一种四苯乙烯-二苯甲酮-咔唑衍生物、晶体及其制备方法与应用与流程
本发明涉及有机发光材料领域,更具体地,涉及一种四苯乙烯-二苯甲酮-咔唑衍生物、晶体及其制备方法与应用。
背景技术:
有机发光二极管(oled)因其重量轻,柔软性好,工作温度范围宽,反应时间短,亮度和对比度高,可视角宽等优点,在平板显示、智能手机以及固体发光等领域有着巨大的应用潜力。
发光材料是器件中最终承担发光功能的物质,因此发光材料的发光效率、发光寿命和发光色度等性质都对oled的性能产生直接的影响。作为oled中的发光材料应该具备如下条件:1)具有高效率的固态荧光,无明显的浓度猝灭现象;2)具有良好的化学稳定性和热稳定性,不与电极和载流子传输材料发生反应;3)容易形成致密的非晶态薄膜并且不易结晶;4)具有适当的发光波长;5)具有良好的导电特性及一定的载流子传输能力。
由于含四苯乙烯衍生物的发光材料聚集会导致荧光猝灭(acq效应),从而降低了发光强度,并且其制备难度大,这些问题限制了该类发光材料在发光器件制备领域中的规模化应用。
因此,需要制备一种能够削弱acq效应并且发光强度高的发光材料。
技术实现要素:
本发明为克服上述现有技术所述的发光材料具有acq效应和发光强度较低的缺陷,提供一种四苯乙烯-二苯甲酮-咔唑衍生物。由四苯乙烯-二苯甲酮-咔唑衍生物制备的晶体具有独特的aie效应、高的发光强度、良好的热稳定性和良好的溶解性。本发明的另一目的在于提供上述四苯乙烯-二苯甲酮-咔唑衍生物制得的四苯乙烯-二苯甲酮-咔唑衍生物晶体。
本发明的另一目的在于提供上述四苯乙烯-二苯甲酮-咔唑衍生物的制备方法。
本发明的还一目的在于提供上述四苯乙烯-二苯甲酮-咔唑衍生物晶体的制备方法。
本发明的再一目的在于提供上述四苯乙烯-二苯甲酮-咔唑衍生物晶体在发光材料、发光器件或智能材料中的应用。
为解决上述技术问题,本发明采用的技术方案是:
一种四苯乙烯-二苯甲酮-咔唑衍生物,所述四苯乙烯-二苯甲酮-咔唑衍生物的名称为(4-(9h-咔唑-9-基)苯基)(4-(1,2,2三苯基乙烯基)苯基)甲酮,结构式如式(ⅰ)所示:
本发明提供了一种四苯乙烯-二苯甲酮-咔唑衍生物,由该四苯乙烯-二苯甲酮-咔唑衍生物制得的晶体具有独特的层状结构,一方面由于咔唑与苯甲酰基之间引入桥联的苯环,形成大的共轭平面;另一方面,虽然相邻分子π共轭平面之间的距离不是很大
本发明同时保护一种四苯乙烯-二苯甲酮-咔唑衍生物晶体,所述四苯乙烯-二苯甲酮-咔唑衍生物晶体由上述四苯乙烯-二苯甲酮-咔唑衍生物制备得到。
优选地,所述四苯乙烯-二苯甲酮-咔唑衍生物晶体的晶体结构参数如下:空间群为p-1,z=2,
本发明同时保护上述四苯乙烯-二苯甲酮-咔唑衍生物的制备方法,包括如下步骤:
s1.将二苯甲酮、锌粉和四氯化钛进行mcmurry偶联反应,处理后得到1,1,2,2-四苯乙烯;
s2.将步骤s1.中的1,1,2,2-四苯乙烯与对氟苯甲酰氯进行傅-克反应,处理后得到(4-氟苯基)(4-(1,2,3-三苯基乙烯基)苯基)甲酮;
s3.将步骤s2.中的(4-氟苯基)(4-(1,2,3-三苯基乙烯基)苯基)甲酮与咔唑进行反应,处理后得到四苯乙烯-二苯甲酮-咔唑衍生物。
优选地,步骤s1.中所述二苯甲酮、锌粉与四氯化钛的摩尔比为(1~1.1)∶(2~2.2)∶(4~4.4)。更优选地,步骤s1.中所述二苯甲酮、锌粉与四氯化钛的摩尔比为1∶2∶4。
优选地,步骤s1.中所述mcmurry偶联反应的溶剂为无水四氢呋喃。
优选地,步骤s1.中所述mcmurry偶联反应的条件是在惰性气体的保护下先进行冰浴搅拌,然后再加热回流。因为四氯化钛遇水会剧烈反应,放出大量的热,因此必须严格管控滴加四氯化钛的的速率,并用冰浴降温。滴加完后还必须再冰浴搅拌一段时间,然后再暖至室温进行加热回流。
优选地,步骤s1.中所述惰性气体为氮气、氩气或氦气。更优选地,步骤s1.中所述惰性气体为氮气。优选地,步骤s1.中所述冰浴搅拌的温度为-10~0℃,时间30~60min。更优选地,步骤s1.中所述冰浴搅拌的温度为-10~-5℃,时间30~60min。进一步优选地,步骤s1.中所述冰浴搅拌的温度为-10℃,时间30min。
优选地,步骤s1.中所述加热回流的温度为88~93℃,时间为18~20h。更优选地,步骤s1.中所述加热回流的温度为90℃,时间为20h。
优选地,步骤s1.中所述处理为冷却、猝灭、萃取、干燥、浓缩、分离。将反应后的溶液冷却至室温后,用体积比为1∶1稀盐酸猝灭反应。所得混合液用二氯甲烷萃取三次,合并三次萃取所得的有机相;然后用无水硫酸钠干燥,再减压浓缩有机相得到粗产品;最后用四氢呋喃与石油醚作为洗脱剂进行硅胶柱层析分离出1,1,2,2-四苯乙烯。
优选地,步骤s2.中所述1,1,2,2-四苯乙烯与对氟苯甲酰氯的摩尔比为(1~1.2)∶(1.2~1.5)。更优选地,步骤s2.中所述1,1,2,2-四苯乙烯与对氟苯甲酰氯的摩尔比为1∶1.3。
优选地,步骤s2.中所述傅-克反应的溶剂为二硫化碳。优选地,步骤s2.中所述傅-克反应的条件是在惰性气体的保护下进行常温搅拌。优选地,步骤s2.中所述惰性气体为氮气、氩气或氦气。优选地,步骤s2.中所述傅-克反应的时间为5~7h。更优选地,步骤s2.中所述傅-克反应的时间为7h。
优选地,步骤s2.中所述处理为冰浴搅拌、萃取、洗涤、干燥、浓缩、分离。反应完成后,先在冰浴下加去离子水搅拌;因为三氯化铝容易潮解,由于水合会放热,遇水可能会爆炸,所以要在冰浴下加入去离子水。再用二氯甲烷萃取三次,合并三次萃取所得有机相,用稀氢氧化钠洗涤,分去水相,有机相用无水硫酸镁干燥,再减压浓缩有机相得到粗产品;最后用四氢呋喃与石油醚作为洗脱剂进行硅胶柱层析分离出(4-氟苯基)(4-(1,2,3-三苯基乙烯基)苯基)甲酮。
优选地,步骤s3.中所述(4-氟苯基)(4-(1,2,3-三苯基乙烯基)苯基)甲酮与咔唑的摩尔比为(1~1.2)∶(1.5~1.8)。更优选地,步骤s3.中所述(4-氟苯基)(4-(1,2,3-三苯基乙烯基)苯基)甲酮与咔唑的摩尔比为1∶1.5。
优选地,步骤s3.中所述反应的溶剂为n,n-二甲基甲酰胺。
优选地,步骤s3.中所述反应的条件是在惰性气体的保护下进行加热回流。优选地,步骤s3.中所述惰性气体为氮气、氩气或氦气。更优选地,步骤s3.中所述惰性气体为氮气。优选地,步骤s3.中所述加热回流的温度为110~115℃,时间为12~15h。更优选地,步骤s3.中所述加热回流的温度为115℃,时间为15h。
优选地,步骤s3.中所述处理为冷却、蒸馏、萃取、干燥、蒸馏、分离。冷却收集得到黄色浑浊液,将浑浊液减压蒸馏除去n,n-二甲基甲酰胺,所剩固体用二氯甲烷萃取三次,合并三次所得有机相,用无水硫酸镁干燥,再减压蒸馏有机相得到粗产品;最后用四氢呋喃与石油醚作为洗脱剂进行硅胶柱层析分离出四苯乙烯-二苯甲酮-咔唑衍生物,即得到(4-(9h-咔唑-9-基)苯基)(4-(1,2,2三苯基乙烯基)苯基)甲酮。
本发明还保护上述四苯乙烯-二苯甲酮-咔唑衍生物晶体的制备方法,包括如下步骤:
m1.将四苯乙烯-二苯甲酮-咔唑衍生物加热至200~300℃、冷却、用溶剂溶解,加入正己烷,分层得混合液;
m2.将m1.的混合液中的四苯乙烯-二苯甲酮-咔唑衍生物结晶,经后处理得到四苯乙烯-二苯甲酮-咔唑衍生物晶体。
本发明提供的含四苯乙烯-二苯甲酮-咔唑衍生物晶体的制备方法中,先将四苯乙烯-二苯甲酮-咔唑衍生物加热至200~300℃,然后降温冷却到室温,再用溶剂溶解。在这个温度范围内进行加热能够使四苯乙烯-二苯甲酮-咔唑衍生物发生相变,从亚稳状态达到一个稳定的状态,有利于培养得到晶型更好的晶体。如果温度过低,难以使其发生相变,而温度过高的话,容易使物质分解变质。优选地,步骤m1.中所述将四苯乙烯-二苯甲酮-咔唑衍生物加热至200℃。
然后以0.5~1.0ml/min的速度缓慢滴加正己烷。由于四苯乙烯-二苯甲酮-咔唑衍生物在正己烷中溶解度较低,缓慢加入正己烷有利于晶体的析出。优选地,步骤m1.中所述溶剂为四氢呋喃。优选地,步骤m1.中所述四氢呋喃与正己烷的体积比为1∶1。
优选地,步骤m2.中所述结晶的温度为20~30℃。如果温度过低,不利于四氢呋喃与正己烷溶剂的挥发,不利于晶体的析出;而温度过高,溶剂挥发过快,容易长出针状的多晶,晶型不好。更优选地,步骤m2.中所述结晶的温度为25℃。发明人研究发现,挥发温度为25℃时,四苯乙烯-二苯甲酮-咔唑衍生物能够获得晶型更好的晶体,实现了四苯乙烯-二苯甲酮-咔唑衍生物晶体的可控制备。优选地,步骤m2.中所述后处理为过滤、洗涤、烘干。洗涤时,使用正己烷为洗涤剂。
本发明还保护上述四苯乙烯-二苯甲酮-咔唑衍生物晶体在发光材料、发光器件或智能材料中的应用。
四苯乙烯-二苯甲酮-咔唑衍生物晶体具有高荧光性、良好的热稳定性以及其很好的发光性能,并且其具有独特的聚集诱导发光(aie)效应,可以削弱器件制备过程中因浓度增大而引起的聚集荧光淬灭(acq)效应,故可以成为oled中的发光材料。该四苯乙烯-二苯甲酮-咔唑衍生物晶体在制备发光材料、发光器件或智能材料等应用方面具有显著的经济价值。
与现有技术相比,本发明的有益效果是:
本发明提供了一种四苯乙烯-二苯甲酮-咔唑衍生物,该四苯乙烯-二苯甲酮-咔唑衍生物制备的晶体具有高的发光强度、良好的热稳定性、独特的aie效应和良好的溶解性,可作为一种性能好、成本较低、空间结构高度扭曲的新型可溶性发光分子。该四苯乙烯-二苯甲酮-咔唑衍生物晶体在制备发光材料、发光器件或智能材料等应用方面具有显著的经济价值。同时,本发明实现了含四苯乙烯-咔唑衍生物晶体的可控制备。
此外,可采用湿法加工制作发光器件,避免了复杂的制备工艺和设备的使用,制备成本低廉,原料来源广泛,可实现大规模的生产,具有广阔的商业化前景。
附图说明
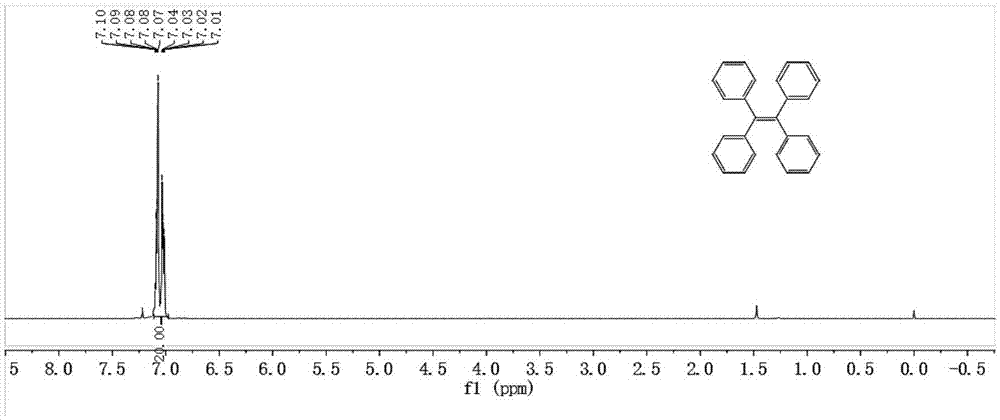
图1为实施例1制备得到的中间体ⅰ的氢谱图。
图2为实施例1制备得到的中间体ⅱ的氢谱图。
图3为实施例4制备得到的四苯乙烯-二苯甲酮-咔唑衍生物晶体的氢谱图。
图4为实施例4制备得到的四苯乙烯-二苯甲酮-咔唑衍生物晶体的质谱图。
图5为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物的晶体结构图。
图6为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物的晶体分子间作用力结构图。
图7为实施例4制备得到的四苯乙烯-二苯甲酮-咔唑衍生物晶体的c…h…π堆积图。
图8为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物的紫外可见吸收光谱图。
图9为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物在不同浓度的溶液中的aie光谱图。
图10为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物的循环伏安图。
图11为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物的热重分析图(tga)。
其中,图中tpe-bz-c为四苯乙烯-二苯甲酮-咔唑衍生物的简称。
具体实施方式
下面结合具体实施方式对本发明作进一步的描述,但本发明的实施方式不限于此。实施例中的原料均可通过市售得到;除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
实施例1
一种四苯乙烯-二苯甲酮-咔唑衍生物,由以下方法制备得到:
s1.中间体ⅰ的制备
a、称取1.82g二苯甲酮和1.315g锌粉于250ml的圆底烧瓶中,加入150ml无水四氢呋喃溶解,得到混合液;
b、将步骤a得到的混合液在氮气的保护下进行磁力搅拌、冷却至0℃,注入4.4ml四氯化钛,冰浴搅拌30min(一般情况下,冰浴搅拌的温度可以为-10~0℃,在实施例1中为-10℃),暖至室温,90℃加热回流20h,随着反应的进行,反应溶液的颜色逐渐加深,由一开始的浅褐色转为黑色,反应溶液呈不透明浑浊状态;
c、反应溶液冷却至室温后,用1∶1稀盐酸(20ml盐酸:20ml去离子水)猝灭反应。所得混合液用二氯甲烷萃取三次,合并三次萃取所得的有机相;然后用无水硫酸钠干燥,再减压浓缩有机相得到粗产品;
d、用四氢呋喃:石油醚作为洗脱剂进行硅胶柱层析分离出1.47g中间体ⅰ,产率为90%。该中间体ⅰ的化学名称为1,1,2,2-四苯乙烯。
反应方程式如下:
s2.中间体ⅱ的制备
a、称取0.3g1,1,2,2-四苯乙烯于50ml的圆底烧瓶中,加入10ml的二硫化碳溶解,得到混合液;
b、将步骤a得到的混合液在氮气的保护下进行磁力搅拌,加入0.16g三氯化铝,最后称取0.19g对氟苯甲酰氯于5ml的二硫化碳中,再缓慢滴加至上述反应溶液,常温搅拌7h,随着反应物的加入,反应溶液的颜色逐渐加深,由一开始的浅黄色转为红褐色,反应溶液呈不透明浑浊状态;
c、反应完成后,先在冰浴下加去离子水搅拌,再用二氯甲烷萃取萃取三次,合并三次萃取所得有机相,用稀氢氧化钠洗涤,分去水相,有机相用无水硫酸镁干燥,再减压浓缩有机相得到粗产品;
d、用四氢呋喃:石油醚作为洗脱剂进行硅胶柱层析分离出0.38g中间体ⅱ,产率为80%。该中间体ⅱ的化学名称为(4-氟苯基)(4-(1,2,3-三苯基乙烯基)苯基)甲酮。
反应方程式如下:
s3.四苯乙烯-二苯甲酮-咔唑衍生物的制备
将0.1mmol上述中间体ⅱ溶于3mldmf中于50ml圆底烧瓶中,再加入0.15mol咔唑,最后加入0.3mmolt-buok,在115℃加热15h,冷却收集得到黄色浑浊液,将浑浊液减压蒸馏除去dmf,所剩固体用二氯甲烷萃取三次,合并三次所得有机相,用无水硫酸镁干燥,再减压蒸馏有机相得到粗产品;用四氢呋喃:石油醚作为洗脱剂进行硅胶柱层析分离出0.48g产物,产率为80%。该产物的化学名称为(4-(9h-咔唑-9-基)苯基)(4-(1,2,2三苯基乙烯基)苯基)甲酮。
反应方程式如下:
实施例2
一种四苯乙烯-二苯甲酮-咔唑衍生物,其制备方法与实施例1的制备方法相比,区别在于,实施例2的步骤s1.中冰浴搅拌的温度为-5℃,时间60min,回流加热时间为18h;
其他原料用量及操作步骤与实施例1一致。
实施例3
一种四苯乙烯-二苯甲酮-咔唑衍生物,其制备方法与实施例1的制备方法相比,区别在于,实施例3的步骤s2.中傅-克反应的时间为5h;步骤s3.中加热回流的温度为110℃,时间为12h;
其他原料用量及操作步骤与实施例1一致。
实施例4
一种四苯乙烯-二苯甲酮-咔唑衍生物晶体,由以下方法制备得到:
m1.将实施例1制得的四苯乙烯-二苯甲酮-咔唑衍生物加热至200℃、冷却到室温、用四氢呋喃溶解,以0.5ml/min的速度滴加正己烷,分层得混合液;
m2.将m1.的混合液中的四苯乙烯-二苯甲酮-咔唑衍生物在25℃下结晶,经过滤、正己烷洗涤、烘干后,得到四苯乙烯-二苯甲酮-咔唑衍生物晶体。
实施例5
一种四苯乙烯-二苯甲酮-咔唑衍生物晶体,由以下方法制备得到:
m1.将实施例2制得的四苯乙烯-二苯甲酮-咔唑衍生物加热至300℃、冷却到室温、用四氢呋喃溶解,以1.0ml/min的速度滴加正己烷,分层得混合液;
m2.将m1.的混合液中的四苯乙烯-二苯甲酮-咔唑衍生物在20℃下结晶,经过滤、正己烷洗涤、烘干后,得到四苯乙烯-二苯甲酮-咔唑衍生物晶体。
表征及性能测试
对实施例1~5得到的四苯乙烯-二苯甲酮-咔唑衍生物及其晶体进行表征及性能测试。
(1)核磁共振:布鲁克400mhz超导核磁共振仪。
采用核磁共振扫描了中间体ⅰ、中间体ⅱ以及四苯乙烯-二苯甲酮-咔唑衍生物晶体的氢信号并对其氢信号进行了指认,实施例1与实施例4的结果如图1~3所示。所得结果与其他实施例相一致。
从图1可知,所述中间体ⅰ的δ(ppm)为7.46,7.42,7.30。从图2可知,中间体ⅱ的δ(ppm)为7.39,7.72,7.69,7.56,7.46,7.42,7.30。从图3可知,四苯乙烯-二苯甲酮-咔唑衍生物晶体的δ(ppm)为7.94,7.58,8.19,8.55,7.83,7.35,7.50,7.20,7.16,7.88,7.69,7.42,7.30,7.56,7.46,7.83,7.88,7.42,7.30。
(2)质谱:液质联用仪lcms-2020。将四苯乙烯-二苯甲酮-咔唑衍生物晶
体溶于乙腈配成浓度为1mg/ml的溶液进行测试。
图4为实施例4制备得到的四苯乙烯-二苯甲酮-咔唑衍生物晶体的质谱图,从图中可以看到,四苯乙烯-二苯甲酮-咔唑衍生物的相对分子质量为602.3,与所合成的晶体的相对分子质量一致。实施例4所得结果与其他实施例相一致。
(3)单晶x-射线衍射:
如图5~7所示,单晶x-射线衍射数据表明,四苯乙烯-二苯甲酮-咔唑衍生物晶体的晶体结构参数如下:空间群为p-1,z=2,
从图5~7中可以看出,四苯乙烯-二苯甲酮-咔唑衍生物晶体具有独特的层状结构,咔唑与苯甲酰基之间引入桥联的苯环形成大的共轭平面;相邻分子π共轭平面之间的距离不是很大
(4)紫外可见吸收光谱:岛津紫外可见光分光光度计uv-2700。将四苯乙烯-二苯甲酮-咔唑衍生物溶于thf中配成1×10-3mol/l的母液,测试时,稀释成1×10-5mol/l。
图8为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物在1×10-5mol/l的四氢呋喃中的紫外可见吸收光谱图。从图8可知,四苯乙烯-二苯甲酮-咔唑衍生物的主要吸收峰位置为340nm。
(5)aie光谱:fls980荧光仪。
保持测试溶液中四苯乙烯-二苯甲酮-咔唑衍生物的浓度为1×10-5mol/l,调节测试溶液中四氢呋喃和水的比例。先将四苯乙烯-二苯甲酮-咔唑衍生物溶于四氢呋喃中配成1×10-3mol/l的母液,维持测试溶液总体积为3ml。例如:水含量为90%时,各成分加入的量为母液∶水∶四氢呋喃=30ul∶2700ul∶270ul。
图9为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物在不同浓度的溶液中的aie光谱图。分别测试了四苯乙烯-二苯甲酮-咔唑衍生物在水含量为1%~99%的四氢呋喃-水溶液中的荧光光谱,箭头指示方向为10条荧光图线所对应的溶液的水含量依次增加方向。由图9中可以看出,四苯乙烯-二苯甲酮-咔唑衍生物的发射波长为496nm;当水含量低于90%时,四苯乙烯-二苯甲酮-咔唑衍生物在溶液中的荧光强度并没有太明显的变化,1%~80%的荧光光谱几乎重合;而当水含量超过90%后,对应荧光强度均发生大幅度增强,可知四苯乙烯-二苯甲酮-咔唑衍生物存在明显的aie现象。
(6)循环伏安:电化学工作站pgstat302。
图10为实施例1制备得到的四苯乙烯-二苯甲酮-咔唑衍生物的循环伏安图。将四苯乙烯-二苯甲酮-咔唑衍生物溶于乙腈中配成1mg/ml的溶液,在电化学工作站下通过循环伏安法测得四苯乙烯-二苯甲酮-咔唑衍生物的还原电位为e=1.25395v。
(7)热重分析:高温同步热分析仪sta409pc,升温速率:10k/min;温度范围:常温~800℃;气体保护:氮气;
在氮气保护下,对四苯乙烯-二苯甲酮-咔唑衍生物进行了热重分析。从图11中可知,四苯乙烯-二苯甲酮-咔唑衍生物的5%失重温度达到了353.6℃,说明四苯乙烯-二苯甲酮-咔唑衍生物具有良好的热稳定性,具有制备性能良好的oled的潜力。
综上所述,本发明提供的四苯乙烯-二苯甲酮-咔唑衍生物晶体具有独特的aie效应、高的发光强度、良好的热稳定性和良好的溶解性,可作为一种性能好、成本较低、空间结构高度扭曲的新型可溶性发光分子。该四苯乙烯-二苯甲酮-咔唑衍生物晶体在制备发光材料、发光器件或智能材料等应用方面具有显著的经济价值。同时,本发明实现了四苯乙烯-二苯甲酮-咔唑衍生物晶体的可控制备。
此外,可采用湿法加工制作发光器件,避免了复杂的制备工艺和设备的使用,制备成本低廉,原料来源广泛,可实现大规模的生产,具有广阔的商业化前景。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种3-(4-苯基-1h-咪唑-5-基)-1h-吲哚衍生物及其制备方法和应用
- 2-烷氧基-4-((1e,6e)-7-(4-氧代苯基)-3,5-二氧-1,6-庚二烯-1-基)苯酚金属盐衍生物 ...的制作方法
- N-取代苯基丙烯酰胺衍生物及其制备方法和用图
- (2e, 6e)-2-(3,5-二甲氧基苯基亚甲基)-6-(4-氯苯基亚甲基)环己酮及其衍生物的新应用
- 杀真菌的3-{苯基[(杂环基甲氧基)亚氨基]甲基}-杂环衍生物的制作方法
- 作为甲酰肽受体调节剂的苯基氨基甲酸酯衍生物的制作方法
- 杀真菌的3-{苯基[(杂环基甲氧基)亚氨基]甲基}-杂环衍生物的制作方法
- 作为甲酰肽受体调节剂的苯基氨基甲酸酯衍生物的制作方法
- 2-[2-(2,4-二氟苯基)烯丙基]-1,3-丙二酸二乙酯的合成方法
- 一种合成2-[2-(2,4-二氟苯基)烯丙基]-1,3-丙二酸二乙酯的方法
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺