流出物通过在放射化学探测器中的一个250uL流动池收集,作为液体闪烁体。高效液相色谱法检测过程繁琐,且易受干扰。
常用的化学发光法测定酯酶主要包括以下几种:(1)液相化学发光法:①鲁米诺化学发光体系:在生物酶作用下与O2生成过氧化氢,生成的过氧化氢和碱性溶液发生反应,产生波长为425nm的光,间接测定酶含量;②光泽精化学发光体系:在生物酶作用下与O2生成过氧化氢的基质,与碱性溶液发生反应生成激发态的N-甲基吖酮,产生最大发射波长470nm的光;(2)生物化学发光法,将酶反应的专一性和化学发光的高灵敏性巧妙结合,产生连续的冷光辐射。化学发光法选择性较差、发光体系相对较少。
近年来,荧光法作为一种新型的检测手段备受关注,其具有选择性好和灵敏度高以及即时检测、响应快、设备简单等特点。同时,荧光化合物在化学结构上易于设计、修饰和改进,能满足不同检测样品的需要。因此,荧光法非常适合于羧酸酯酶的分析检测。中国专利(申请号:201210282674.6)制备了带正电荷小分子探针3,4,9,10-四-(4-三甲基胺丁基氧-羰基)-苝,该探针能产生很强的荧光信号,加入聚阴离子,将不同溶度的羧酸酯酶与底物硫代乙酰胆碱反应,得到水解产物,再加入探针,得到混合溶液。在442nm的激发光照射下,可在460-650nm波长范围发射荧光,从而实现了对羧酸酯酶的荧光增强型检测;但是该荧光探针与聚阴离子容易聚集,发生荧光淬灭,不利于工业生产领域中的检测应用;另外,利用该方法实现对酶的检测的步骤较繁琐。研究论文(RSC Analyst,2012,137,716-721)报道了一种基于试卤灵(9-羟基-3-异吩恶嗪酮)衍生物的荧光探针,该探针在585nm处几乎没有荧光;体系中有羧酸酯酶存在时,其可催化探针分子的羧酸酯键断裂,导致苯甲基部分断开,释放试卤灵,在550nm的激光照射下,该探针分子在585nm处发射橙红色荧光,实现了对羧酸酯酶的荧光增强型检测;但该荧光探针易受外界因素干扰,有时难以实现对酶的准确分析。综上所述,本领域急需发展一种抗干扰性强、准确度高、简便且高效的羧酸酯酶检测方法。
技术实现要素:
为了解决以上现有技术的缺点和不足之处,本发明的首要目的在于提供一种用于检测羧酸酯酶的荧光探针即荧光化合物。
本发明的另一目的在于提供上述荧光探针的制备方法。
本发明的再一目的在于提供上述荧光探针的应用。所述荧光探针在羧酸酯酶检测中的应用。
本发明目的通过以下技术方案实现:
一种用于检测羧酸酯酶的荧光探针,该探针的荧光化合物为2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈,其具有如下结构式:
所述用于检测羧酸酯酶的荧光探针的制备方法,包括如下步骤:
(1)将2-甲基-4H-1-苯并吡喃-4-酮和丙二腈溶于乙酸酐中,搅拌加热反应,去除溶剂,加入水,回流反应,分离纯化,得到2-(2-甲基-4H-1-苯并吡喃-4-基)-丙二腈;
(2)将4-氨基苯甲醇溶于混合溶液中,所述混合溶液为强碱溶液、有机溶剂1与水的混合物,冷却,加入二碳酸二叔丁酯,搅拌反应,纯化,得到反应产物;向反应产物中加入有机溶剂2以及二氧化锰,继续搅拌反应,分离纯化,得到4-叔丁氧羰氨基苯甲醛;
(3)将步骤(1)所得2-(2-甲基-4H-1-苯并吡喃-4-基)-丙二腈和步骤(2)所得4-叔丁氧羰氨基苯甲醛溶于有机溶剂3中,加入催化体系,搅拌加热反应,过滤,纯化,得到固体产物;向固体产物中加入有机溶剂4溶解,再加入三氟乙酸,搅拌反应,分离纯化,得到2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈;
(4)将4-羟基苯甲醇溶于有机溶剂,冷却,在惰性气体的保护下,加入含有碱性有机化合物的乙酰氯有机溶液,搅拌反应,分离纯化,得到乙酸-4-羟甲基苯酯;
(5)将步骤(3)所得2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈和三光气溶于有机溶剂5,在惰性气体的保护下,逐滴加入吡啶的有机溶液,低温反应,室温反应,减压旋干,得到反应产物;在惰性气体的保护下,向反应产物中加入由步骤(4)所得乙酸-4-羟甲基苯酯配成的有机溶液,搅拌反应,去除溶剂;加入碱性有机化合物,继续反应,分离纯化,得到用于检测羧酸酯酶的荧光探针(2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈)。
步骤(1)中所述搅拌加热反应的温度为140℃-143℃,搅拌加热反应的时间为10-12h;所述回流反应的温度为95℃-100℃,时间为0.5-1小时。
步骤(2)中所述有机溶剂1为1,4-二氧六环;所述强碱溶液为氢氧化钠水溶液,强碱溶液的浓度为1M;所述冷却的温度为-5~0℃;步骤(2)中所述搅拌反应的时间为1.5-6小时,搅拌反应的温度为室温;步骤(2)中所述纯化是指将反应后的体系减压旋干去除有机溶剂,用乙酸乙酯或二氯甲烷萃取,有机相用水、饱和氯化钠溶液进行清洗,再用干燥剂(如:无水硫酸钠)干燥,旋蒸除去溶剂;所述有机溶剂2为三氯甲烷,所述继续搅拌反应的温度为室温,反应时间为6-12小时。
步骤(3)中所述有机溶剂3为乙腈。
步骤(3)中所述催化体系为催化剂、有机碱以及干燥剂组成。
所述干燥剂为硫酸镁、有机碱为哌啶、催化剂为冰醋酸。
步骤(3)中所述搅拌加热反应的温度为70℃-73℃,反应时间为24-26小时。
步骤(3)中所述纯化是指过滤后,将滤饼用有机溶剂(如:二氯甲烷)溶解,减压旋干。
步骤(3)中所述有机溶剂4为二氯甲烷。
步骤(3)中所述搅拌反应的温度为室温,反应的时间为1-2小时。
步骤(4)中所述有机溶剂为乙酸乙酯。
步骤(4)中所述冷却的温度为-5~0℃。
步骤(4)中所述碱性有机化合物为三乙胺,所述含有碱性有机化合物的乙酰氯有机溶液中有机溶剂为乙酸乙酯。
步骤(4)中所述搅拌反应的时间为5-8小时。
步骤(5)中所述有机溶剂5为二氯甲烷;步骤(5)中所述吡啶的有机溶液中有机溶剂为二氯甲烷。
步骤(5)中所述低温反应的条件为-5℃~0℃反应1-2小时,所述室温反应的时间为2-4小时。
步骤(5)中所述由步骤(4)所得乙酸-4-羟甲基苯酯配成的有机溶液中有机溶剂为二氯甲烷。
步骤(5)中所述搅拌反应的条件为于室温下搅拌反应12-14小时。
步骤(5)中所述碱性有机化合物为三乙胺。
步骤(5)中所述继续反应的条件为于室温下搅拌反应12-14小时。
步骤(1)中所述2-甲基-4H-1-苯并吡喃-4-酮和丙二腈的摩尔比为(0.7-0.72):1;所述丙二腈和水的摩尔体积比为1mmol:(0.97-1)mL;所述丙二腈和乙酸酐的摩尔体积比为1mmol:(0.80-1.2)mL。
步骤(2)中所述二碳酸二叔丁酯和4-氨基苯甲醇的摩尔比为(1.1-1.5):1;二氧化锰与二碳酸二叔丁酯的摩尔比为(1.5-1.8):1。
步骤(2)中所述4-氨基苯甲醇与有机溶剂1的摩尔体积比为1mmol:(0.6-0.7)mL;所述4-氨基苯甲醇与水的摩尔体积比为1mmol:(0.6-0.7)mL;所述有机溶剂2与4-氨基苯甲醇的用量比为(0.9-1.3)mL:1mmol。
步骤(2)中所述4-氨基苯甲醇与强碱溶液的摩尔体积比为1mmol:(0.98-1.1)mL。
步骤(3)中所述2-(2-甲基-4H-1-苯并吡喃-4-基)-丙二腈与4-叔丁氧羰氨基苯甲醛的摩尔比为1:(1-1.2)、所述2-(2-甲基-4H-1-苯并吡喃-4-基)-丙二腈与三氟乙酸的摩尔体积比为1mmol:(5.0-6.0)mL;
步骤(3)中所述2-(2-甲基-4H-1-苯并吡喃-4-基)-丙二腈与催化体系中催化剂的摩尔体积比为(2.05-2.55)mmol:1mL。步骤(3)中所述催化体系中催化剂、有机碱和干燥剂的用量比1mL:(1.6-2)mL:(17.5-21.3)mmol。
步骤(4)中所述4-羟基苯甲醇与所述含有碱性有机化合物的乙酰氯有机溶液中乙酰氯的摩尔比为1:(0.99-1.12),所述4-羟基苯甲醇与碱性有机化合物的摩尔比为1:(0.73-1.07)。
步骤(5)中所述2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈、三光气、吡啶、乙酸-4-羟甲基苯酯的摩尔比为1:(2.5-3):(9-10):(0.8-0.9)。
步骤(1)中所述分离纯化步骤为:反应液冷却至室温,用二氯甲烷萃取,所得有机层用水、饱和氯化钠溶液进行清洗,有机相再干燥,旋蒸除去有机溶剂,所得固体经硅胶柱层析纯化;步骤(2)所述分离纯化步骤为:反应液过滤除去反应体系中的固体,所得滤液旋蒸除去溶剂,再经硅胶层析柱纯化;步骤(3)所述分离纯化步骤为:旋蒸除去溶剂,所得固体经硅胶柱层析纯化;步骤(4)所述分离纯化步骤为:反应液过滤,用乙酸乙酯洗,旋蒸除去溶剂,所得固体经硅胶层析柱纯化;步骤(5)所述分离纯化步骤为:反应液旋蒸除去溶剂,所得固体经硅胶层析柱纯化。
本发明所得产物荧光探针2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈,分子式为C30H21N3O5,相对分子质量为503.5。该化合物光稳定性好,无毒。
本发明的荧光探针即荧光化合物的合成路线图如图1所示。
所述荧光探针在羧酸酯酶检测中的应用,对羧酸酯酶进行定性和定量分析。该探针在羧酸酯酶存在条件下会与其快速发生反应,引起分子内部不同基团之间的电子发生转移,从而产生分子内电荷转移效应,使DCM母体(2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈部分)荧光信号增强,其在470nm的激发光照射下,在670nm左右发射强烈的红色荧光。本发明荧光化合物可用于生物体中羧酸酯酶的定性和定量分析。
本发明的探针在羧酸酯酶存在的条件下会快速发生水解反应,使探针末端羧酸酯键断裂,裸露出的羟基触发自消除分解链,发生1,6-消除反应致使探针分子产生分子内电荷转移效应,释放出DCM染料分子增强荧光信号。因此该探针可实现对羧酸酯酶的荧光增强型检测,随着羧酸酯酶浓度的增加,红色荧光逐渐增强。这种荧光增强型的检测模式较为直观,荧光发射明显且易于观测,提高了检测精度和准确性。在一定的浓度下,荧光强度与检测物浓度存在较好的线性关系,可用于定量检测。
相对于现有技术,本发明具有如下优点及有益效果:
(1)本发明的探针化合物对羧酸酯酶检测具有较好的抗干扰性,常见的其他离子或化合物无法催化探针中的羧酸酯键的断裂,因此荧光信号不会增强,说明该探针具有很好的抗干扰性,可特异性用于羧酸酯酶的检测。
(2)本发明的探针可实现对羧酸酯酶的荧光增强型检测,随着羧酸酯酶浓度的增加,红色荧光逐渐增强;这种荧光增强型的检测模式较为直观,荧光发射明显且易于观测,提高了检测精度和准确性;在一定的浓度下,荧光强度与检测物浓度存在较好的线性关系,可用于定量检测。
(3)本发明所得荧光探针的检测体系构建了一种准确性高的检测羧酸酯酶的方法,其使用方便,便于推广应用。
(4)本发明探针分子合成工艺较为简便,产率较高。
附图说明
图1为本发明的荧光探针(即荧光化合物)的合成路线图;
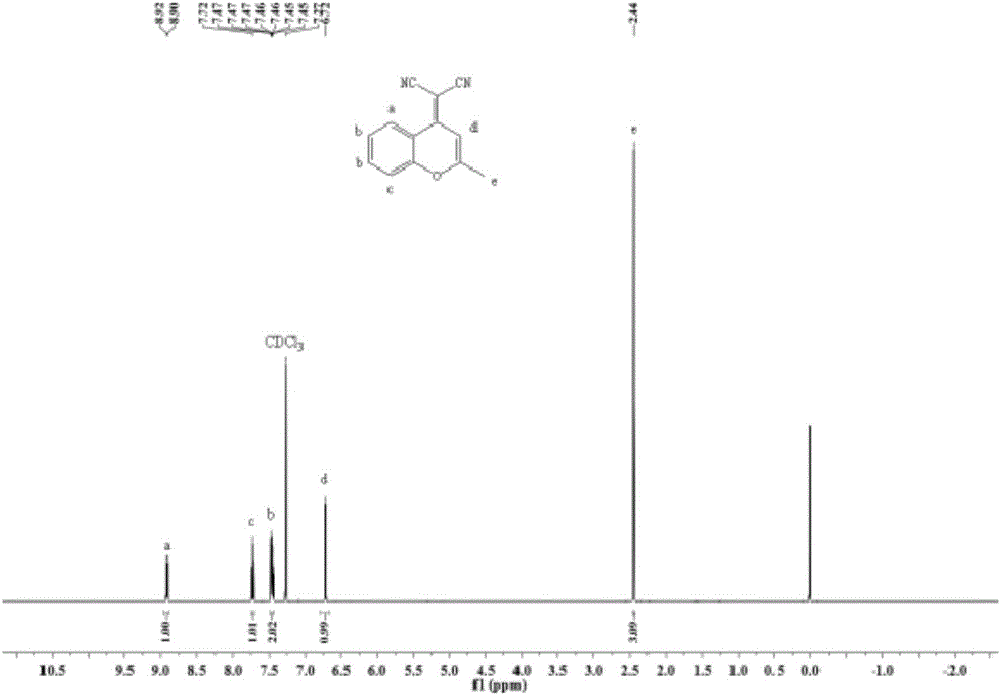
图2为实施例1中2-(2-甲基-4H-1-苯并吡喃-4-基)-丙二腈的核磁共振氢谱图;
图3为实施例1中2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈的核磁共振氢谱图;
图4为实施例1中乙酸-4-羟甲基苯酯的核磁共振氢谱图;
图5为实施例1中2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈的核磁共振氢谱图;
图6为实施例1的荧光探针对羧酸酯酶响应不同时间的荧光光谱图;
图7中(a)为实施例1的荧光探针对不同浓度羧酸酯酶响应的荧光光谱图,(b)为实施例1的荧光探针的荧光强度比值(I666/I606)与不同浓度羧酸酯酶之间的关系图;
图8为实施例1的荧光探针与羧酸酯酶反应前(n)和反应后(l)的荧光强度比值(I666/I606)与pH之间的关系图;
图9为实施例1的荧光探针抗干扰性测试柱状图即探针的荧光强度比值(I666/I606)与不同离子、化合物之间的关系图(1.空白样,2.钠离子,3.钙离子,4.镁离子,5.钾离子,6.过氧化氢,7.羟基自由基,8.葡萄糖,9.丝氨酸,10.胎牛血清,11.谷氨酸,12.精氨酸,13.谷氨酰胺,14.维他命C,15.羧酸酯酶)。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
(1)将2304mg 2-甲基-4H-1-苯并呋喃-4-酮(14.4mmol)和1320mg丙二腈(20mmol)溶于20mL乙酸酐中,搅拌加热控制温度为143℃反应12小时,反应完后,减压旋干去除溶剂,然后加入20mL水,加热回流1小时,控制温度为100℃;冷却至室温后,用二氯甲烷萃取,所得有机层再用水、饱和氯化钠各洗一次,有机相用无水硫酸钠干燥,过滤;旋转蒸发除去有机溶剂,所得固体经硅胶柱层析提纯(石油醚:二氯甲烷,V/V=2:3),得到白色固体产物2-(2-甲基-4H-二氢苯并吡喃-4-基)-丙二腈1857mg(产率为62%);通过核磁共振氢谱对该产物进行表征,核磁共振氢谱图如图2所示;
(2)将2460mg 4-氨基苯甲醇(20mmol)溶于14mL 1,4-二氧六环、14mL水和22mL 1M氢氧化钠溶液的混合溶液,冷却到0℃,加入6540mg二碳酸二叔丁酯(30mmol),在室温下搅拌6小时,减压旋干去除有机溶剂,用乙酸乙酯或二氯甲烷萃取,有机相用水、饱和氯化钠溶液各洗一次,用无水硫酸钠干燥,旋蒸除去溶剂;然后加入20mL三氯甲烷溶解,分批加入4696mg二氧化锰(54mmol),室温下搅拌反应12小时,反应完后,过滤,减压旋干,所得固体经硅胶柱层析提纯(石油醚:乙酸乙酯,V/V=10:1),得到白色固体产物4-叔丁氧羰氨基苯甲醛3933mg(产率为89%),直接用于下一步反应;
(3)将728mg 2-(2-甲基-4H-1-苯并吡喃-4-基)-丙二腈(3.5mmol)和923mg 4-叔丁氧羰氨基苯甲醛(4.2mmol)溶于40mL乙腈中,加入3570mg硫酸镁(29.75mmol)、3.4mL哌啶和1.7mL冰醋酸,搅拌加热控制温度为73℃反应26小时,反应完后,冷却至室温过滤,用乙腈洗涤滤饼,收集滤饼,用二氯甲烷溶解,减压旋干;然后加入50mL二氯甲烷溶解、20.8mL三氟乙酸,室温下搅拌反应2小时,旋蒸除去二氯甲烷和三氟乙酸,所得固体经硅胶柱层析提纯(石油醚:二氯甲烷,V/V=2:3),得到褐色固体产物2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈713mg(产率为65.5%);通过核磁共振氢谱对该产物进行表征,核磁共振氢谱图如图3所示;
(4)将1950mg 4-羟基苯甲醇(14mmol)溶于25mL乙酸乙酯中,冷却到0℃,在氮气保护下,加入2.1mL三乙胺(14.9mmol)和1.11mL 14mol/L乙酰氯(15.5mmol)的乙酸乙酯溶液,搅拌8小时,反应完后,反应液过滤,用乙酸乙酯洗,旋蒸除去溶剂,所得固体经硅胶层析柱提纯(己烷:乙酸乙酯,V/V=3:2),得到固体乙酸-4-羟甲基苯酯795mg(产率为40%);通过核磁共振氢谱对该产物进行表征,核磁共振氢谱图如图4所示;
(5)将62mg 2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈(0.2mmol)和178.2mg三光气(0.6mmol)溶于5mL二氯甲烷中,在氮气保护下,逐滴加入145μL 12.4mol/L吡啶(1.8mmol)的二氯甲烷溶液,搅拌并控制温度在0℃反应2小时,而后在室温下反应4小时,减压旋干;在氮气保护下,用注射器加入1.875mL 0.096mol/L乙酸-4-羟甲基苯酯(0.18mmol)二氯甲烷溶液,室温搅拌14小时,旋蒸除去溶剂;加入5mL三乙胺,室温搅拌14小时,反应完后,旋干溶剂,所得固体经硅胶层析柱提纯(石油醚:乙酸乙酯,V/V=5:1)得到荧光探针即橙色固体荧光化合物2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈69mg(产率为80%);通过核磁共振氢谱对该产物进行表征,核磁共振氢谱图如图5所示。
从图2的测试结果可知:1H NMR(600MHz,CDCl3):δ8.91(d,J=8.3Hz,1H),7.72(ddd,J=8.5,7.2,1.4Hz,1H),7.48-7.43(m,2H),6.72(s,1H),2.44(s,3H);其中,8.91ppm、7.72ppm、7.45ppm处对应为苯环上质子特征峰,6.72ppm处对应的为六元氧杂环烯基质子特征峰,2.44ppm处对应的为甲基特征峰;通过核磁的分析可以确定所合成的产物为目标中间体。
从图3的测试结果可知:1H NMR(600MHz,HDMSO):δ8.70(d,J=8.1Hz,1H),7.87(t,J=7.7Hz,1H),7.73(d,J=8.2Hz,1H),7.61(d,J=15.7Hz,1H),7.56(t,J=7.6Hz,1H),7.47(d,J=8.4Hz,2H),7.05(d,J=15.7Hz,1H),6.83(s,1H),6.61(d,J=8.4Hz,2H),6.01(s,2H)。其中,8.70ppm、7.87ppm、7.73ppm、7.61ppm、7.56ppm、7.47ppm、7.05ppm、处对应为苯环上质子特征峰,6.83ppm处对应的为六元氧杂环烯基质子特征峰,6.61ppm处对应的为乙烯基的特征峰,6.01ppm处对应的为氨基特征峰;通过核磁的分析可以确定所合成的产物为目标中间体。
从图4的测试结果可知:1H NMR(600MHz,CDCl3):δ7.33(dt,J=9.2,2.2Hz,2H),7.08-7.03(m,2H),4.60(d,J=2.6Hz,2H),2.76(s,1H),2.29(t,J=2.5Hz,3H)。其中,7.33ppm、7.08-7.03ppm处对应为苯环上质子特征峰,4.60ppm对应的是亚甲基上质子的特征峰,2.76ppm是羟基质子的特征峰,2.29ppm处对应的为甲基特征峰;通过核磁的分析可以确定所合成的产物为目标中间体。
从图5的测试结果可知:1H NMR(600MHz,DMSO):δ10.11(s,1H),8.73(d,J=8.3Hz,1H),7.92(t,J=7.8Hz,1H),7.79(d,J=8.4Hz,1H),7.70(t,J=10.6Hz,3H),7.61(t,J=7.7Hz,1H),7.56(t,J=9.4Hz,2H),7.49(d,J=8.5Hz,2H),7.37(d,J=16.0Hz,1H),7.16(d,J=8.5Hz,2H),6.98(s,1H),5.18(s,2H),2.27(s,3H)。其中,10.11ppm处对应的为氨基特征峰,8.73ppm、7.92ppm、7.79ppm、7.70ppm、7.61ppm、7.56ppm、7.49ppm、7.37ppm处对应为苯环上质子特征峰,7.16ppm处对应的为乙烯基的特征峰,6.98ppm处对应的为六元氧杂环烯基质子特征峰,5.18ppm对应的是亚甲基上质子的特征峰,2.27ppm处对应的为甲基特征峰。此外,还通过质谱对本实施例制备的荧光探针进行了辅助证明,MS(ESI):m/z 676.5[M-H]-。通过核磁和质谱的分析可以确定所合成的产物目标荧光化合物。
实施例2
(1)将5552mg 2-甲基-4H-1-苯并呋喃-4-酮(34.7mmol)和3262mg丙二腈(49.4mmol)溶于40mL乙酸酐中,搅拌加热控制温度为140℃反应10小时,反应完后,减压旋干去除溶剂,然后加入48mL水,加热回流0.5小时,控制温度为95℃;冷却至室温后,用二氯甲烷萃取,所得有机层再用水、饱和氯化钠各洗一次,有机相用无水硫酸钠干燥,过滤;旋转蒸发除去有机溶剂,所得固体经硅胶柱层析提纯(石油醚:二氯甲烷,V/V=2:3),得到白色固体产物2-(2-甲基-4H-二氢苯并吡喃-4-基)-丙二腈5485mg(产率为76%);
(2)将1500mg 4-氨基苯甲醇(12.18mmol)溶于7.3mL 1,4-二氧六环、7.3mL水和12mL 1M氢氧化钠溶液的混合溶液,冷却到-5℃,加入2930mg二碳酸二叔丁酯(13.4mmol),在室温下搅拌1.5小时,减压旋干去除有机溶剂,用乙酸乙酯或二氯甲烷萃取,有机相用水、饱和氯化钠溶液各洗一次,用无水硫酸钠干燥,旋蒸除去溶剂;然后加入15mL三氯甲烷溶解,分批加入1748mg二氧化锰(20.1mmol),室温下搅拌反应6小时,反应完后,过滤,减压旋干,所得固体经硅胶柱层析提纯(石油醚:乙酸乙酯,V/V=10:1),得到白色固体产物4-叔丁氧羰氨基苯甲醛2388mg(产率为88.7%);
(3)将582mg 2-(2-甲基-4H-二氢苯并吡喃-4-基)-丙二腈(2.8mmol)和619mg 4-叔丁氧羰氨基苯甲醛(2.8mmol)溶于30mL乙腈中,加入2806mg硫酸镁(23.38mmol)、2.2mL哌啶和1.1mL冰醋酸,搅拌加热控制温度为70℃反应24小时,反应完后,冷却至室温过滤,用乙腈洗涤滤饼,收集滤饼,用二氯甲烷溶解,减压旋干;然后加入45mL二氯甲烷溶解、14mL三氟乙酸,室温下搅拌反应1小时,旋蒸除去二氯甲烷和三氟乙酸,所得固体经硅胶柱层析提纯(石油醚:二氯甲烷,V/V=2:3),得到荧光探针即褐色固体产物2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈567mg(产率为65.1%);
(4)将200mg 4-羟基苯甲醇(1.64mmol)溶于2mL乙酸乙酯中,冷却到-5℃,在氮气保护下,加入0.17mL三乙胺(1.2mmol)和0.116mL 14mol/L乙酰氯(1.63mmol)的乙酸乙酯溶液,搅拌5小时,反应完后,反应液过滤,用乙酸乙酯洗,旋蒸除去溶剂,所得固体经硅胶层析柱提纯(己烷:乙酸乙酯,V/V=3:2),得到固体乙酸-4-羟甲基苯酯92mg(产率为39.6%);
(5)将140mg 2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈(0.45mmol)和334mg三光气(1.125mmol)溶于10mL二氯甲烷中,在氮气保护下,逐滴加入325μL 12.4mol/L吡啶(4.05mmol)的二氯甲烷溶液,搅拌并控制温度在-5℃反应1小时,而后在室温下反应2小时,减压旋干,在氮气保护下,用注射器加入3.75mL 0.096mol/L乙酸-4-羟甲基苯酯(0.36mmol)二氯甲烷溶液,室温搅拌12小时,旋蒸除去溶剂;加入15mL三乙胺,室温搅拌12小时,反应完后,旋干溶剂,所得固体经硅胶层析柱提纯(石油醚:乙酸乙酯,V/V=5:1)得到橙色固体荧光化合物2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈137mg(产率为79.5%)。
本实施例所得荧光化合物的中间体以及最终的化合物2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈的表征与实施例1中的结果是相同的。
实施例3
(1)将2825mg 2-甲基-4H-1-苯并呋喃-4-酮(17.65mmol)和1631mg丙二腈(24.7mmol)溶于25mL乙酸酐中,搅拌加热控制温度为141℃反应11小时,反应完后,减压旋干去除溶剂,然后加入24mL水,加热回流0.7小时,控制温度为98℃;冷却至室温后,用二氯甲烷萃取,所得有机层再用水、饱和氯化钠各洗一次,有机相用无水硫酸钠干燥,过滤;旋转蒸发除去有机溶剂,所得固体经硅胶柱层析提纯(石油醚:二氯甲烷,V/V=2:3),得到白色固体产物2-(2-甲基-4H-二氢苯并吡喃-4-基)-丙二腈2525mg(产率为68.8%);
(2)将1355mg 4-氨基苯甲醇(11mmol)溶于7.15mL 1,4-二氧六环、7.15mL水和11.55mL 1M氢氧化钠溶液的混合溶液,冷却到-3℃,加入3127mg二碳酸二叔丁酯(14.3mmol),在室温下搅拌3.5小时,减压旋干去除有机溶剂,用乙酸乙酯或者二氯甲烷萃取,有机相用水、饱和氯化钠溶液各洗一次,用无水硫酸钠干燥,旋蒸除去溶剂;然后加入10mL三氯甲烷溶解,分批加入1990mg二氧化锰(22.88mmol),室温下搅拌反应9小时,反应完后,过滤,减压旋干,所得固体经硅胶柱层析提纯(石油醚:乙酸乙酯,V/V=10:1),得到白色固体产物4-叔丁氧羰氨基苯甲醛2159mg(产率为88.8%);
(3)将874mg 2-(2-甲基-4H-二氢苯并吡喃-4-基)-丙二腈(4.2mmol)和1021mg 4-叔丁氧羰氨基苯甲醛(4.62mmol)溶于30mL乙腈中,加入4249mg硫酸镁(35.4mmol)、3.6mL哌啶和1.8mL冰醋酸,搅拌加热控制温度为71℃反应25小时,反应完后,冷却至室温过滤,用乙腈洗涤滤饼,收集滤饼,用二氯甲烷溶解,减压旋干;然后加入80mL二氯甲烷溶解、23mL三氟乙酸,充分搅拌,室温下反应1.5小时,旋蒸除去二氯甲烷和三氟乙酸,所得固体经硅胶柱层析提纯(石油醚:二氯甲烷,V/V=2:3),得到褐色固体产物2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈853mg(产率为65.3%);
(4)将1116mg 4-羟基苯甲醇(9mmol)溶于15mL乙酸乙酯中,冷却到-2℃,在氮气保护下,加入1.25mL三乙胺(9mmol)和0.643mL 14mol/L乙酰氯(9mmol)的乙酸乙酯溶液,搅拌6.5小时,反应完后,反应液过滤,用乙酸乙酯洗,旋蒸除去溶剂,所得固体经硅胶层析柱提纯(己烷:乙酸乙酯,V/V=3:2),得到固体乙酸-4-羟甲基苯酯514mg(产率为40.2%);
(5)将109mg 2-{2-[4-(4'-氨基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈(0.35mmol)和281mg三光气(0.945mmol)溶于9mL二氯甲烷中,在氮气保护下,逐滴加入267μL 12.4mol/L吡啶(3.325mmol)的二氯甲烷溶液,搅拌并控制温度在-2℃反应1.5小时,而后在室温下反应3小时,减压旋干,在氮气保护下,用注射器加入3.1mL 0.096mol/L乙酸-4-羟甲基苯酯(0.3mmol)二氯甲烷溶液,室温搅拌13小时,旋蒸除去溶剂;加入10mL三乙胺,室温搅拌13小时,反应完后,旋干溶剂,所得固体经硅胶层析柱提纯(石油醚:乙酸乙酯,V/V=5:1),得到荧光探针即橙色固体荧光化合物2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈115mg(产率为79.8%);
本实施例所得荧光化合物的中间体以及最终的化合物2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈的表征与实施例1中的结果是相同的。
性能测试:
对实施例1制备的荧光探针2-{2-[4-(4'-乙酸苯甲酯苯甲氧基-甲酰胺基)-苯乙烯基]-4H-苯并吡喃-4-基}-丙二腈进行性能测试,测试结果如图6~图9所示。
1、测试步骤和条件分别为:取12个5ml样品瓶,分别加入1200uL的PBS、390uL的二甲基亚砜(DMSO)、酯酶400uL(该酯酶母液浓度为1000U/L,是用PH=7.4的PO43-浓度为10mM/L的PBS溶液配制),在水浴锅中搅拌并控制温度为37℃,预热3min,然后分别加入实施例1中所得的荧光探针配置的溶液10uL(该荧光探针母液浓度为1mM,是用二甲基亚砜配制),最后分别测定探针与酯酶反应0min、1min、2min、3min、4min、5min、10min、15min、20min、25min、30min、35min,以470nm为激发波长,分别测定这些样品的荧光强度,得12个样品的荧光发射光谱变化图,结果见图6。图6为实施例1的荧光探针对羧酸酯酶响应不同时间的荧光光谱图。
2、测试步骤和条件分别为:取22个5ml样品瓶,分别加入1600-1000uL的PBS,390uL的二甲基亚砜(DMSO),相对应加入PBS的体积,酯酶分别加入0-600uL(该酯酶母液浓度为1000U/L,是用PH=7.4的PO43-浓度为10mM/L的PBS溶液配制),在水浴锅中搅拌并控制温度为37℃,预热3min,然后分别加入实施例1中所得的荧光探针配置的溶液10uL(该荧光探针母液浓度为1mM,是用二甲基亚砜配制),最后分别测定探针与酯酶反应30min,以470nm为激发波长,分别测定这些样品的荧光强度,得22个样品的荧光发射光谱变化图,见图7(a)。根据图7(a)在666nm和606nm两处荧光强度比值的变化可作出对应的拟合曲线,结果见图7(b)。图7(a)为实施例1的荧光探针对不同浓度羧酸酯酶响应的荧光光谱图,(b)为探针的荧光强度比值(I666/I606)与不同浓度羧酸酯酶之间的关系图。
3、测试步骤和条件分别为:取11个5ml样品瓶,分别加入1600uL的PH分别为2、3、4、5、6、7、7.4、8、9、10、11的PBS、390uL的二甲基亚砜(DMSO)、酯酶400uL(该酯酶母液浓度为1000U/L,是用PH=7.4的PO43-浓度为10mM/L的PBS溶液配制),在水浴锅中搅拌并控制温度为37℃,预热3min,然后分别加入实施例1中所得的荧光探针配置的溶液10uL(该荧光探针母液浓度为1mM,是用二甲基亚砜配制),最后分别测定探针与酯酶反应30min,以470nm为激发波长,分别测定这些样品的荧光强度,得11个样品的荧光发射光谱变化图,根据测得的荧光谱图在666nm和606nm两处荧光强度比值作图,结果见图8。图8为实施例1的探针与羧酸酯酶反应前(n)和反应后(l)的荧光强度比值(I666/I606)与pH之间的关系图。
4、测试步骤和条件分别为:分别取15个5ml样品瓶,分别加入1500uL PBS,390uL的二甲基亚砜(DMSO),酯酶400uL(该酯酶母液浓度为1000U/L,是用PH=7.4的PO43-浓度为10mM/L的PBS溶液配制),再分别加入100uL的PBS,钠离子、钙离子、镁离子、钾离子(以上4种金属离子由PBS缓冲溶液配制,检测浓度为100mM),过氧化氢、羟基自由基、胎牛血清(以上3种化合物由PBS缓冲溶液配制,检测浓度为100mM),葡萄糖、丝氨酸、谷氨酸、精氨酸(以上4种化合物由PBS缓冲溶液配制,检测浓度为50mM),维他命C(以上该种化合物由PBS缓冲溶液配制,检测浓度为10mM),羧酸酯酶(以上该种化合物由PBS缓冲溶液配制,检测浓度为20U/L),在水浴锅中搅拌并控制温度为37℃,预热3min,然后分别加入实施例1中所得的荧光探针配置的溶液10uL(该荧光探针母液浓度为1mM,是用二甲基亚砜配制),最后分别测定探针与酯酶反应30min,以470nm为激发波长,分别测定这些样品的荧光强度,得15个样品的荧光发射光谱变化图,根据测得的荧光谱图在666nm和606nm两处荧光强度比值的柱状图,结果见图9。图9为实施例1的探针抗干扰性测试柱状图:探针的荧光强度比值(I666/I606)与不同离子、化合物之间的关系图(1.空白样,2.钠离子,3.钙离子,4.镁离子,5.钾离子,6.过氧化氢,7.羟基自由基,8.葡萄糖,9.丝氨酸,10.胎牛血清,11.谷氨酸,12.精氨酸,13.谷氨酰胺,14.维他命C,15.羧酸酯酶)。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!