聚乙二醇-聚丙交酯嵌段共聚物的制备方法以及药物载体与流程
本发明涉及生物医药材料技术领域,特别涉及一种聚乙二醇-聚丙交酯嵌段共聚物的制备方法以及药物载体。
背景技术:
聚合物胶束是由两亲性的嵌段聚合物材料在水中达到一定浓度后,自发形成的一种药物传递系统,其中两亲性聚合物材料包括亲水性链和疏水性链,其中亲水性链的空间长度一般大于疏水性链,以便在水中排列成核壳结构。其中,疏水链段形成的疏水核,可容纳水难溶性的药物;而亲水链形成胶束的溶剂化的亲水壳层,可以提高胶束在体内的稳定性。两亲性嵌段聚合物的碳链长,并且相对分子量大,所以胶束的临界胶束浓度非常低,即聚合物材料的浓度即使是很低时也会形成胶束;又由于疏水片段之间的相互作用使胶束的核心紧密而且稳定,故当材料的浓度被稀释到浓度低于临界胶束浓度时,胶束的解聚也是很缓慢的过程,因此,有较好的动力学稳定性,即使经过血液大量稀释,也能将药物定向输送到体内的靶部位,因此,聚合物胶束作为水难溶药物的载体,其优点有以下几点:(1)目前研究的嵌段聚合物多数为生物相容和可降解的材料,便于体内降解和排除,故生物毒性小;(2)亲水性壳层的一些活性基团可连接靶向配体,实现药物的主动靶向;(3)聚合物胶束的粒径较小,容易通过肿瘤的血管壁,达到肿瘤的被动靶向效果;(4)有些粒径大的胶束粒子能够被巨噬细胞吞噬,也可以具有被动靶向能力;(5)聚合物胶束的粒径大小和表面特征经过修饰可避免网状内皮系统识别,延长在体内的作用时间,达到长循环的目的。
聚乙二醇(polyethyleneglycol,peg)因具有两亲性、生物相容性、无免疫原性和低毒性等优点而被广泛用作药物载体。peg含两个活性羟基,用于化学修饰时容易引起不期望的交联,因而目前在peg的药物修饰研究中,大多使用的是只具有一个活性羟基的聚乙二醇单甲醚(mpeg),而不会直接以具有两个活性羟基的peg为原料来制备用于聚合物胶束药物载体的高分子聚合物材料,但是使用mpeg的原料成本远高于使用具有两个活性羟基的peg。
技术实现要素:
本发明的主要目的是提出一种聚乙二醇-聚丙交酯嵌段共聚物的制备方法以及药物载体,旨在提供一种以具有两个活性端基的聚乙二醇作为原材料合成带有羧基端的聚乙二醇-聚丙交酯嵌段共聚物的方法。
为实现上述目的,本发明提出一种聚乙二醇-聚丙交酯嵌段共聚物的制备方法,包括以下步骤:
步骤s10、对聚乙二醇进行重结晶处理,得到重结晶后的聚乙二醇;
步骤s20、以重结晶后的聚乙二醇为原料,合成聚乙二醇羧酸酯,然后将聚乙二醇羧酸酯水解,得到生成有单羧基聚乙二醇和双羧基聚乙二醇的混合物;
步骤s30、分离出所述羧基化聚乙二醇混合物中的单羧基聚乙二醇;
步骤s40、将单羧基聚乙二醇与l-丙交酯在催化剂作用下进行聚合反应,得到聚乙二醇-聚丙交酯嵌段共聚物。
优选地,在步骤s10中,所述聚乙二醇为聚乙二醇2000、聚乙二醇4000或聚乙二醇6000。
优选地,步骤s10具体包括:
将聚乙二醇粉末加入异丙醇中后加热至40~50℃并搅拌,形成聚乙二醇的异丙醇溶液,然后将所述聚乙二醇的异丙醇溶液在3~5℃下静置22~26h后过滤,收集滤饼并干燥成粉末,得到重结晶后的聚乙二醇;其中,所述聚乙二醇的异丙醇溶液中,聚乙二醇的浓度为8~12g/700ml。
优选地,步骤s20中所述的合成聚乙二醇羧酸酯的步骤,具体包括:
步骤s21、将重结晶后的聚乙二醇与无水甲苯混合,形成混合液,然后将所述混合液加热至118~122℃共沸50~70min后,降温至48~52℃;
步骤s22、在氮气保护下,向降温后的所述混合液中加入叔丁醇钾,并使所述叔丁醇钾完全溶解以形成混合溶液,并将所述混合溶液降温至23~27℃;
步骤s23、在氮气保护下,向降温后的所述混合溶液中加入溴乙酸乙酯,混合形成待反应溶液,将所述待反应溶液加热至108~112℃回流反应10~14h,得到形成有固体的反应溶液,分离并除去所述产物溶液中的所述固体,得到反应产物;
步骤s24、将所述反应产物浓缩后加入3~5℃的乙醚中进行剧烈搅拌,然后过滤并收集滤渣,再将所述滤渣干燥成粉末,得到聚乙二醇羧酸酯。
优选地,步骤s20中所述的将所述聚乙二醇羧酸酯水解的步骤,具体包括:
步骤s25、将聚乙二醇羧酸酯溶解于氢氧化钠水溶液中,然后加入氯化钠,得到碱性溶液,将所述碱性溶液加热至43~47℃下保温50~70min后,冷却至20~25℃;
步骤s26、使用稀盐酸调节冷却后的所述碱性溶液的ph值至2.5~3.5后,以二氯甲烷为溶剂对所述碱性溶液进行萃取,收集下层有机相并用硫酸镁干燥,然后过滤所述有机相并收集滤液;
步骤s27、将所述滤液浓缩后加入温度为3~5℃的乙醚中进行剧烈搅拌,然后过滤并收集沉淀物,再对所述沉淀物进行干燥,得到羧基化聚乙二醇混合物。
优选地,步骤s30具体包括:
步骤s31、将所述羧基化聚乙二醇混合物溶解于水中后,加入装有阴离子交换剂的层析柱中,然后进行洗脱并收集洗脱液;
步骤s32、将经碘缸显色视检呈单一同一色点的洗脱液合并后浓缩形成浓缩液,向所述浓缩液中加入氯化钠,然后调节所述浓缩液的ph至2.5~3.5后,以二氯甲烷为溶剂对所述浓缩液进行萃取,收集有机层并用硫酸镁干燥后再次浓缩;
步骤s33、将再次浓缩后的溶液加入温度为3~5℃的乙醚中进行剧烈搅拌,然后过滤并收集沉淀,再对所述沉淀进行干燥,得到单羧基聚乙二醇。
优选地,步骤s31具体包括:
对阴离子交换剂进行预处理后装填入层析柱中,然后将所述羧基化产物溶解于水中的溶液加入所述层析柱内,先使用去离子水进行洗脱,然后依次采用摩尔浓度为6mm、10mm、14mm、18mm以及22mm的碳酸氢铵溶液进行洗脱,收集洗脱液;其中,所述阴离子交换剂为deaesephadexa-25。
优选地,步骤s40具体包括:
步骤s41、将单羧基聚乙二醇、l-丙交酯和催化剂溶解于甲苯中,然后在氮气环境下加热至108~112℃持续反应22~26h,得到生成有白色聚合物的反应液;
步骤s42、向所述反应液中加入乙醚并剧烈搅拌,然后过滤并收集所述白色聚合物,再对所述白色聚合物进行干燥,得到聚乙二醇-聚丙交酯嵌段共聚物。
优选地,在所述步骤s41中:
所述催化剂为辛酸亚锡,且所述辛酸亚锡的添加量为所述l-丙交酯的质量的0.8~1.2%。
本发明还提出一种药物载体,所述药物载体包括聚合物胶束,所述聚合物胶束由如上所述的聚乙二醇-聚丙交酯嵌段共聚物制备而成。
本发明提供的技术方案中,采用具有两个活性羟基的普通聚乙二醇为原料,经过羧基化处理后通过分离纯化,获得单羧基聚乙二醇,然后将单羧基聚乙二醇与l-丙交酯通过聚合反应成功合成了带有羧基端的聚乙二醇-聚丙交酯嵌段共聚物,降低了制备聚乙二醇-聚丙交酯嵌段共聚物的原料成本。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅为本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
图1为本发明实施例中peg2000的红外光谱图;
图2为本发明实施例中制备的单羧基peg的红外光谱图;
图3为本发明实施例中制备的双羧基peg的红外光谱图;
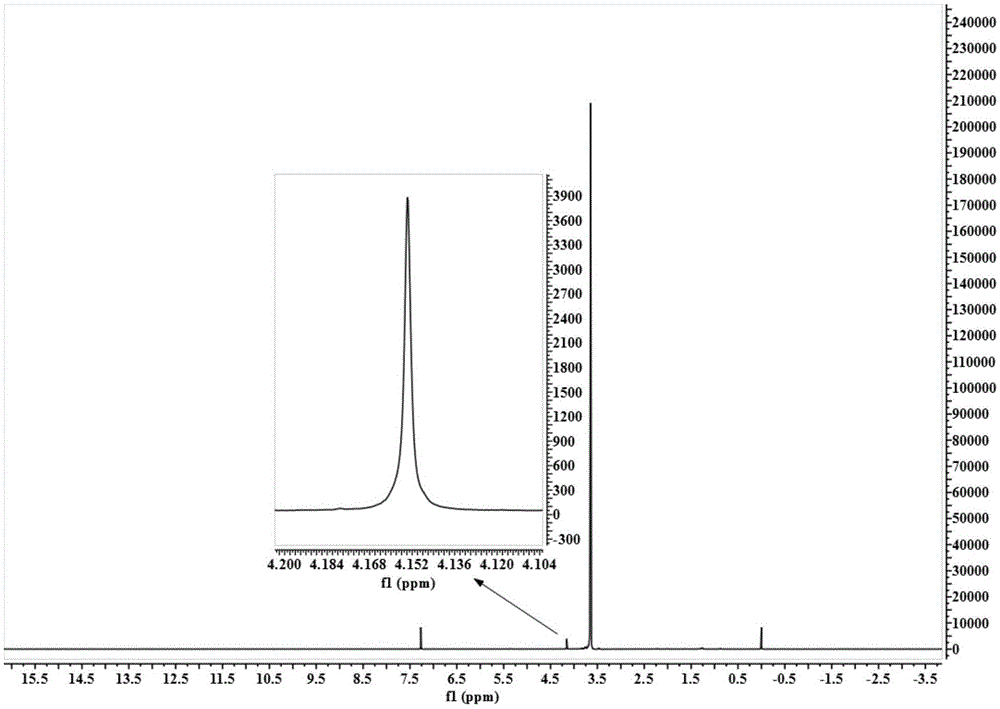
图4为本发明实施例中制备的单羧基peg的1hnmr图;
图5为本发明实施例中制备的双羧基peg的1hnmr图;
图6为本发明实施例中制备的双羧基peg的红外光谱13cnmr图;
图7为本发明实施例中制备的peg-pla嵌段共聚物的红外光谱图;
图8为本发明实施例中制备的peg-pla嵌段共聚物的1hnmr图;
图9为本发明实施例中制备的peg-pla嵌段共聚物的13cnmr图;
图10为本发明实施例中制备的peg-pla嵌段共聚物的临界胶束浓度的测算曲线图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
聚乳酸(polylacticacid,pla),又称为聚丙交酯,因其具有生物相容性和生物可降解性而一直作为药物载体的研究热点。乳酸有d(+)和l(-)两种旋光异构体,等量的d(+)和l(-)两种异构体混合而成的d,l-乳酸不具有旋光性,因此有3种不同旋光性的乳酸。丙交酯从旋光性看有4种:(l)两个l-乳酸分子脱水形成的丙交酯称为l,l-丙交酯(或简称为l-丙交酯);(2)两个d-乳酸分子脱水形成的丙交酯称为d,d-丙交酯(或简称为d-丙交酯);(3)一个l-乳酸分子和一个d-乳酸分子脱水形成的丙交酯称为内消旋d,l-丙交酯(又称meso-丙交酯);(4)等量的l-和d-丙交酯(熔点均为96℃)混合,形成外消旋d,l-丙交酯(熔点127℃,以下简称d,l-丙交酯)。pla(包括plla、pdla、pdlla)在体内的代谢终产物是co2和h2o,plla是l-丙交酯聚合而成,因其具有更好的生物相容性,其应用更为广范。
聚乙二醇(polyethyleneglycol,peg)因具有两亲性、生物相容性、无免疫原性和低毒性等优点而被广泛用作药物载体。peg含两个活性羟基,用于化学修饰时容易引起不期望的交联,因而目前在peg的药物修饰研究中,大多使用的是只具有一个活性羟基的聚乙二醇单甲醚(mpeg),但是mpeg的成本比peg的成本高,使得以mpeg作为原料制备而成的聚合物胶束药物载体的成本较高,不利于推广使用。
本发明提出一种peg-pla嵌段共聚物的制备方法,以具有两个活性羟基的普通peg为原料,经过羧基化将peg制成单羧基peg,再将l-丙交酯与单羧基peg合成带羧基端的peg-pla,羧基端可作为活性反应基团连接叶酸等主动靶向基团,达到主动靶向的作用,进而可以用于能够实现药物靶向的药物载体。在本发明提供的peg-pla嵌段共聚物的制备方法的一实施例中,所述peg-pla嵌段共聚物的制备方法包括以下步骤:
步骤s10、对peg进行重结晶处理,得到重结晶后的peg;
在本实施例中,所述聚乙二醇(polyethyleneglycol,peg)为具有两个活性端基的普通peg,优选为peg2000、peg4000或peg6000。在本发明的其他实施例中,也可以选用peg5000或peg3000等型号的peg。以下将以所述peg选用peg2000为例进行说明。
重结晶是将晶体溶于溶剂或熔融以后,又重新从溶液或熔体中结晶的过程,可以使不纯净的物质获得纯化,或使混合在一起的盐类彼此分离。通过先对peg进行重结晶处理,可以去除peg原料中的杂质,提高peg的纯度,有利于提高后续合成产物的纯度。进一步地,在本实施例中,对peg进行重结晶处理可以采用以下方式:将peg2000粉末加入异丙醇中后加热至40~50℃(优选为水浴加热)并搅拌,使peg2000完全溶解于异丙醇中而形成peg2000的异丙醇溶液,然后将peg2000的异丙醇溶液在3~5℃下静置22~26h后过滤(在本发明实施例中更优选为在4℃下静置24h),收集滤饼并干燥成粉末,得到重结晶后的peg2000;其中,所述peg2000的异丙醇溶液中,所述peg2000的浓度为8~12g/700ml,也即,每8~10g的peg2000对应加入700ml的异丙醇进行溶解。
所述滤饼的干燥方式有多种,可以通过鼓风干燥、真空干燥或微波干燥等方式进行,以将所述滤饼干燥成粉末状为准,在本实施例中,所述滤饼的干燥方式为:将所述滤饼先放入温度设置为42~48℃的鼓风干燥器中干燥22~26h,然后再将所述滤饼转移至温度设置为38~42℃的真空干燥箱中干燥46~50h,所述滤饼即被干燥成粉末状,完成所述滤饼的干燥处理。
步骤s20、以重结晶后的peg为原料,合成peg羧酸酯,然后将所述peg羧酸酯水解,得到生成有单羧基peg和双羧基peg的混合物;
peg2000经过重结晶处理后,再通过羧基化处理合成带有单羧基和双羧基的peg,在本实施例中,所述羧基化处理包括以重结晶后的peg为原料合成peg羧酸酯,然后再将peg羧酸酯水解,生成羧基化peg混合物,所述混合物中同时含有单羧基peg和双羧基peg。
进一步地,在本实施例中,步骤s20中所述的合成peg羧酸酯的步骤,具体包括:
步骤s21、将重结晶后的peg与无水甲苯混合,形成混合液,然后将所述混合液加热至118~122℃共沸50~70min后,降温至48~52℃;
步骤s22、在氮气保护下,向降温后的所述混合液中加入叔丁醇钾,并使所述叔丁醇钾完全溶解以形成混合溶液,并将所述混合溶液降温至23~27℃;
步骤s23、在氮气保护下,向降温后的所述混合溶液中加入溴乙酸乙酯,混合形成待反应溶液,将所述待反应溶液加热至108~112℃回流反应10~14h,得到形成有固体的反应溶液,分离并除去所述产物溶液中的所述固体,得到反应产物;
步骤s24、将所述反应产物浓缩后加入3~5℃的乙醚中进行剧烈搅拌,然后过滤并收集滤渣,再将所述滤渣干燥成粉末,得到peg羧酸酯。
在步骤s21至步骤s24中,主要发生的反应过程如下:
优选地,在步骤s21至步骤s24中,重结晶后的peg2000与无水甲苯的固液比为25g/120ml,所述叔丁醇钾的添加量为2.512g(也即22mmol),所述溴乙酸乙酯的添加量为4.7ml(也即22mmol)。在步骤s21至步骤s23中,重结晶后的peg与无水甲苯混合后,加入叔丁醇钾和溴乙酸乙酯进行回流反应的过程可以在油浴锅中进行,不仅能达到加热至100℃以上的温度要求,且加热过程中的温度变化较为缓和,避免了温度的急剧变化。在步骤s24中,所述滤渣的干燥方式同样可以采用鼓风干燥、真空干燥、微波干燥等方式,在本实施例中优选为采用真空干燥,具有干燥效率高、不会影响被干燥物质本身性能的优点,具体做法为:将所述滤渣放入温度设置为38~42℃的真空干燥箱内干燥10~14h,即将所述滤渣干燥成为粉末状。
在本发明提供的其他实施例中,步骤s20中所述的合成peg羧酸酯的过程中也可以采用例如高锰酸钾等强氧化剂进行,但是其反应过程不易控制,且peg链容易氧化断裂,而导致peg的聚合度发生变化;反应过程中所采用的溶剂也可以用四氢呋喃或者三氯甲烷等替换,并对应调整反应温度和反应时间,例如以四氢呋喃为反应溶剂时,反应温度为60~70℃、反应时间为24h以上,反应耗时较长,且出于对溶剂的毒性及挥发性的考虑,优选毒性相对较低的甲苯作为反应溶剂,其反应温度为108~112℃、反应时间为10~14h,缩短了反应所需要的时间。
更进一步地,在本实施例中,步骤s20中所述的将所述peg羧酸酯水解的步骤,具体包括:
步骤s25、将peg羧酸酯溶解于氢氧化钠水溶液中,然后加入氯化钠,得到碱性溶液,将所述碱性溶液加热至43~47℃下保温50~70min后,冷却至20~25℃;
步骤s26、使用稀盐酸调节冷却后的所述碱性溶液的ph值至2.5~3.5后,以二氯甲烷为溶剂对所述碱性溶液进行萃取,收集下层有机相并用硫酸镁干燥,然后过滤所述有机相并收集滤液;
步骤s27、将所述滤液浓缩后加入温度为3~5℃的乙醚中进行剧烈搅拌,然后过滤并收集沉淀物,再对所述沉淀物进行干燥,得到peg的羧基化产物。
在步骤s25至步骤s27中,主要发生的反应过程如下:
优选地,在步骤s25中,所述氢氧化钠水溶液的当量浓度为1n(也即,所述氢氧化钠水溶液的摩尔浓度为1mol/l);在步骤s26中,所述稀盐酸的当量浓度为6n(也即,所述稀盐酸的摩尔浓度为6mol/l),使用二氯甲烷进行萃取的次数优选为3次;在步骤s27中,所述沉淀物的干燥方式同样可以采用鼓风干燥、真空干燥、微波干燥等方式,在本实施例中优选为采用真空干燥,具有干燥效率高、不会影响被干燥物质本身性能的优点,具体做法为:将所述沉淀物放入温度设置为38~42℃的真空干燥箱内干燥10~14h即可。
步骤s30、分离出所述混合物中的单羧基peg;
对peg进行羧基化之后,产物中生成有带单羧基的peg和带双羧基的peg,分别为单羧基peg和双羧基peg,此时,需要通过分离和纯化处理,从而获得单羧基peg,以用于后续与l-丙交酯的聚合反应,而双羧基peg则可用作其他反应的活性基团。优选地,在本实施例中,采用离子交换层析的方法从peg的羧基化产物中分离并纯化单羧基peg,具体操作包括以下步骤:
步骤s31、将所述羧基化peg混合物溶解于水中后,加入装有阴离子交换剂的层析柱中,然后进行洗脱并收集洗脱液;
所述阴离子交换剂包括带阴离子基团如deae(二乙基胺乙基)和qae(四级胺乙基)等的阴离子交换剂,在本实施例中优选为二乙基胺乙基葡聚糖凝胶a-25(即deaesephadexa-25),对于单羧基peg的分离效率更高。
离子交换层析的操作一般包括交换剂预处理、装层析柱、加样以及洗脱,可以按照现有技术中离子交换层析的操作方法进行,也可以按照离子搅拌层析的基本原理、以及交换剂的选择来设计具体的操作方法。在本实施例中,以deaesephadexa-25作为阴离子交换剂,通过层析柱进行离子交换层析的操作流程为:对阴离子交换剂进行预处理后装填入层析柱中,然后将所述羧基化产物溶解于水中的溶液加入所述层析柱内,先使用去离子水进行洗脱,然后依次采用摩尔浓度为6mm、10mm、14mm、18mm以及22mm的碳酸氢铵溶液进行洗脱,收集洗脱液。
进一步地,在本实施例中,以deaesephadexa-25作为阴离子交换剂,通过层析柱进行离子交换层析的具体操作步骤包括:(1)阴离子交换剂预处理:将deaesephadexa-25干粉25g缓缓加入700ml的沸水中,并用玻璃棒搅拌至deaesephadexa-25分散均匀后停止搅拌,静置3h,使deaesephadexa-25自然膨胀并冷却、分层,然后弃去上层的大部分水及上层漂浮物,保留下层膨胀后的交换剂,即完成阴离子交换就的预处理。(2)层析柱装填:将层析柱垂直固定于滴定铁架上,柱底放入一小团棉花,关闭层析柱的出液口,然后向层析柱内加入约100ml的去离子水,并打开阀门排出气泡,排完气泡后关闭阀门,此时,在层析柱底端保留约5ml的一段液体;用玻璃棒缓慢搅拌膨胀后的交换剂以形成匀浆,将所述匀浆沿着层析柱内壁一次性倒入层析柱内,然后打开出液口,使交换剂在层析柱内自由沉降,至交换剂上留有月5cm的澄清液体时关闭阀门。(3)加样:将15g羧基化peg混合物加入小烧杯内,并加入20ml的去离子水中,使羧基化peg混合物溶解形成羧基化peg的水溶液;然后打开层析柱的阀门,直至上层清液约为1cm时,关闭阀门,并将羧基化peg的水溶液缓慢倒入层析柱内,每次使用大约5ml去离子水润洗小烧杯并将润洗液加入层析柱内。(4)洗脱:先用100ml的去离子水洗脱,然后依次使用100ml的摩尔浓度分别为6mm、10mm、14mm和18mm的碳酸氢铵梯度溶液洗脱,最后再用300ml的摩尔浓度为22mm的碳酸氢铵溶液洗脱,用试管收集洗脱液,每管收集约20ml。
经过离子交换层析后,羧基化peg混合物被分离为单羧基peg和双羧基peg,然后通过碘缸显色视检筛选出其中的单羧基peg并进行纯化,即可得到单羧基peg。
步骤s32、将经碘缸显色视检呈单一同一色点的洗脱液合并后浓缩形成浓缩液,向所述浓缩液中加入氯化钠,然后调节所述浓缩液的ph至2.5~3.5后,以二氯甲烷为溶剂对所述浓缩液进行萃取,收集有机层并用硫酸镁干燥后再次浓缩;
步骤s33、将再次浓缩后的溶液加入温度为3~5℃的乙醚中进行剧烈搅拌,然后过滤并收集沉淀,再对所述沉淀进行干燥,得到单羧基peg。
将经碘缸显色视检呈单一同一色点的洗脱液分别收集并合并,然后经过浓缩、酸化、有机溶剂萃取、纯化和干燥处理后,对应得到单羧基peg和双羧基peg,从而有效地将单羧基peg从羧基化peg混合物中分离出来,分离率高且操作简便。优选地,在步骤s32中,使用二氯甲烷进行萃取的次数优选为3次;在步骤s33中,所述沉淀的干燥方式同样可以采用鼓风干燥、真空干燥、微波干燥等方式,在本实施例中优选为采用真空干燥,具有干燥效率高、不会影响被干燥物质本身性能的优点,具体做法为:将所述沉淀放入温度设置为38~42℃的真空干燥箱内干燥过夜即可。
步骤s40、将单羧基peg与l-丙交酯在催化剂作用下进行聚合反应,得到peg-pla嵌段共聚物。
在催化剂作用下,使单羧基peg与l-丙交酯发生开环聚合反应得到peg-pla嵌段共聚物之后,还需要对反应产物进行纯化,因此,在本实施例中,步骤s40包括peg-pla嵌段共聚物的合成以及纯化,具体包括以下步骤:
步骤s41、将单羧基peg、l-丙交酯和催化剂溶解于甲苯中,然后在氮气环境下加热至108~112℃持续反应22~26h,得到生成有白色聚合物的反应液;
在本实施例中,所述催化剂优选为辛酸亚锡,且所述辛酸亚锡的添加量为所述l-丙交酯的质量的0.8~1.2%,所述单羧基peg与所述l-丙交酯反应生成peg-pla嵌段聚合物的反应过程可以在dean-start装置中进行,反应过程如下:
步骤s42、向所述反应液中加入乙醚并剧烈搅拌,然后过滤并收集所述白色聚合物,再对所述白色聚合物进行干燥,得到peg-pla嵌段共聚物。
本发明提供的技术方案中,采用具有两个活性端基的普通peg为原料,经过羧基化处理后通过分离纯化,得到较大量的单羧基peg和双羧基peg,工艺方法简单、成本低廉、分离率和产物得率均较高,然后将单羧基peg与l-丙交酯通过聚合反应成功合成了peg-pla嵌段共聚物,其合成方法简单、产率高,且不需要特殊的纯化方法即可获得纯度较高的产物,所合成的聚合物的临界胶束浓度小,可作为制备聚合物胶束药物载体的材料,降低了用于制备聚合物胶束药物载体的高分子聚合物材料的成本,而且,所合成的聚合物在用作聚合物胶束药物载体时,其羧基端可作为活性反应基团连接叶酸等主动靶向基团,达到主动靶向的作用,实现药物的主动靶向。
得到的高分子聚合物的临界胶束浓度小,可作为制备聚合物胶束药物载体的材料,相比于采用mpeg为原料进行合成,降低了用于制备聚合物胶束药物载体的高分子聚合物材料的成本。
本发明还提出一种药物载体,所述药物载体包括聚合物胶束,所述聚合物胶束由如上所述的peg-pla嵌段共聚物制备而成。所述聚合物胶束的制备方法参考现有技术进行,在此不做赘述。在本发明提供的药物载体的一实施例中,所述聚合物胶束为采用上述peg-pla嵌段共聚物的制备方法制备而成的peg-pla嵌段共聚物溶解于水中形成的peg-pla水溶液,其临界胶束浓度(criticalmicellesconcentration,cmc)值可达2.64×10-3g/l,其cmc值远小于常用的低分子量表面活性剂的cmc值,所形成的胶束在体液中不易缔合、稳定性好,适宜于用作聚合物胶束药物载体,实现药物的主动靶向,尤其适合于作为静脉输送药物的载体。
以下结合具体实施例和附图对本发明的技术方案作进一步详细说明,应当理解,以下实施例仅仅用以解释本发明,并不用于限定本发明。
实施例
(1)peg重结晶
精密称取peg2000粉末(麦克林)30g放入圆底烧瓶,然后向圆底烧瓶中加入2100ml异丙醇,盖上塞子,在45℃水浴锅中振荡,使peg2000完全溶解;待peg2000完全溶解后,放入温度设置为4℃的冰箱中静置24h后取出,然后立即用布氏漏斗过滤,收集乳白色滤饼;将滤饼放入鼓风干燥器中,在45℃下干燥24h,再将滤饼转移到真空干燥箱中,在40℃下干燥48h。称量干燥后的滤饼粉末质量,按下式计算重结晶收率:
重结晶收率(%)=重结晶后得到的peg质量/重结晶前称取的peg质量×100
按照上述计算公式计算出peg2000的重结晶收率为84.70%,而且在重结晶的过程中,重结晶后过滤的速度很慢,且过滤刚开始时容易漏液造成回收的peg2000的量减少,收率偏低。
(2)合成peg羧酸酯
精密称取上述步骤(1)得到的peg200025g放入圆底烧瓶,然后向圆底烧瓶中加入120ml无水甲苯,置于油浴锅中并磁力搅拌,加热至120℃共沸1h并通入氮气;将油浴温度设置为50℃,待温度降至设置温度后,向圆底烧瓶中加入2.512g(22mmol)叔丁醇钾,等待30min,使固体完全溶解;再将油浴温度设置为25℃,待温度降至设置温度后,加入4.7ml溴乙酸乙酯(22mmol),混匀后,加热至110℃回流反应12h;待反应完毕后,过滤除去反应产生的固体,用30ml二氯甲烷冲洗滤渣,并将滤液于70℃旋转蒸发,浓缩至大约60ml;将浓缩液缓慢倒入300ml的温度为5℃的乙醚中,同时剧烈搅拌,然后过滤并收集滤渣,将滤渣在40℃温度下真空干燥12h,得到浅黄色固体粉末,即为peg羧酸酯,产量为25.81g。
(3)水解peg羧酸酯
将上述步骤(2)得到的浅黄色固体粉末溶解于50ml的naoh水溶液(1n)中,再加入10gnacl,置于45℃中水浴1h,然后冷却至室温,用约8ml的6n稀盐酸酸化溶液,并搅拌,用ph试纸检测,至ph为3;然后每次用60ml的二氯甲烷萃取,共萃取3次,收集下层有机相,用mgso4干燥有机相;过滤有机相,然后于30℃旋转蒸发,浓缩至约50ml;将浓缩液缓缓加入300ml的4℃冷乙醚中,并剧烈搅拌,然后过滤并收集沉淀物,将沉淀物在40℃温度下真空干燥24h,得到生成有单羧基peg和双羧基peg的混合物,产量为23.52g,为白色固体粉末。
(4)单羧基peg的制备
①阴离子交换剂预处理:将700ml去离子水用电炉加热至沸腾,然后称取deaesephadexa-25干粉(叶源生物科技有限公司)25g,缓缓加入沸水中,并用玻璃棒缓慢搅拌,至deaesephadexa-25干粉分散均匀后停止搅拌,静置3h,使其自然膨胀,冷却,分层,再倾去上层的大部分水及上层漂浮物,保留下层约250ml膨胀后的交换剂。
②层析柱装填:将层析柱垂直固定于滴定铁架上,向柱底放入一小团棉花,关闭出液口,向层析柱中加入约100ml去离子水并打开阀门排气泡,待排完气泡后,关闭阀门,使柱子底端保留约5cm的一段液体;用玻璃棒缓慢搅匀膨胀后的交换剂至形成匀浆,将匀浆沿着层析柱内壁一次性倒入柱内,打开柱子出液口,使交换剂在柱内自由沉降,至交换剂上留有约5cm的澄清液体时关闭阀门。
③加样:精密称取15g上述步骤(3)得到的羧基化peg混合物,转移至50ml小烧杯中,加入20ml去离子水,使羧基化peg混合物溶解形成羧基化peg水溶液;打开层析柱阀门,直至上层清液约1cm时,关闭阀门,并将羧基化peg水溶液缓慢加入层析柱内,每次用大约5ml去离子水润洗小烧杯,并将润洗液加入柱内。
④洗脱:先用100ml的去离子水洗脱,然后依次使用100ml的摩尔浓度为6mm、10mm、14mm和18mm的碳酸氢铵梯度溶液洗脱,最后用300ml的摩尔浓度为22mm的碳酸氢铵溶液洗脱,用试管收集洗脱液,每管收集约20ml。
⑤薄层色谱法检测洗脱液组份:分别精密称取10mgpeg2000粉末和羧基化peg混合物,转移到离心管中,分别编号为1号样品和2号样品;分别向1号样品和2号样品的管中加入1ml去离子水,振摇至固体完全溶解,形成1号样品溶液和2号样品溶液;配制异丙醇/25%氨水=5/1(v/v)的展开剂,用配制的1号样品溶液和2号样品溶液以及收集在试管中的洗脱液点样,干燥后,放入碘缸显色视检。
⑥单羧基peg的分离和纯化:将收集的含有单一同一个色点的洗脱液分别合并,各浓缩至约50ml;向加入浓缩液中加入20g氯化钠,酸化浓缩液至ph为3,然后每次使用50ml的二氯甲烷萃取,共萃取3次,收集有机层,将收集的有机层用mgso4干燥,然后再次浓缩至约50ml,将再次浓缩后的浓缩液缓慢加入300ml的5℃乙醚中搅拌,过滤并收集沉淀,将沉淀在40℃温度下真空干燥过夜,即对应获得单羧基peg(peg-ch2-cooh)和双羧基peg(cooh-ch2-peg-ch2-cooh)。其中,层析柱的加样量为15g的羧基化peg混合物,经过离子交换层析后,单羧基peg的产量为5.86g,得率为39.1%,双羧基peg的产量为6.12g,得率为40.8%,单羧基peg和双羧基peg的总得率为79.9%。
通过傅立叶红外光谱仪(670ft-ir,thermonicoletnexustm)对上述步骤(4)制得的单羧基peg和双羧基peg进行结构表征,分别以peg2000和制得的peg-ch2-cooh和cooh-ch2-peg-ch2-cooh作为表征对象,采用kbr压片法进行检测,检测结果如图1至图3所示,图1至图3依次为peg2000、peg-ch2-cooh和cooh-ch2-peg-ch2-cooh的红外光谱图。由图1至图3可知,peg无端羧基,故在1700cm-1无吸收峰;peg-ch2-cooh在1700cm-1附近有吸收峰,为羰基吸收峰,而cooh-ch2-peg-ch2-cooh在1700cm-1附近有两个羰基吸收峰,且羰基吸收峰比peg-ch2-cooh的强。
通过核磁共振波谱仪(brukerbiospingmbh)对上述步骤(4)制得的单羧基peg和双羧基peg进行结构表征,采用氘代氯仿作为溶剂进行检测,检测结果如图4至图6所示,图4为peg-ch2-cooh的1hnmr图(氢谱图),图5和图6分别为cooh-ch2-peg-ch2-cooh的1hnmr图(氢谱图)和13cnmr图(碳谱图)。由图4和图5可知,peg-ch2-cooh的重复单元o-ch2-ch2-o亚甲基质子峰化学位移为3.6;端基羧乙基的亚质子峰化学位移为4.11。hooc-ch2-peg-ch2-cooh的重复单元o-ch2-ch2-o亚甲基质子峰化学位移为3.64;端基羧乙基的亚质子峰化学位移为4.15。hooc-ch2-peg-ch2-cooh的重复单元o-ch2-ch2-o亚甲基碳化学位移为77.35、77.03、76.71、71.37和70.57,端基羧乙基的亚甲基碳化学位移为68.89,基羧极端的碳化学位移为171.52。
综合图1至图6中对实施例制备的单羧基peg和双羧基peg的结构检测结果,说明本实施例成功制得了带有单羧基端的peg-ch2-cooh和带有双羧基端的cooh-ch2-peg-ch2-cooh,且peg-ch2-cooh得以从羧基化peg混合物中有效分离出来,以供后续与l-丙交酯进行进一步的合成。
(5)peg-pla嵌段聚合物的制备
①合成peg-pla嵌段聚合物:搭建dean-start装置后,将peg-ch2-cooh、l-丙交酯(70mmol)与辛酸亚锡(1%的l-丙交酯的重量)溶解于75ml的甲苯中,加热至60℃,待l-丙交酯完全溶解后,在氮气环境下,加热至110℃持续反应24h后停止加热,得到生成有peg-pla嵌段聚合物的反应液,冷却至常温。
②将冷却的反应液加入相当于反应液体积10倍的乙醚中,剧烈搅拌后过滤,收集的固体白色聚合物,真空干燥数日,即制得peg-pla嵌段聚合物(pla-peg-cooh)。
通过傅立叶红外光谱仪和核磁共振波谱仪对制备的peg-pla嵌段聚合物的结构进行表征,表征结果如图7至图9所示,图7至图9依次为pla-peg-cooh的红外光谱图、1hnmr图(氢谱图)和13cnmr图(碳谱图)。
图7中,自上至下依次为pla-peg-cooh、l-丙交酯(la)和peg-ch2-cooh的红外光谱,由图7可知,实施例制备的pla-peg-cooh的特征峰有:pla的c=o伸缩振动峰,位于1754.905cm-1;peg的c-c-o-c伸缩振动峰,位于1091.512cm-1;pla-peg-cooh的末端羧基c=o伸缩振动峰,位于1635.34cm-1;而实施例中制备的peg-ch2-cooh的特征峰为在1708.622cm-1处的末端羧基c=o伸缩振动峰。
由图8和图9可知,pla-peg-cooh的peg重复单元o-ch2-ch2-o亚甲基质子峰化学位移为3.67;pla-peg-cooh的pla重复单元的甲基质子化学位移为1.59,pla的ch化学位移为5.19。pla-peg-cooh的peg重复单元o-ch2-ch2-o亚甲基碳峰化学位移为69.02,其羧基碳化学位移为178.11;pla-peg-cooh的pla重复单元的甲基碳及叔碳化学位移分别为16.66和69.02;pla-peg-cooh的酯键羰基碳化学位移为169.61。
综合图7至图9中的表征结果,说明l-丙交酯与peg-cooh的羟基发生了开环聚合,而羧基羟基未发生反应,成功制备出带有一个羧基端的peg-pla嵌段聚合物。
采用凝胶渗透色谱仪,用dmf(二甲基甲酰胺)做流动相测定实施例制备的pla-peg-cooh的分子量,测定结果为:pla-peg-cooh的分子量为mw=2.4×104da。
以芘为荧光探针,用荧光方法测定pla-peg-cooh的临界胶束浓度(criticalmicellesconcentration,cmc),具体方法为:
取本实施例制备的pla-peg-cooh,分别配制浓度为1.0×10-1~1.0×10-7g/l的pla-peg-cooh水溶液待测。取10μl用丙酮配制的3.0×10-3m的芘贮备液,加入10ml的离心管中,待丙酮挥发完全,精密量取5ml待测溶液加入离心管中,充分震荡,在室温下避光静置24h,使得芘在待测溶液中的分布达到平衡,即得含芘浓度为6.0×10-6m的各浓度的待测液。以393nm为发射波长,记录激发波长在300~360nm区间的荧光强度,激发和发射的狭缝宽度分别为2nm和5nm。以338nm处与333nm处的荧光强度比值对胶束分散液浓度的对数作图来测算cmc,cmc值被定义为荧光强度发生突变时所对应的聚合物浓度,图10为以聚合物胶束的浓度的对数值为横坐标,以不同浓度pla-peg-cooh水溶液的芘在激发波长338nm处和333nm处的荧光强度比值为纵坐标作图所得的曲线。
由图10所示的结果表明,当pla-peg-cooh浓度低于cmc时,溶液中没有胶束存在,芘溶解在水中,i338/i333变化很小,曲线几乎为水平直线;当浓度达到cmc后,共聚物在溶液中逐渐形成胶束,芘会从水相中转移到亲油的胶束内部,由于芘所处的环境发生了改变,其荧光光谱也随之发生了改变,曲线急剧上升,i338/i333值增大。根据低浓度时i338/i333值的数据点所拟合的直线与高浓度时i338/i333值显著增大时的的数据点所拟合的直线,两条直线的交点的横坐标为cmc的对数值,由此计算出该聚合物的cmc值为2.64×10-3g/l。
综上所述,本发明实施例提供的peg-pla嵌段共聚物的制备方法,工艺简单,且所制得的peg-pla嵌段共聚物的产率和纯度均较高,其其重均分子量为2.4×104da,cmc为2.64×10-3g/l,表明所制备的聚合物具有一定的亲水性,能够形成稳定的胶束,其cmc远小于常用的低分子量表面活性剂的cmc,形成的胶束在体液中不易解缔合,稳定性好,适宜于用作聚合物胶束药物载体,实现药物的主动靶向,尤其适合于作为静脉输送药物的载体;而且,所制备的聚合物在用作聚合物胶束药物载体时,其羧基端可作为活性反应基团连接叶酸等主动靶向基团,达到主动靶向的作用,实现药物的主动靶向。
以上仅为本发明的优选实施例,并非因此限制本发明的专利范围,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!