能应用于互作蛋白筛选的在植物细胞中完整表达的改造的DH5αBirA基因及其构建方法与流程
本发明涉及一种真核表达载体构建技术,尤其涉及一种能在真核细胞中完整表达的改造的DH5α BirA基因及其构建方法,以及改造后的BirAG基因在植物蛋白生物素化中的应用和在筛选植物互作蛋白中的应用。
背景技术:
大肠杆菌存在着生物素连接酶BirA,它能将靶标蛋白生物素化,使其带上生物素标记。BirA主要是利用了biotin-N-hydroxysuccinimide或类似的一些酰化物,通过生物素依靠的羧化酶和磷酸戊糖途径中的脱羧酶(Chapman-Smith et al.,1999),对蛋白的氨基酸基团进行修饰。该酶十分专一,在大肠杆菌中,只能生物素化几个特定的蛋白质。
研究人员发现,如果将BirA的第118位的(R)突变成甘氨酸(G),这种BirA突变体(BirA*)的亲和能力会比野生型的BirA低很多(Kwon et al.,2000),导致的结果是BirA*可以生物素化在空间上与之靠近的蛋白,尤其是相互作用的蛋白(Choi-Rhee et al.,2004;Cronan,2005),可以利用BirA*这一特性来检测生物体中蛋白互作的情况。目前该技术已被成功应用于原核生物、哺乳动物、人等细胞中(Gupta GD et al.,2015;Firat-Karalar EN et al.,2015;Batsios P et al.,2016)。
以大肠杆菌DH5α菌种为材料,克隆了DH5α BirA片段,并构建了DH5αBirA*片段,检测发现,DH5α BirA*在高等生物中的表达是存在可变剪接现象的,且剪切区域位于BPL_lipA/B蛋白结构域,这对DH5α BirA*在细胞中发挥作用造成影响。
技术实现要素:
本发明的目的是,通过基因工程技术,对野生的DH5α BirA基因或DH5αBirA*基因进行改造,使之能在真核细胞中完整表达,并确定改造后的突变体可以在植物中工作,并应于互作蛋白的筛选。
为达到以上技术目的,本发明采用的技术方案如下:
首先,提供一种能在植物细胞中完整表达的改造的DH5α BirA基因,所述改造的DH5α BirA基因与野生的DH5α BirA基因的差异在于编码第118位氨基酸的序列以及剪接识别位点的AG区域。
优选地,所述改造的DH5α BirA基因能在水稻细胞中完整表达。
更优地,所述改造的DH5α BirA基因序列为SEQ ID No.1所示。
其次,提供一种重组真核表达载体,其包含如前所述的改造的DH5αBirA基因。
可选择地,所述真核表达载体为pCAMBIA1390载体、pUbi载体或pUbi-EGFP载体。
再次,提供一种如前所述的重组真核表达载体的构建方法,其包括以下步骤:
获得野生的DH5α BirA基因;
对所述野生的DH5α BirA基因的可变剪接的剪切位点进行点突变,以获得能完整表达的改造的DH5α BirA基因。
优选地,对所述野生的DH5α BirA基因的可变剪接的剪切位点进行点突变之前,对所述野生的DH5α BirA基因进行点突变以获得突变的DH5αBirA*基因。
具体地,对所述野生的DH5α BirA基因第352位碱基进行点突变使所编码的蛋白质的第118为甘氨酸,以获得突变的DH5α BirA*基因。
进一步地,对所述野生的DH5α BirA基因进行剪切位点的点突变和/或第352位碱基的点突变之后,将已进行点突变的片段进行特异性扩增,与未进行点突变的扩增片段进行连接,以获得所述改造的DH5α BirA基因。
更进一步地,将所述方法改造的BirAG基因与增强启动子的基因序列、待研究的蛋白的基因序列进行连接,以形成所述待研究的蛋白质的蛋白互作研究载体。
还提供一种植物细胞中蛋白质互作的检测方法,其采用如前所述的能在植物细胞中完整表达的改造的DH5α BirA基因,或采用如前所述的重组真核表达载体。
可选择地,提供一种植物细胞中蛋白质互作的检测方法,其所采用的重组真核表达载体采用如前所述的重组真核表达载体的构建方法所构建。
结果表明BirAG具有生物素化植物蛋白的能力,并能够生物素化与BirAG融合的目的蛋白相互作用的蛋白。以上结果表明所改造的BirAG具有筛选目的蛋白互作蛋白的能力。
与现有技术相比较,本发明具有如下优势:
(1)本发明的能在植物细胞中完整表达的改造的DH5α BirA基因及其构建方法,对野生的DH5α BirA基因或DH5α BirA*基因的剪切位点进行了改造,并对剪切位点附近的部分密码子进行优化,使之更符合真核生物中的表达模式,使改造后的BirAG基因可以在植物细胞中表达,并具有生物素化一系列植物细胞内源蛋白的能力。
(2)本发明的能在植物细胞中完整表达的改造的BirAG基因及其构建方法,将所述改造后的BirAG基因与真核表达载体进行重组,可以在植物细胞中增强表达,能显著提高受体细胞生物素化蛋白数量,BirAG与目的蛋白融合后,可以生物素化与目的蛋白相互作用的蛋白。因此所改造的BirAG可以用于互作蛋白的筛选。
附图说明
图1为野生型DH5α BirA、DH5α BirA*突变体、改造的BirAG突变体的序列的比较。
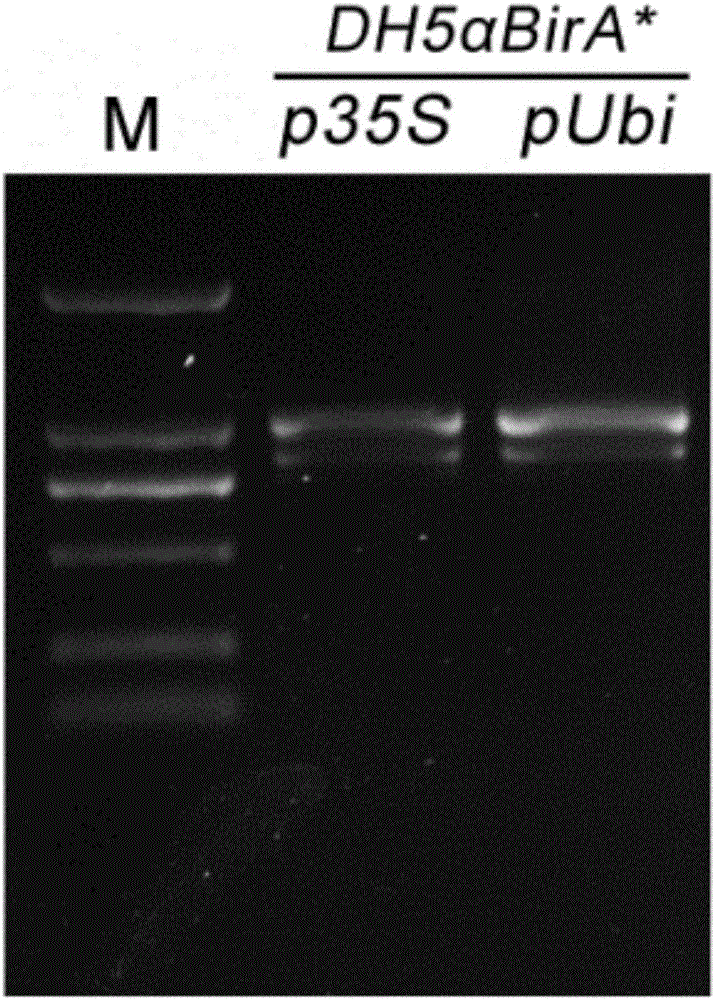
图2为p35S::DH5α BirA*和pUbi::DH5α BirA*瞬时转化水稻原生质体后RT-PCR扩增BirA*片段电泳图。
图3为扩增出的两个条带分别测序的结果。
图4为改造后的p35S::BirAG和pUbi::BirAG瞬时转化水稻原生质体后RT-PCR扩增BirAG片段电泳图。
图5为p35S::BirAG::EGFP和pUbi::BirAG::EGFP瞬时转化水稻原生质体后激光共聚焦显微镜观察结果。
图6为p35S::BirAG和pUbi::BirAG瞬时转化水稻原生质体后提取总蛋白,以HRP-Streptavidin为抗体的Western-blot结果图。
图7为WT和pUbi::BirAG::OsFD2瞬时转化水稻原生质体后鉴定到的生物素化蛋白的比较。
图8为pUbi::BirAG::OsFD2瞬时转化水稻原生质体后所鉴定到的蛋白与WT组进行比较,随机挑选出10个pUbi::BirAG::OsFD2组中鉴定到的特有的蛋白进行BiFC验证的结果。其中鉴定到的三个与OsFD2有互作的蛋白的BiFC结果图。
具体实施方式
以下结合附图和具体实施方式对本发明作进一步详细描述。
(一)DH5α BirA*的获得。
为构建DH5α BirA*,我们以大肠杆菌DH5α为材料进行PCR扩增,获得了DH5α BirA片段,并采用重叠延伸法构建DH5α BirA*突变体(突变了第352位碱基,C→G),分别构建了两个载体,为p35S::DH5α BirA*、pUbi::DH5αBirA*。
具体步骤如下:
(1)从大肠杆菌中获得DH5α BirA片段
DH5α BirA(SEQ ID No.2)的来源材料为大肠杆菌DH5α。通过PCR的方式,以BirAFw(上游引物,见SEQ ID No.3)和BirARe(下游引物,见SEQ ID No.4)为引物,直接将大肠杆菌菌落加入PCR体系中进行扩增,聚合酶Blend Taq由TOYOBO公司提供,具体PCR体系如下:
所得产物与T载体相连接,连接所用缓冲液Solution I由TAKARA公司提供,具体连接体系如下:
将连接产物转化大肠杆菌,具体过程为5μl连接产物加入解冻的大肠杆菌JM109感受态中,冰浴30分钟,42℃热激90秒,立即冰浴5分钟以上,加入800μl LB培养液后在37℃培养箱以160rpm/min的转速培养45~60分钟,将菌液涂布于含有抗性的(T载体为氨苄抗性)抗性板上,37℃倒置培养12~16小时。挑取单菌落进行PCR检测,PCR体系与上述PCR体系相同。最后通过测序的方式获得正确的带DH5α BirA片段载体的大肠杆菌。
(2)DH5α BirA*片段的获得。
对测序正确的含DH5α BirA载体的工程菌,采用重叠延伸法构建DH5αBirA*片段(突变了第352位碱基,C→G),具体实施方法为设计两个点突变引物BirAMF(上游引物,见SEQ ID No.5)和BirAMR(下游引物,见SEQ ID No.6),这两个引物与BirA的第352位碱基上都为错配。通过扩增BirAFw-BirAMR和BirAMF-BirARe两个片段(PCR体系与上述PCR体系相同),利用Axygen公司的胶回收试剂盒进行切胶回收,后将两样品等量混合,稀释100倍后以BirAFw和BirARe为引物进行扩增(PCR体系与上述PCR体系相同),将产物与T载体相连,转化大肠杆菌后挑取单菌落进行PCR检测并测序(连接与转化体系与上述体系相同),获得正确的带DH5α BirA*的载体。
(二)DH5α BirA*突变体在真核生物中存在可变剪接。
以水稻原生质体为材料,对DH5α BirA*在细胞中的表达模式进行了检测。将p35S::DH5α BirA*、pUbi::DH5α BirA*载体瞬时转化水稻原生质体,提取原生质体RNA并进行逆转录,扩增DH5α BirA*片段,参考图2,电泳结果显示能扩增出两条带。分别回收这两条带进行测序,参考图3,结果显示水稻对DH5α BirA*片段进行了剪接,剪切序列长165bp,该剪接情况为常见真核生物剪接模式(exon/GU-intron-AG/exon),所剪切区域位于BPL_lipA/B蛋白结构域。
(三)改造突变体BirAG在真核生物中能完整表达。
为防止DH5α BirA*在真核生物中发生可变剪接,对所述剪切区域的位点进行了改造,并对剪切位点附近的部分密码子进行优化,使之更符合真核生物中的表达模式。由于在可变剪接的剪切位点附近后SphI酶切位点,因此利用了这个酶切位点进行改造。
具体实施方法为利用重叠延伸法将DH5α BirA*改造成BirAG突变体,设计BirASphI引物(见SEQ ID No.7),扩增BirAFw-BirASphI片段,同时利用SphI和BglII酶切DH5α BirA*载体,将BirAFw-BirASphI片段与该载体相连,转化大肠杆菌后挑取单菌落进行检测并测序,获得正确的带改造的DH5α BirA基因的载体,即带BirAG基因的重组载体。
进一步地,构建了四个载体p35S::BirAG、p35S::BirAG::EGFP、pUbi::BirAG、pUbi::BirAG::EGFP,将p35S::BirAG、pUbi::BirAG载体转入原生质体中,提取RNA并逆转录,扩增BirAG片段,参考图4,电泳结果显示两个样品只扩增出1条带,表明BirAG在水稻中不发生剪接。
更进一步地,将p35S::BirAG::EGFP、pUbi::BirAG::EGFP载体转入水稻原生质体中,观察它们的表达及亚细胞定位,参考图5,结果发现载体均能正确表达且表达产物定位于整个细胞。
参考图1,右边的箭头处为第352位碱基突变位点,突变体DH5α BirA*和BirAG的核酸均为G,矩形框内为BirAG改造序列,其中左边箭头处为剪切的AG识别位点,BirAG中为改造为G碱基,另外剪切位点附近的碱基也进行了优化,使之更适合于真核细胞中表达,本实施例中,针对水稻细胞进行改造。
(四)真核表达载体的构建
为验证BirAG是否可以在细胞中发挥作用,生物素化水稻中的蛋白,以水稻为材料,将p35S::BirAG、pUbi::BirAG载体转入原生质体中,提取蛋白,并以HRP-Streptavidin为抗体,利用western-blot技术进行检测。
为确定融合BirAG的待研究蛋白可以生物素化与其空间上靠近的蛋白,我们以水稻FD2蛋白为研究对象,构建了pUbi::BirAG::OsFD2载体,并将其转入原生质体,对照组转入的为ddH2O。提取原生质体蛋白后通过Streptavidin磁珠将生物素化的蛋白进行富集,最后利用质谱进行蛋白测序。
具体步骤如下:
(1)将扩增出来的DH5α BirA*片段和BirAG片段都与pCAMBIA1390载体相连。由于所述设计的BirAFw和BirARe分别带有BglII和BamHI酶切位点,因此具体实施方式是将上述构建正确的带DH5α BirA*和BirAG载体均用BglII和BamHI酶切3小时后并用Axygen胶回收试剂盒进行回收目的片段(1000bp左右),将pCAMBIA1390载体用BamHI酶切3小时后65℃热变性处理15分钟,分别与DH5α BirA*片段和BirAG片段进行连接,转化大肠杆菌,鉴定,测序。具体方法同上,载体酶切体系如下,酶为TAKARA公司提供:
片段酶切体系如下:
(2)pUbi::BirAG::OsFD2载体的构建方法为:从植物总RNA中克隆OsFD2的CDS片段,所用引物为OsFD2Fw(上游引物,见SEQ ID No.8)和OsFD2Re(下游引物,见SEQ ID No.9),与构建好的pUbi::BirAG载体均用BamHI和EcoRI酶切3小时后,将它们均用65℃热变性处理15分钟,最后进行连接、转化大肠杆菌、鉴定及测序,所用体系同上。
(3)pUbi::DH5α BirA*::EGFP和pUbi::BirAG::EGFP载体的构建方法为:从植物总RNA中克隆OsFD2的CDS片段,所用引物为OsFD2Fw和OsFD2Re(SEQ ID No.8&No.9),与构建好的pUbi::BirAG载体均用BamHI和EcoRI酶切3小时后,将它们均用65℃热变性处理15分钟,最后进行连接、转化大肠杆菌、鉴定及测序,所用体系同上。
(五)提取RNA并逆转录
RNA的提取方法:采用OMEGA Plant RNA Kit试剂盒,提取总RNA。实验步骤如下:
(1)取水稻材料100mg,液氮中迅速研磨成粉末,趁液氮尚未挥发完全,将粉末转移到含有500μl Buffer RB/β-巯基乙醇的1.5ml灭菌离心管,剧烈振荡;
(2)14000×g室温离心5min;
(3)转移上清至gDNA Filter Column置于2ml离心管中,10000×g离心2min,取滤液。然后加入0.5倍体积的无水乙醇,上下颠倒离心管,混匀;
(4)把步骤3全部溶液转移至HiBind RNA Mini Column置于2ml离心管中,10000×g离心1min,倒掉下清,把柱子放回离心管中。
(5)加入配好的50μl DNaseI反应液(39μlRNase-free水+5μl DNaseI buffer+6μl DNaseI)至柱子内膜中央。室温反应5min;
(6)加入400μl溶液RWC,室温再放置10min,10000×g离心1min,弃废液;
(7)加入500μl溶液RNA Washing BufferII,10000×g离心1min,弃废液;
(8)重复步骤七洗涤一次;
(9)小心取出柱子,弃去收集管废液,将柱子放回同一根柱子中,10000×g离心1min;
(10)小心取出柱子,放入1.5ml的RNase-free离心管中,在柱子内膜中央小心加入50μl DEPC水,50℃放置2min;
(11)10000×g室温离心1min,收集RNA样品;
(12)取2μl样品2%琼脂糖凝胶电泳,120V恒压5min,紫外灯下观察。样品保存于-80℃;
逆转录及cDNA合成,实验步骤如下:
(1)取1μg水稻总RNA,加入1μl的50μM Oligo(dT)18,补RNase-free水至6μl;
(2)70℃保温10min后迅速放置冰上急冷2min以上;
在上述管中配制下列反转录反应液:
(4)42℃保温1h,以随机引物作为反转录引物时应先进行30℃,10min。然后再在42℃保温1h;
(5)70℃保温15min后冰上冷却,cDNA产物-20℃保存。
(六)原生质体的制备、转化及亚细胞定位观察
获得原生质体所用水稻种子均去壳后进行消毒处理,具体消毒方式为75%酒精浸泡10秒,用无菌水清洗1次,用含Tween-20的2.5%NaClO清洗30分钟,再用无菌水清洗5次以上,最后将每20颗种子播种于1/2MS培养基上培养8—12天。
原生质体制备的方法为:取幼苗30-40株,放在一张干净的A4白纸上,用刀片切去叶部和根部,保留茎段并切成0.5mm细丝,迅速转移至20ml0.6M甘露醇中,黑暗放置10分钟。去掉甘露醇,加入20mL酶解液,置摇床70rpm/min,黑暗中酶解5h。向锥形瓶中加入20ml W5,剧烈晃动10秒,将原生质体过滤到离心管中,用W5洗3-5次,用1ml MMG缓冲液,悬浮原生质体,冰浴30分钟。
转化原生质体的方法为:将MMG溶液中的原生质体调至终浓度约为2x106个/mL,将要转化的处理标记好2ml离心管,每管加入100μl原生质体和5-10μg质粒,轻轻混匀,静置5min。向每管缓慢加入110μl 40%的PEG-Ca2+溶液,轻轻混匀,26℃孵育15min,缓慢加入440μl缓冲液W5终止反应,轻轻颠倒离心管混匀。1500rpm离心3min,吸去上清,加入500μl缓冲液WI,轻轻混匀后转入24孔培养板,26℃培养6~16h。
取10μl培养的细胞,在激光共聚焦显微镜下在激发光下进行观察。
上述方法所需溶液配方为:
(1)40mM MES:0.853g MES溶于100mL双蒸水中,KOH调pH 5.7,过滤除菌。
(2)1M甘露醇:36.44g加热溶解于200mL双蒸水中,高压灭菌。
(3)W5缓冲液
(4)MMG
(5)WI缓冲液
(6)酶解液(现配现用)
(7)40%PEG(现配现用)
(七)总蛋白提取
提取蛋白的方法如下:
(1)在液氮中研磨样品,加入600ul蛋白提取液,振荡混匀。
(2)超声处理2分钟(超声2秒,间隙5秒),功率20%。
(3)加入12ul Triton X-100,4℃放置10分钟。
(4)加入600ul 50mM Tris-HCl(pH 7.4),振荡混匀,超声处理2分钟(超声2秒,间隙5秒),功率20%。
(5)4℃离心13200rpm 10分钟,取上清于新的1.5mL离心管,置于-20℃保存。
蛋白提取液配方如下:
(八)SDS-PAGE电泳及western-blot
SDS-PAGE实验步骤如下:
(1)将玻璃板,样品梳等用洗涤剂洗净,用ddH20冲洗数次,再用乙醇擦拭,晾干。
(2)参照说明书安装玻璃板。
(3)按如下体系配置12%的分离胶,按顺序依次混匀各成分,加入TEMED后迅速灌至两块玻璃板中间的间隙中。
(4)给浓缩胶留出足够的空间(离玻璃板上沿2cm左右),在上层加满水压平胶面,室温放置。
(5)聚合完成(约30min),倒掉上层的水,用吸水纸吸尽边缘的水。
(6)按如下体系配置4%的浓缩胶,按顺序依次混匀各成分,加入TEMED后,将混合液加入分离胶上层,插入梳子,室温放置30-40min,待胶完全凝固。
(7)将凝胶固定在电泳装置上,在内外槽中加入1×Tris-Gly电泳缓冲液。
(8)将等体积的样品和2×SDS凝胶上样缓冲液混匀,95℃水浴加热5min使蛋白变性,10000rpm离心3min,取20μl上样;
(9)80V电泳30min,待蛋白样品进入分离胶,120V继续电泳至染料前沿即将跑出凝胶。
转膜步骤如下:
(1)将胶从玻璃板上取下来,在去离子水中漂洗一下,去掉胶的一角作为记号。
(2)将胶放在转移换成液中浸泡30min。
(3)剪一块合适大小的PVDF膜,同时在一个角上做好标记,放入纯甲醇中浸泡15s,然后放去转移缓冲液中至少5min。将海绵垫,滤纸等转膜工具也放入转移缓冲液中浸泡。
(4)准备转膜“三明治”:按照海绵垫-滤纸-胶-膜-滤纸-海绵垫的顺序组装“三明治”(膜放在靠正极一侧,层与层之间不能留有气泡)。
(5)将夹子放入转移槽中,4度60V转移2h
免疫反应步骤如下:
(1)转移结束后,将膜取下来,放入1×TBST中浸泡一下,然后将膜放入TBST+Milk,室温下摇床上摇动封闭处理30min。
(2)倒掉封闭液,用1×TBST将膜洗2-3遍,每遍5-8min,将抗体用TBST+Milk稀释至适当浓度备用。
(3)倒掉TBST,加入抗体溶液室温孵育1h。
(4)倒掉抗体溶液,用用1×TBST将膜洗2-3遍,每遍5-8min,然后进行化学放光反应。
底物显色步骤如下:
采用的是BeyoECL Plus(超敏ECL化学发光试剂盒),BeyoECL Plus工作液的配制:等体积混合适量BeyoECL Plus A液和B液,室温放置备用。工作液宜在临检测前配制。
(1)用平头镊将膜取出,用吸水纸略吸去过多的液体(切勿接触膜的蛋白面),然后置于一洁净保鲜膜上。
(2)根据膜的大小,按每10平方厘米膜加1ml BeyoECL Plus工作液的比例,滴加BeyoECL Plus工作液到膜上,确保使工作液均匀覆盖在膜上,放置1-2分钟。
(3)取膜,弃BeyoECL Plus工作液,用吸水纸略吸去过多的液体。将膜放在两片保鲜膜中间,随后进行压片检测。
(4)压片检测:将膜固定于片夹内。暗室内压片1分钟,立即显影定影,根据结果再调整压片时间。
上述方法所需溶液配方为:
(1)30%Acr/Bis混合溶液(100ml):称取29g丙稀酰胺和1g bis丙稀酰胺,用温热的去离子水溶解定容到100ml,过滤,4摄氏度避光保存一个月。
(2)1.5M Tris-HCl(pH 8.8):称取18.17g Tris碱,加去离子水充分搅拌溶解,拥抱过浓盐酸调节pH值至8.8,将溶液定容至100ml,高温高压灭菌后,室温保存。
(3)1.0M Tris-HCl(Ph 6.8):称取12.114g Tris碱,加去离子水充分搅拌溶解,拥抱过浓盐酸调节pH值至6.8,将溶液定容至100ml,高温高压灭菌后,室温保存。
(4)10%SDS:称取10g SDS,用蒸馏水水浴加热至50℃溶解,定容到100ml,室温保存。
(5)10%AP:称取过硫酸铵0.1g,加入1ml去离子水溶解,贮存于4℃(有效期为2周左右)。
(6)5×Tris-Gly电泳缓冲液(室温保存)
(7)2×SDS凝胶加样缓冲液:Tris-HCl pH6.8(100mM);SDS(4%);溴酚兰(0.2%);甘油(20%);β-巯基乙醇(200mM),不含硫代试剂的加样Buffer可在室温保存。β-巯基乙醇临用前加入。
(8)考马斯亮蓝R-250染色液
(9)蛋白脱色液
(10)膜转移缓冲液:分别称取Tris碱3.02g,Glycine 14.4g,加入去离子水搅拌溶解,定容至800ml后,加入200ml甲醇。
(11)10×Tris-Buffered saline
(12)1×TBST Buffer
(13)1×TBST Buffer+5%Milk
(九)质谱鉴定蛋白
(1)磁珠预处理:
取600ul链霉亲和素磁珠(sphero公司,SVM-30-10)于磁力架上,吸出上清,加入600ul缓冲液Q,超声处理2分钟(超声时间2秒,间隔5秒),功率20%。再用缓冲液Q清洗磁珠2次。
(2)样品与磁珠混合:将样品加入磁珠中,于旋转器上4℃旋转过夜。
(3)将样品置于磁力架上,吸出上清,磁珠随后分别用缓冲液W1清洗2次,缓冲液W2清洗1次,W3清洗1次,缓冲液W4清洗2次,50mM NH4HCO3(电泳纯,pH8.3)清洗6次。加入20ul溶解于电泳纯的NH4HCO3的0.2ug/ul的TPCK胰蛋白酶,置于37℃16小时,吸出上清于新的离心管中,真空浓缩仪干燥,干粉进行质谱鉴定。
上述方法所需溶液配方为:
(1)缓冲液Q
(2)缓冲液W1
(3)缓冲液W2成分
(4)缓冲液W3成分
(5)缓冲液W4成分
参考图6,WT为不加质粒进行原生质体转化,BirAG为加了带有BirAG片段的质粒进行原生质体转化。p35S表示35S启动子,pUbi表示Ubi启动子。p35S::BirAG和pUbi::BirAG样品的条带数均多于野生型。结果显示BirAG能明显提升水稻蛋白生物素化的情况。
参考图7,WT为不加质粒进行原生质体转化,pUbi::BirAG::OsFD2为加了带有BirAG::OsFD2片段的质粒进行原生质体转化,提取培养过夜的原生质体的总蛋白,利用链霉亲和素磁珠吸附被生物素标记的蛋白,并通过蛋白质谱进行鉴定。测序结果发现pUbi::BirAG::OsFD2组与ddH2O组相比生物素化蛋白数目明显增多,也表明BirAG能显著提高水稻生物素化蛋白数量。(十)BiFC鉴定蛋白互作事件
(1)将两个待检测的蛋白分别与BiFC载体的N端和C端融合,构建N端载体和C端载体。酶切和连接的体系同上述。
(2)载体转化大肠杆菌,转化子鉴定,测序,并进行大肠杆菌培养和质粒提取。实验方法同上述。
(3)制备原生质体,并将N端载体和C端载体转化到原生质体中瞬时表达。制备及转化原生质体方法同上述。
(4)将培养的细胞转移到载玻片上,轻轻盖上盖玻片,在共聚焦显微镜观察原生质体中的发光情况。
参考图8,BF为明场,YFP为黄色荧光通道,Chl为叶绿体自发荧光,Merge以上三图的融合。结果表明,在随机挑选的10个pUbi::BirAG::OsFD2组特有的蛋白中,Q5VMJ3、Q5JMX3、Q0JLX8为鉴定到的与OsFD2有相互作用的蛋白。以上结果表明BirAG融合目的蛋白后可以应用于植物细胞中,进行蛋白互作的鉴定。
综上所述,本发明能在植物细胞中完整表达的改造的DH5α BirA基因能在植物细胞中完整表达,所述改造的BirAG基因及其构建方法为检测蛋白质相互作用技术实现条件。此外,本发明表明改造的DH5α BirA可以在植物细胞中发挥作用,可用于植物中互作蛋白的鉴定,
上述实施例为本发明较佳的实施方式,但并不仅仅受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,均包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!