一种超敏感引物及其设计方法和应用与流程
本发明属于分子生物学、核酸扩增领域以及核酸分子检测领域,具体涉及一种钳制聚合酶链反应的超敏感引物及其设计方法和应用,该引物通过一定的设计要求可以有效的放大钳铗的阻断效应,尤其涉及通过选择性的放大有突变的基因而进行基因突变检测的多种检测技术领域。
背景技术:
尽管近来在癌症治疗方面有了很大的进展,尤其是高效特异性的重组单克隆抗体类药物的迅速增加,但由癌症造成的死亡率在全球范围和大多数发达和发展中国家仍处在第一位。做好癌症发病的预防和早期进行人群筛查是有效的长期策略,但如何改善和增进癌症的治疗疗效是目前能够提高治愈率和延缓复发的关键所在。随着靶向药物(优点是疗效特异和副作用小)的不断涌现,完善有效的个体治疗方案也要根据具体该病人的发病原因和基因组的遗传特征,再加上药敏试验进行确定和及时的调整,以避免造成肿瘤形成耐药和扩散。也就是说我们需要一种或多种检测方法来协助临床医生选择有效的药物,还需要有较目前更加敏感和特异的检测方法来尽早的告诉医生药物是否有效和发生转移/复发的可能性,以便及早调整药物和密切观察病情进展。还有是在相反的情况,有些病人在肿瘤发病早期得到了没有必要的过度治疗,这也是要避免的,以减少对机体免疫力的损害或由放射造成的新的癌症细胞产生。
大多数的病人在确定患了癌症以后,会利用现有的检测手段确定是否有转移,并及时的采取手术切除和后续的放疗/化疗。不适合做手术的,则会采取积极的化疗和/或放疗。确定化疗/放疗的方案,要根据肿瘤的类型(病理诊断)和发病的病理机制,再加上主治医生以往的治疗经验和病人的身体状况来制定。为了减少化疗药物的副作用,越来越多的针对肿瘤某一传导通路特异的药物(如只阻断肿瘤细胞的生长,而对正常细胞影响很少)被制药公司大量开发了出来。随之伴随的检测方法也要跟上来,协助药物的选择,结合起来使得治疗效果得到明显提高。然而,癌症病人的长期存活率和治愈率并未得到大的改观,其主要原因是肿瘤细胞会产生耐药,另外手术和术后附加的化疗/放疗并没有将弥散或转移的癌症细胞完全清除和杀死,而目前用的监测手段和技术(主要依赖影像如ct检查)不够敏感,使得现用药物对肿瘤细胞不再有抑制作用没能得到及时发现,以致于换药太迟、肿瘤得到扩散/转移而不可控制。
分子病理诊断的临床应用将会进一步扩展经典病理学诊断的内涵,将传统的形态诊断外延到肿瘤发生易感性、基因与染色体变化、基因路径与基因治疗、生物学行为评估、对药物治疗反应的评价、临床预后的预测等医疗全过程之中。
基因检测,即取检测者的肿瘤组织、唾液、尿液、胸水或血液等,经提取和扩增其基因信息后,通过相应的分子生物学技术,对被检测者细胞中的dna分子的基因信息进行检测,分析他的基因状态,从而对肿瘤的诊断或治疗提供一定的帮助。随着分子生物学的发展和对肿瘤发生发展机制的深入认识,病理诊断已经不再局限于形态学。基因检测为肿瘤的鉴别诊断提供精准和便捷的科学依据,已成为肿瘤个性化及精准治疗的前提。分子靶向治疗已经成为当前肿瘤治疗的趋势,分子靶向药物针对特异的分子靶点,因此患者在采用靶向治疗前必须对相应的分子靶点进行基因检测,省去患者尝试各种治疗方法的时间,提升治疗效果。另外,肿瘤的基因检测出的突变或其它异常可以对肿瘤的预后、生存期长短及不同个体设计切实可行的治疗方案等,为医生提供可依据的信息。即使是同一种肿瘤,其致病因素和体内突变的基因都是不一样的,每一个患者的肿瘤都有自己独特的生物特征,这就是肿瘤的特异性,这种特异性要求“一药一病”,对每个患者“区别对待”,这就是肿瘤的个性化治疗。个性化治疗,目前已被公认为提高肿瘤患者生存期和生活质量的最佳途径。而制定个性化治疗方案、提高疗效,临床医生最需要精准的分子病理诊断报告。
循环肿瘤dna(ctdna),是指肿瘤细胞体细胞dna经脱落或者当细胞凋亡后释放进入循环系统,随着基因测序的飞速发展,目前,已能在血液中对其进行检测并计数。ctdna来自肿瘤细胞的体细胞突变,不同于遗传突变的是,其存在于体内每个细胞。因此,ctdna是一种特征性的肿瘤生物标记物,并且还可以被定性、定量和追踪。随着肿瘤分子生物学研究的进展,循环血游离dna的检测及其生物学指标的研究,将有可能为临床肿瘤的早期诊断、预后判定及跟踪随访等提供一系列方便、快捷、特异、无创或微创和分子生物学检测手段。与组织活检相比,ctdna检查微创小,并允许对治疗反应实时监测来评估治疗反应以及可能出现的耐药性。我们还可以使用ctdna活检作为肿瘤标志物而评估早期癌症的残留病灶,并确定我们能否告诉术后的患者,他们是否还需要额外的辅助治疗。液体活检可能永久改变活检方式,包括对治疗方案的反应、抗药性的出现,将来甚至还能用于早期诊断。液体活检只需要患者的血液、尿液、唾液等,无需传统的肿瘤组织(手术或活检),可以减少诊断过程中对患者身体的损伤,其检测肿瘤复发要比日常用的ct和核磁共振平均早10个月。这样及早的发现复发或转移,病人可以提前得到治疗或对产生耐药的肿瘤提前改变治疗方案,提高治愈率和延长生存时间并减少盲目无效的治疗所造成的医疗费用。
肿瘤的基因检测已在临床应用,但敏感性和特异性因所用的检测技术而有所差异。检测技术应用的选择和标准化和规范化是保证检测结果的灵敏性和准确性的重要保证。现有的有效检测基因突变的技术手段包括:
1)扩增阻滞突变系统(arms)-pcr法
扩增阻碍突变系统(amplificationrefractorymutationsystem,arms)是pcr技术应用的发展,也称等位基因特性pcr(allele-specificpcr,as-pcr)等,用于对已知突变基因进行检测,是目前实验室常用的基因突变检测方法。arms-pcr法检测灵敏度高,可检测肿瘤细胞中突变比例为1%甚至更低的突变基因,但只能检测已知的突变类型,不能发现一些新的、未知的突变;如果检测的突变位点或类型较多,则随着引物数目增加出现非特异性结合的概率也相应增加;当检测位点较多时,对dna样本量的需求增加。
2)xna钳制pcr技术
钳制pcr是另一发展起来的针对特异性放大突变基因的新技术。它是基于pna、lna、bna等改型的核苷酸链(统称为xna)与dna结合有更强的亲和力。pna是一种全新的dna类似物,即以中性的肽链酰胺2-氨基乙基甘氨酸键取代了dna中的戊糖磷酸二酯键骨架,其余的与dna相同,pna可以通过watson-crick碱基配对的形式识别并结合dna或rna序列,形成稳定的双螺旋结构。由于pna不带负电荷,与dna之间不存在静电斥力,因而结合的稳定性和特异性都大为提高;不同于dna间的杂交,pna与dna的杂交几乎不受杂交体系盐浓度影响,与dna分子的杂交能力远优于dna/dna或dna/rna,表现在很高的杂交稳定性、优良的特异序列识别能力、不被核酸酶和蛋白酶水解。对于错配的pna/dna,哪怕只有一个碱基的错配,也会造成其融解温度下降9-10℃左右。目前,肽核酸作为一种非常有用分子生物学工具已在疾病的诊断、治疗等领域得到应用。pna钳制有效抑制样本在pcr反应中占绝大多数野生型基因的扩增,从而提高突变基因的检测灵敏度以及降低检测下限。灵敏度可由普通的实时定量pcr的1%降到0.1%。但要做液体活检,灵敏度要求在0.01%或更低。
3)数字pcr(digitalpcr)
数字pcr是一种核酸分子绝对定量技术。相较于qpcr,数字pcr能够直接数出dna分子的个数,对起始样品绝对定量,目前的应用包括稀有等位基因检测、基因表达绝对定量、不需要核酸标准品等。灵敏度可达0.01%,高特异性,可检测复杂背景下的靶标序列;可高度耐受pcr反应抑制剂;不必依赖对照品或标准品,可对目标拷贝数直接进行精确的鉴定,分析微小的浓度差异;实验数据分析便捷,检测结果以阴性、阳性判读,数据分析自动化;可统计突变率,通过统计分析可得出靶点的突变率。但数字pcr已不再有放大的功能。
4)新一代(或二代)测序(nextgenerationsequencing,ngs)
ngs又称大规模平行测序(mps)或二代测序,包含多种可以一次性产生大量数字化基因序列的测序技术,是继sanger测序的革命性进步,采用平行测序的理念,能够同时对上百万甚至数十亿个dna分子进行测序,实现了大规模、高通量测序的目标。有定量功能,样品中dna被测序的次数反映了样品中这种dna的丰度。检测灵敏度和测序丰度相关。一般ngs在肿瘤体细胞突变检测时,检测灵敏度为10%。增加丰度,检测成本也相应提高。已知的与肿瘤相关驱动基因数量有限,疾病表型和基因型的关系还有赖于生物信息的解读,目前ngs应用于肿瘤细胞突变检测的标准化和质量控制尚未形成共识。
技术实现要素:
虽然现有技术公开了多种检测基因突变的技术,但所采用的扩增手段其引物均为与野生型或突变型基因完全匹配的引物对,以此来保证扩增的特异性和灵敏度。即使如此,pcr扩增手段中,尤其是即时定量pcr,无论是用arms法还是针对野生型xna钳制技术,检测肿瘤突变基因的敏感性也只能达到0.1%,对于有些突变检测,如液体活检,以及丰度本就不高的突变基因检测,如循环肿瘤dna(ctdna)突变检测,现有技术的扩增手段达不到所需的灵敏度。从实时定量pcr的试验结果看,一般来说,如pna钳铗在好的情况下只能抑制十个循环(δct)野生型的放大(相当于0.1%的检测灵敏度),而作为液体活检的技术,则需要提高到近20个δct,才有希望检测到占十万分之一以下的突变型的可能性。
此外,具体的,对于xna钳制pcr技术的使用,由于受突变碱基周围序列的限制,提高钳铗的效率主要方式是通过延长xna的长度,但其后果是降低了特异性和可能对突变型也出现阻断效应。另外一种方式,就是相对降低被阻断引物的亲和力而不影响特异性,这可以通过设计不同的上游引物来达到目的,这往往需要进行多条引物的筛选。基于上述,xna钳制pcr技术的具体实践过程中,由于受突变碱基所处位置和前后碱基序列的限制,再加上设计钳铗的限制(如pna,最多允许6gs连续在一起,自身互补在4个碱基一下,gc不超过60%以免溶解度过低),常常遇到针对一个具体的突变位点,设计不出很好的能够阻断相应野生型的pna钳铗的情况,这限制了xna钳制pcr技术的应用。
针对上述种种,本发明通过综合因素的配合和设计可以达到高灵敏度和高特异性的扩增手段和突变位点或单核苷酸多型性(snp)的检测。
本发明的目的之一在于提供一种用于钳制pcr的超敏感引物及其设计方法。通过新的引物设计手段,使得钳铗的阻断作用更易影响干扰基因(如本发明采用的野生型基因)的扩增,xna钳铗更容易的影响上游引物的匹配,使得干扰基因的扩增变得极为不易发生,而对于目标基因(如突变基因)而言,上游引物与基因的匹配不受影响,特异性增高。
本发明的目的之二在于提供一种钳制pcr技术。该聚合酶链式反应手段可以更为有效的扩增所需要的目标基因。
本发明的目的之三在于提供一种富集突变基因的方法。
本发明的目的之四在于提供一种发现或鉴别新的基因突变的方法。
本发明的目的之五在于提供一种检测基因突变的方法。
为实现上述发明目的,具体的,本发明涉及以下技术方案:
本发明公开了一种用于钳制pcr的超敏感引物组,所述引物组中至少一个引物被设计为:
(1)引物由3段序列组成,由5’端至3’端依次为5’端配对区域、不匹配区域、3’端配对区域,5’端配对区域和3’端配对区域与目标序列完全互补,而不匹配区域与该段目标序列无法匹配互补;
(2)引物的3’端与钳铗对应匹配的dna序列邻近,或引物的3’端配对区域与钳铗对应匹配的dna序列有数个碱基的配对;
(3)引物满足目标序列pcr扩增的特异性。
pcr扩增所用的引物是人工合成的两段寡核苷酸,一个与感兴趣区域一端的一条dna模板链互补,另一个与感兴趣区域另一端的另一条模板链互补。通常引物的长度依需扩增的目标基因的大小而不同,本文的引物长度符合常规扩增特异性的要求,引物长度可在18-50bp范围内选择,优选的实施方案中,引物的长度在25-48bp范围内,更为优选的实施方案中,引物的长度为30-45bp,例如引物的长度可以为31bp、32bp、33bp、34bp、35bp、36bp、37bp、38bp、39bp、40bp、41bp、42bp、43bp、44bp等。
本发明所述引物中,3段序列中不匹配区域占引物序列的1/2以下,不能影响引物与目标基因的紧密结合,优选的,不匹配区域占引物序列的1/3及以下;不匹配区域的长度也不能过小且3’端配对区域不能过长,因为本发明设计超敏感引物的其中一个主要目的在于,通过在引物中间设计有一段与目标序列不匹配的引物(引物两头的序列与目标基因靶点相匹配),使得引物的3'末端序列在结合到目标基因靶点时不稳定,更容易被钳铗(xna)替换掉,从而xna钳铗更容易的影响上游引物的匹配,使得非目标基因的扩增变得极为不易发生,例如不匹配区域占引物序列的1/5及以上,优选的技术方案中,不匹配区域占引物序列的1/4及以上,3’端配对区域长度占引物序列的1/4及以下、1/6及以上,优选的,3’端配对区域长度占引物序列的1/5及以下、1/6及以上。
在本发明一个的实施方案中,所述引物的不匹配区域占引物序列1/3-1/4,3’端配对区域长度占引物序列的1/4-1/6。
具体到引物序列的碱基数,本发明所述引物3段序列中5’端配对区域、不匹配区域、3’端配对区域的碱基数分别为18-22bp、9-15bp、6-9bp;更为优选的实施方案中,5’端配对区域、不匹配区域、3’端配对区域的碱基数分别选择为20bp、8-14bp、7-8bp。
除了在引物中合理设计不匹配区域以及不匹配区域和3’端配对区域的长度外,本发明所述引物是用于钳制pcr扩增反应的,其与钳铗(xna)结合使用,因此需要考虑钳铗(xna)对于引物的影响因素。
引物3’端是pcr扩增延伸开始的地方,因此引物3’端序列的错配或结构的限制会影响扩增特异性与效率。本发明需要将引物设计为3’端与钳铗配对dna序列邻近,或引物的3’端配对区域与钳铗对应匹配的dna序列有数个碱基的配对(即引物的3’端配对区域与钳铗序列有数个“碱基”的重叠),以使得所述引物成为超敏感引物,增强钳制效应,从而反映在pcr扩增的灵敏度。由于钳铗(xna)的空间大小有限,因此引物的3’端应与钳铗配对dna序列邻近,如引物的3’端配对区域对应的dna序列与钳铗对应匹配的dna序列之间的距离相差不超过5个碱基,如相差5个碱基,相差4个碱基,相差3个碱基,相差2个碱基,相差超过1个碱基或引物的3’端配对区域对应的dna序列与钳铗对应匹配的dna序列相接。通过将引物的3’端配对区域设计为与钳铗序列有数个碱基的重叠,可以直接的使得钳铗(xna)阻碍引物的3’端配对,由于受突变碱基所处位置和前后碱基序列的限制以及设计钳铗的限制,该数个碱基的配对位于突变位点前,且数个碱基的配对为连续配对。
所述引物不匹配区域,可以采用随机的不配对碱基序列(但需要满足其他条件,如引物设计的一般要求和tm等),相对优选的是,采用c:c,g:gora:g,尽量避开a:a或t:g的使用。
本发明所述的引物组,可以为用于钳制pcr扩增目标基因的一对引物,也可以为扩增多个目标基因的多对引物。
本发明所述的钳铗(xna),是指pna、lna、bna等与dna结合有更强的亲和力的改型的肽核甘酸或改进的核苷酸链。
pna(peptidenucleicacid,肽核酸)是一种全新的dna类似物,于1991年由dr.nielsen,dr.egholm,dr.berg和dr.buchardt发明。该分子的特点是以中性的肽链酰胺2-氨基乙基甘氨酸键取代了dna中的戊糖磷酸二酯键骨架,其余的与dna相同。pna可以通过watson-crick碱基配对的形式识别并结合dna或rna序列,形成稳定的双螺旋结构。由于pna不带负电荷,与dna和rna之间不存在静电斥力,因而结合的稳定性和特异性都大为提高;不同于dna或dna、rna间的杂交,pna与dna或rna的杂交几乎不受杂交体系盐浓度影响,与dna或rna分子的杂交能力远远优于dna/dna或dna/rna,表现在很高的杂交稳定性、优良的特异序列识别能力、不被核酸酶和蛋白酶水解等。
lna(lockednucleicacid,锁核酸)是一种核酸类似物,和普通核酸分子区别在于其碳环的2′氧原子和4′碳原子位置引入亚甲基桥形成锁状结构(其单体结构如式i所示)。它是一种特殊的双环状核苷酸衍生物,结构中含有一个或多个2’-o,4’-c-亚甲基-β-d-呋喃核糖核酸单体,核糖的2′-o位和4′-c位通过不同的缩水作用形成氧亚甲基桥、硫亚甲基桥或胺亚甲基桥,并连接成环形,这个环形桥锁定了呋喃糖c3′-内型的n构型,降低了核糖结构的柔韧性,增加了磷酸盐骨架局部结构的稳定性。由于lna与dna/rna在结构上具有相同的磷酸盐骨架,故其对dna、rna有很好的识别能力和强大的亲和力。与其他寡核苷酸类似物相比,lna有很多优点:1、和dna、rna互补的双链有很强的热稳定性(δtm=3~8℃);2、抗3′脱氧核苷酸酶降解的稳定性;3、lna-dna杂交物能激活rnaseh;4、水溶性好,自由穿入细胞膜,易被机体吸收;5、体内无毒性作用;6、高效的自动寡聚化作用,合成方法相对简单,部分或完全修饰的lna寡核酸链可用氨基磷酸法在dna自动合成仪上合成。
本发明所述的引物,满足一般pcr扩增的特异性要求,引物的特异性都经primerblast的计算机软件与目标基因做比较,显示只与目标基因匹配,且pcr扩增放大后的产物与目标基因一致,例如将pcr扩增放大后的产物进行琼脂糖凝胶电泳,在凝胶电泳上也只显示一条带,或者扩增后的产物进行测序分析,扩增产物序列除引物序列外与目标基因序列完全一致且唯一。
此外,本发明所述引物为用于钳制pcr扩增的引物,其称之为引物,自然也满足常规引物设计的一般要求,目前,引物设计原则包括:(1)引物的tm值不能过高或过低,过高(高于72℃)或过低(低于35℃)都不利于引发扩增反应,上下游引物(或称之为前引物、后引物)的tm值相差不能过大,最好相差不能大于5℃;(2)g+c含量应在40%-60%,4种碱基分布/配要均匀,避免出现4个以上碱基同一重复、序列反向重复(发夹结构)、和序列简单重复的二级结构;(3)一对引物间不能有3base或3base碱基以上的连续反向互补,特别是引物间3’末端的反向互补;(4)引物的3’末端碱基,尤其是最末和倒数第2个碱基应正确与靶配对,尽可能使每个引物的3’最末碱基为g/c,但不能为nngc或nncg末端(所谓gc/cg夹),亦不能为特异性差的t结尾。值得注意的是,为了进一步增强钳制pcr扩增的特异性和扩增效率,优选的实施方案中,本发明所述引物的tm值低于反向引物的tm3-5℃,具体的实施方案中,所述引物的tm一般在55-60℃之间。
本发明公开了一种钳制pcr扩增方法,所述扩增方法包括(1)设计或获得上述用于钳制pcr的超敏感引物组,设计或获得钳铗(xna);(2)获得扩增所需的模板,进行钳制pcr扩增反应。
为了进一步提高钳制pcr对于目标基因的扩增效率和特异性,本发明钳制pcr进一步采用分阶段的策略,具体而言,钳制pcr扩增反应阶段,开始的3-5个pcr循环放大在低温下进行,以使得超敏引物与要放大的目标序列充分结合;随后的循环放大反应则提升引物结合的温度,该过程只让超敏引物与前面放大合成的新的核苷酸链结合。通过该分阶段扩增的方式,可以有效节约模板用量,并且提高目标基因的丰度,降低检测难度。
该钳制pcr扩增反应可以用于富集突变基因使用。例如,如图1所示,钳制pcr扩增反应开始的3-5个pcr循环放大是在低温(60℃至50℃)下进行,让超敏引物与要放大的目标序列结合,但随后的十个左右的循环则提升引物结合的温度(60℃或以上,或高至反向引物可耐受的温度),只让超敏引物与前面放大合成的新的核苷酸链结合(即突变型,因为野生型收到阻断,前期没有得到放大)。这个预放大的过程,可以同时放大多个靶标,相互之间没有干扰,从而节约标本的用量,还可降低试验的费用和操作步骤。预放大后的样本,随后可用即时定量pcr(qpcr)或微滴数字化pcr(ddpcr)来做单项的检测和定量,所用的内围引物(nestedprimer)则取用超敏引物的3'端的序列,即只识别在预放大中被超敏引物放大过的片段,保持其特异性。经过这样的预放大,12-15轮后突变型的比例就有百万分之一增加到0.03-0.2%。由此再由数字pcr检测则完全在其检测的灵敏度范围之内。另一方面需要考虑的是,即使拿出十分之一的预放大产物做下一步的检测,也至少有400个突变型靶点,非常容易检测出来。
本发明上述钳制pcr扩增方法也同样适用于发现或鉴别新的基因突变的过程。通过所述钳制pcr扩增,或进一步富集突变基因后,进行扩增产物的测序,便于发现或鉴别出新的基因突变。
本发明所述的基因突变,是指基因在结构上发生碱基对组成或排列顺序的改变,所述基因突变包括常见的点突变,也包括基因缺失突变、移码突变、插入突变、融合突变等。点突变,如一种嘌呤被另一种嘌呤取代或一种嘧啶被另一种嘧啶取代,或者嘌呤取代嘧啶或嘧啶取代嘌呤的突变;移码突变(translocation),指dna片段中某一位点插入或丢失一个或几个碱基对时,造成插入或丢失位点以后的一系列编码顺序发生错位的一种突变;缺失突变(deletion),基因也可以因为较长片段的dna的缺失而发生突变;插入突变(insertion),一个基因的dna中如果插入一段外来的dna,那么它的结构便被破坏而导致突变。
此外,本发明公开了一种检测基因突变的方法,其包括采用上述超敏感引物组进行扩增的过程。
检测基因突变的方法,可以采用以下一种或多种手段进行:
该检测方法,进行如下一种或多种检测进行突变基因检测:
(1)采用上述钳制pcr扩增或进一步富集突变基因后,进行扩增产物的测序,所述测序手段包括但不限于sanger测序、ngs测序手段;
(2)利用上述超敏感引物组和钳铗进行实时定量pcr(qpcr)检测或微滴数字化pcr(ddpcr)检测;
(3)利用上述钳制pcr扩增进行预防大(包括多靶点)后再进行实时定量pcr(qpcr)或微滴数字化pcr(ddpcr)检测。
本领域技术人员明了,并非所有的基因突变均与疾病相关,本发明所述检测基因突变的方法包括非疾病诊断和/治疗部分。所述突变基因检测的样本可以来源于细胞或组织器官等,由于本发明检测方法灵敏性和特异性的提高,也可以检测来自于血液、尿液、唾液或其他组织液的生物样本,尤其是对于目标突变丰度本身不高的样本的检测,如ctdna突变检测。
本发明相应的检测基因突变的方法应用过程中,对应的产品为检测试剂盒,作为pcr检测试剂盒,其核心为所使用的引物,具体的,本发明的检测试剂盒包括上述的超敏感引物组和钳铗(xna)。此外,检测试剂盒包括常规钳制pcr扩增反应所需要聚合酶、dntps、mg2+、染料(qpcr或dpcr检测试剂盒选用)等。
更为具体的,本发明公开了一种检测egfr基因突变的试剂盒,该试剂盒包括:
正向引物:ctgggcatctgcctata
反向引物:tctttgtgttcccggacatagtc(seqidno.2),
pna钳铗:nh2-ctcatcacgcagct-cooh(seqidno.3)。
本发明的技术方案取得了以下效果:(1)通过超敏感引物的设计,使得钳铗(xna)的阻断作用更易影响非突变基因的扩增,xna钳铗更容易的影响上游引物的匹配,使得非靶点基因(如野生型或一种亚型)的扩增变得极为不易发生,而对于目标基因(如突变基因或其它亚型)而言,上游引物与基因的匹配不受影响,钳制pcr扩增的效率和特异性明显提高,使其可以满足液体活检的需要;(2)克服了arms-pcr法检测时仅能检测已知突变类型的限制,并且不需要设计多条引物,对于样本的模板量需求较少,意味着临床检测时只需要很少的临床样本就可以检测多个靶基因的检测,从而节约标本的用量,还可降低试验的费用和操作步骤;(3)本发明所述钳制pcr扩增方法进一步结合微滴数字pcr(ddpcr)可以有效的针对活体样本进行检测和定量,可以检测出检测出1:1万倍的突变,扩展了微滴数字pcr检测的应用范围;(4)本发明所述超敏感引物可以应用于:a、选择性的放大有突变的基因,用于测序(一代和二代),提高检测突变基因的几率;b、用于基因缺失和融合的检测;c、直接用于qpcr或ddpcr的检测;d、作为预放大,选择性的放大有突变的一个或多个基因(multiplex),然后用qpcr或ddpcr检测。
附图说明
图1本发明所述超敏感引物设计及超敏钳制pcr示意图
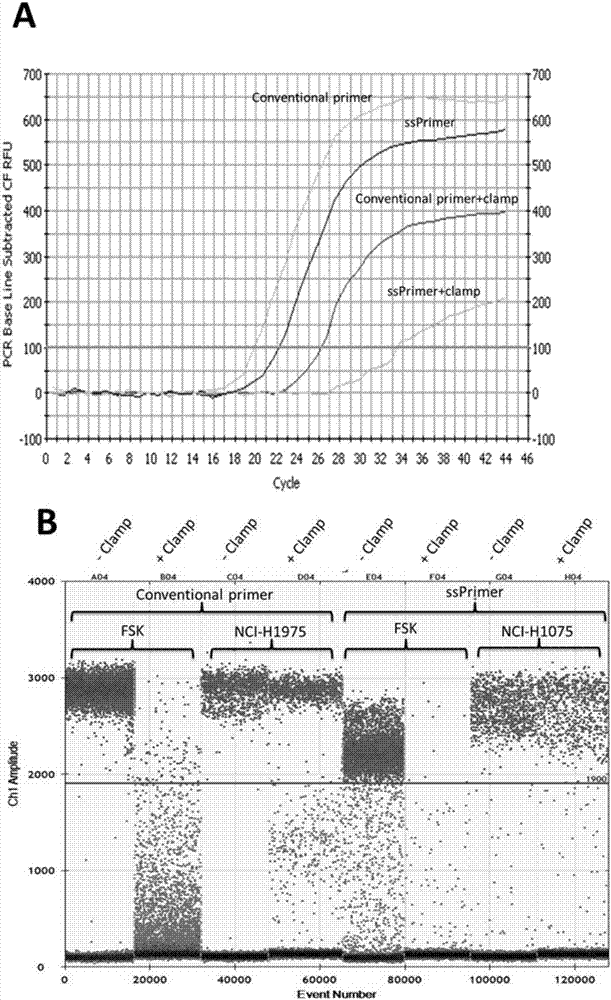
图2常规钳制pcr和超敏钳制pcr比较实验。a图为实时定量pcr(qpcr),b图为微滴式数字pcr(ddpcr)。扩增反应中,基因组dna用量为0.04to1μg(原代鳞状上皮细胞fsk只含野生型egfr,扩增时基因组dna用量为100ng;细胞株nci-h1975是杂合子,含有egfrt790m突变和野生型两种,扩增时基因组dna用量为40ng),pna添加浓度为0.1μm。
图3超敏钳制pcr检测突变的灵敏度实验。分别40ng的fsk和nci-h1975按照不同比例混合进行微滴式数字pcr(ddpcr)实验,分别400ng的fsk和nci-h1975按照不同比例混合进行即时定量pcr(qpcr)实验(为进行预富集放大),pna添加浓度为0.1μm。
图4超敏钳制pcr后用微滴数字pcr检测。a图为没有预放大的情况,b图为预放大后再用微滴数字pcr检测的情况。
具体实施方式
实施例一
针对超敏感引物的设计,我们首先在上皮生长因子受体(egfr)的t790m的检测上做了验证,并与常规的引物做比较。引物的特异性都经primerblast的计算机软件与人类基因组做过比较,显示只与目标基因完全匹配。pcr放大后的产物在凝胶电泳上也只显示一条带。常规正向引物的序列是:
tcacctccaccgtgcagctc(seqidno.4),以保证基因多型性都得到机会放大),反向引物的序列是:tctttgtgttcccggacatagtc(seqidno.2,放大的片段长度是75bp),超敏正向引物经反复测试后最后选用的序列是:
ctgggcatctgcctata
试验结果:如图2(a)所示,pna(0.1μm)这一浓度的只能部分抑制常规pcr(6个循环的不同,加和不加pna之间)。若改用超敏引物(其它不变),同样浓度的pna则可抑制10个循环的放大,表明其抑制超敏引物的效应明显加强。同样的试验用ddpcr重复,得出同样的结论。如图2(b)所示,pna(0.1μm)没能将用常规引物放大的野生型的信号全部压倒底线。但同样浓度的pna(0.1μm)在用超敏引物做的pcr放大,则将全部的野生型控制在底线,没有丝毫的放大。在用nci-h1975(杂合子)做的同样的试验中,针对野生型得到了同样的结果,而对突变型没有任何影响。这些结果显示超敏引物的确对pna介导的阻断作用更敏感,分析原因是由于它的3'末端在结合到靶点时不稳定,更容易被pna替换掉。当将pna的浓度增加到1.5μm,两个试验之间的差距就更为明显。在使用常规引物的试验中,在加与不加钳铗之间只看到十个循环的差别。如图3所示,在一系列的混合样本(nci-h1975与fsk)的试验中,当使用超敏引物,pna在同样的浓度可以导致完全的抑制。其它不同的混合比例,则得到相应的推迟曲线,在1:10万的情况下仍可以检测的到。充分显示出超敏引物对目标突变基因的特异性,因此有希望应用到标本中只含有一个突变基因的情况下。
我们也进一步测试在一个预放大富集反应中加两对(egfr790和858)引物和相应的钳铗(检测egfrl858r基因突变的正向引物:
acaccgcagcatgtcgcacgagtgagccttgggc(seqidno.6),反向引物:ccttactttgcctccttctg(seqidno.7),pna钳铗:
nh2-tgggctggccaa-cooh(seqidno.8),内围正向引物:
acgagtgagccttgggc(seqidno.9)),每个靶点都得到了相应的富集,而且相互之间没见到有任何干扰。这意味着只需要很少的临床标本,就可以检测到多个靶基因的检测。
实施例二
用微滴数字pcr检测:如图4a所示,在没有预放大的情况下,微滴数字pcr尽管灵敏度高,但要检测出1:1万倍用野生型稀释后的个别突变基因也很受限,下结论会很勉强。而预放大富集后,只需用1/10的反应量。我们可以选择内围引物做微滴数字(ggagtaatagctgcagctc,seqidno.5)pcr。这个因为同超敏引物(seqidno.1)3'末端的19个碱基(斜体部分)完全匹配,它只会放大从预放大中得到的产物,以保证进一步pcr的特异性和降低噪音。
做微滴数字pcr的反应成分与步骤完全按照生产商的指示。使用美国bio—rad公司的qxl00tm型微滴式数字pcr系统(包括微滴发生板、微滴发生器、pcr仪、微滴分析仪)检测。所有样本进行微滴式数字pcr,pcr反应体系:2xddpcr探针反应混合液、引物(250nm)、pna钳铗(100nm-1.5μm)、探针(125nm),模板dna(1-2μl预防大产物),去离子水补足至20μl。将配置充分混匀的反应混合液加入8通道微滴发生板,每通道加入70μl油滴,将微滴发生板放入微滴发生器,将反应混合液制成约20,000个微滴。将微滴转移入96孔pcr反应板,用铝箔热封,置入热循环仪(t100,bio-rad)中进行pcr,pcr反应条件:95℃10min;95℃30s,70℃20s,60℃60s,45个循环;98℃10min。将pcr反应后的96孔pcr反应板放入微滴分析仪(qx100,bio-rad),仪器会自动顺序吸取每个样品的微滴并随微滴读取油逐一通过双色检测器,有荧光信号的微滴为阳性,无荧光信号的为阴性,软件记录每个样品里阳性和阴性微滴数。
经预放大后再用微滴数字pcr检测,即使在1:10万与野生型稀释后,也可以轻易地检测出来(图4b)。
尽管这项技术适用于检测多个突变基因的预放大(multiplex),但并不作为用来筛查癌症的首选方法.在临床实际应用中,针对活检或手术后的肿瘤标本,首先建议用二代测序法对常见的突变基因的位点测序。然后根据其结果,选用已有的或设计新的检测引物和探针及钳铗,对5个左右的位点做血液、尿液、唾液等的检测,随着时间进行检测。
以上所述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案作出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
sequencelisting
<110>济南徕富尚圣生物科技有限公司
<120>一种超敏感引物及其设计方法和应用
<130>
<160>9
<170>patentinversion3.5
<210>1
<211>36
<212>dna
<213>正向引物
<400>1
ctgggcatctgcctataggagtaatagctgcagctc36
<210>2
<211>23
<212>dna
<213>反向引物
<400>2
tctttgtgttcccggacatagtc23
<210>3
<211>14
<212>dna
<213>pna
<400>3
ctcatcacgcagct14
<210>4
<211>20
<212>dna
<213>正向引物
<400>4
tcacctccaccgtgcagctc20
<210>5
<211>19
<212>dna
<213>正向引物
<400>5
ggagtaatagctgcarctc19
<210>6
<211>34
<212>dna
<213>正向引物
<400>6
acaccgcagcatgtcgcacgagtgagccttgggc34
<210>7
<211>20
<212>dna
<213>反向引物
<400>7
ccttactttgcctccttctg20
<210>8
<211>12
<212>dna
<213>pna
<400>8
tgggctggccaa12
<210>9
<211>17
<212>dna
<213>正向引物
<400>9
acgagtgagccttgggc17
- 还没有人留言评论。精彩留言会获得点赞!