聚(乙二醇)甲基丙烯酸酯微凝胶、制备方法和用途与流程
本发明涉及聚(寡聚(乙二醇)甲基丙烯酸酯)微凝胶、用于在水性介质中制备所述微凝胶的方法、以及所述微凝胶在各种应用领域(如光学、电子学、传感器、化妆品、药学和医学诊断)中的用途。
这些微凝胶具有单分散性、ph响应性和温度响应性的优点,并具有能够掺入有机分子或无机颗粒的优点。由这些微凝胶(任选地填充有无机纳米颗粒)的胶体溶液制备的膜具有非常优越的光学性能和机电性能。
背景技术:
存在若干种温度响应性微凝胶的化学成分。主要的化学成分是基于聚(n-异丙基丙烯酰胺)(pnipam),而不常见的化学成分则是基于聚(n-乙烯基己内酰胺)(pvcl)或基于聚(寡聚(乙二醇)甲基丙烯酸酯)。
已在文献中进行了描述的用于合成聚(寡聚(乙二醇)甲基丙烯酸酯)微凝胶的各种途径包括诸如二(乙二醇)甲基醚甲基丙烯酸酯单体(m(eo)2ma,有些疏水性)和五(乙二醇)甲基醚甲基丙烯酸酯单体(m(eo)5ma,有些亲水性)的单体的组合。
由温度响应性核(基于p(m(eo)2ma-co-m(eo)5ma))和壳(基于聚(寡聚(乙二醇)甲基丙烯酸酯)和聚(丙烯酸)的混合物(p(m(eo)2ma-co-m(eo)5ma-co-aa)))构成的温度响应性和ph响应性的微凝胶的合成已由chi,c.,t.cai和z.hu,oligo(ethyleneglycol)-basedthermoresponsivecore-shellmicrogels.langmuir,2009.25:p.3814-3819进行了描述。这些微凝胶具有带有疏水性核和亲水性壳的核/壳结构。
已经建议将生物活性分子掺入基于聚(寡聚(乙二醇)甲基丙烯酸酯)的微凝胶中。
例如,在zhou等,engineeringoligo(ethyleneglycol)-basedthermosensitivemicrogelsfordrugdeliveryapplications.polymer,2010.51:p.3926-3933;以及wang等,preparationofbiocompatiblenanocapsuleswithtemperature-responsiveandbioreducibleproperties,journalofmaterialschemistry,2011.21:p.15950中分别提出了用于递送活性成分的温度响应性微凝胶,所述温度响应性微凝胶由聚(m(eo)2ma)核和聚(m(eo)2ma-co-m(eo)5ma)壳以及聚(m(eo)2ma-co-oegma)纳米胶囊(通过将聚合物接枝到牺牲(sacrificial)二氧化硅颗粒上获得)组成。
含有无机纳米颗粒的已知杂化(hybrid)(或纳米复合)温度响应性微凝胶是基于聚(n-异丙基丙烯酰胺)(pnipam)的微凝胶。这些微凝胶具有非生物相容性的缺点。
用于制备这些材料的第一种方法是在纳米颗粒的存在下合成微凝胶。由于聚合的复杂性,这种策略使得难以制造这些杂化微凝胶。掺入的纳米颗粒的比例通常较小,并且微凝胶是多分散性的。此外,它通常产生其中的纳米颗粒均匀分布的核-壳型结构。
第二种方法是首先合成由离子基团官能化的温度响应性微凝胶。接着,通过纳米颗粒的前体盐的共沉淀掺入无机纳米颗粒。申请wo2004/081072描述了例如具有丙烯酸钠(-coo-na+)阴离子单元的pnipam微凝胶以及各种纳米颗粒(如磁性(fe3o4)颗粒、金(au)颗粒和量子点(cdte、cds)颗粒)的前体盐的原位共沉淀。
第三种方法是通过溶剂转移掺入无机纳米颗粒。合成疏水性无机纳米颗粒并将其分散在有机相中。将温度响应性微凝胶加入到含有纳米颗粒的溶液中,然后将所有物质转移到水溶液中,这将纳米颗粒包封在微凝胶中。该方法已经成为了使用聚(n-异丙基丙烯酰胺)微凝胶和量子点(cds)纳米颗粒用于光致发光应用的专利(us7914710)的主题。
第四种也是最后一种方法是合成具有离子电荷的微凝胶并在微凝胶表面吸附相反电荷的纳米颗粒。具有2-氨基乙基甲基丙烯酸酯(aema)阳离子基团的聚(苯乙烯-共-n-异丙基丙烯酰胺)胶乳的制备已经在申请wo1997/045202中进行了描述。表面带负电荷的磁性γ-fe2o3纳米颗粒通过静电相互作用被吸附在胶乳的表面。然后通过在杂化胶乳的表面合成pnipam新外壳,将纳米颗粒困于结构中。
最近,已经将带有正电荷的纳米颗粒(tio2)掺入通过包含丙烯酸根(coo-)基团的单元而带电的pnipam微凝胶结构中(us8158005)。
在某些领域中,微凝胶、特别是具有磁性性能的温度响应性微凝胶的使用需要以薄膜的形式制备。
由预先分散在水相中的微凝胶组成的薄膜的形成是困难的合成过程,因为它需要调和两个相互矛盾的因素。分散在水相中的微凝胶通过防止微凝胶聚集或沉降的排斥电荷而稳定。然而,微凝胶必须彼此相互作用以形成微凝胶层,从而形成膜。为了形成它们,必须开发几种方法。
用于在改性表面上使微凝胶自组装的第一种方法是将微凝胶锚定到经预处理以在其表面上产生离子电荷的衬底的表面上。微凝胶具有通常衍生自聚合引发剂或离子共聚单体的离子基团。可以通过与带相反电荷的基团的静电相互作用将微凝胶锚定到衬底的表面。这种技术使得可以在衬底上沉积微凝胶的薄层(单层技术),但也可以通过连续的表面处理来累加这些层(叠层技术)。已经根据该技术将基于聚(n-异丙基丙烯酰胺-共-丙烯酸)或p(nipam-co-aa)的微凝胶沉积在由3-氨基丙基三甲氧基硅烷(aptms)基团接枝的衬底上。在各微凝胶层之后,将具有正电荷的聚合物(聚(烯丙胺盐酸盐)pah或聚(乙烯亚胺)pei)加入到改性衬底中,以在胺的酸度常数的更低的ph下使正电荷再生。
沉积在衬底上的微凝胶膜已被用于递送活性成分,诸如用于治疗糖尿病的胰岛素(nolan,c.m.,m.j.serpe和l.a.lyon,thermallymodulatedinsulinreleasefrommicrogelthinfilms.biomacromolecules,2004.5(5):p.1940-1946)或用于治疗癌细胞的多柔比星(serpe,m.j.等,doxorubicinuptakeandreleasefrommicrogelthinfilms.biomacromolecules,2005.6(1):p.408-413)。
该技术具有控制微凝胶薄膜生产的固有参数(例如膜厚度和微凝胶的结构化)的优点。该方法的缺点在于其生产复杂性,需要使用特定的微凝胶(带电微凝胶)和预处理的衬底。这阻碍了微凝胶在任意表面上的任意直接使用。
用于生产包封胶体颗粒的水凝胶膜的第二种方法是将颗粒包封在水凝胶中以形成柔性的湿膜。该方法主要研究用于光子应用(photonicapplications)。该想法是将组装的颗粒的光学性能与处于湿态的水凝胶的机械性能相结合。在文献中对一些实例进行了描述,实例中主要使用了处于硬球体形式的聚(苯乙烯)颗粒。e.tian等已经将聚(苯乙烯-共-甲基丙烯酸甲酯-共-丙烯酸)颗粒组装成若干结构层,并将所有物质包封在聚(丙烯酰胺)水凝胶中(tian,e.等,colorfulhumiditysensitivephotoniccrystalhydrogel.journalofmaterialschemistry,2008.18:p.1116-1122)。这两种实体的组合使得可以开发光子水凝胶。最近,h.jiang等还将聚(苯乙烯)颗粒包封在聚(乙烯醇)或pva水凝胶中(jiang,h.等,photoniccrystalphandmetalcationsensorsbasedonpoly(vinylalcohol)hydrogel.newjournalofchemistry,2012.36:p.1051-1056)。
最近,h.kim等在磁场的作用下对涂覆有二氧化硅(sio2)的氧化铁(fe3o4)磁性颗粒进行了组装,并将该组装体包封在聚(乙二醇)二丙烯酸酯单体和光引发剂的混合物中。该混合物的光聚合使得可以将颗粒固定于聚(乙二醇丙烯酸酯)树脂中。固定于本体(bulk)中的磁性颗粒具有可由施加的磁场限定的光子性能(us2012/0028834)。该技术具有生产更具柔性的微凝胶膜的优点,因为后者由溶液中的“软”水凝胶而不是固体支持物支撑。然而,这些颗粒的膜的生产仍然是复杂的,并且需要各种聚合步骤,这阻碍了直接使用颗粒期间的任何自发的自组装。
用于形成微凝胶膜的第三种方法是将反应性官能团添加到微凝胶的表面。通过在微凝胶合成期间加入共聚单体来提供该官能性。这些反应性官能团可以彼此形成共价键,或者通过与另一实体反应形成共价键。然后可以在各微凝胶之间形成交联点,整体给出由彼此化学结合的微凝胶组成的膜。该自组装方法主要研究用于光子应用。该自组装方法取决于在微凝胶合成过程中添加的官能团类型。这些研究中的绝大多数着力于基于pnipam的微凝胶的组装。
第一种方法是在pnipam微凝胶中加入聚(丙烯酸)(或paa)。微凝胶的自组装是由于paa的羧酸官能团之间的弱相互作用而发生的。相互作用的总和使得微凝胶能够自组装并能够将介质凝胶化。
还提出了第二种方法,该方法通过向pnipam微凝胶的溶液中加入交联剂、或通过pnipam-co-nma微凝胶(nma:n-羟甲基(methylol)丙烯酰胺或n-羟甲基(hydroxymethyl)丙烯酰胺)的缩聚而产生共价键。通过微凝胶分散体的简单干燥而使微凝胶自组装。膜形成需要交联剂热后聚合(post-polymerization)或nma的热和酸碱催化缩合的额外步骤。
一项研究使用了具有可表面聚合官能团的寡聚(乙二醇)甲基丙烯酸酯衍生微凝胶。这些微凝胶在组装用于光子应用期间通过uv光聚合而交联(us2010/0076105)。然而,膜形成需要约数周的非常长的干燥时间。
技术实现要素:
本发明的目的是提出一种相对于现有技术的微凝胶而言具有以下优点中的至少一个的寡聚(乙二醇)甲基丙烯酸酯微凝胶:单分散性;ph响应性;生物相容性;能够通过吸附带相反电荷的纳米颗粒形成杂化(或纳米复合)微凝胶;能够通过简单干燥过程自组装在若干层中;能够形成透明膜;能够形成粘性和弹性膜;处于膜的形式,能够通过压缩效应(compressioneffect)产生电位;处于膜的形式,能够衍射光而产生颜色。
在本发明的上下文中已经发现,某些聚(寡聚(乙二醇)甲基丙烯酸酯)通过在环境温度下水的蒸发而具有高的成膜能力。特别地,本发明首次提出具有结构性能的处于薄膜形式的磁性杂化微凝胶的自组装。
本发明的微凝胶膜具有完全自支撑(self-supported)的优点。由于微凝胶不被包封或支撑,因此在膜形成过程中,微凝胶和衬底(例如皮肤,微凝胶沉积在衬底上)之间的相互作用是最大的。通过在环境温度下简单干燥而获得膜,并且无需自由基引发剂。任选地含有纳米颗粒的本发明的微凝胶能够自组装在若干层中以形成透明膜。
在本发明的上下文中,还发现聚(寡聚(乙二醇)甲基丙烯酸酯)微凝胶是能够通过机械作用产生电场的聚电解质材料。
具体实施方式
本发明涉及一种聚(寡聚(乙二醇)甲基丙烯酸酯)微凝胶,该微凝胶具有胶体性能,并且由于任选地盐化(salified)-cooh基团的存在而在水中对温度和/或ph变化具有响应性。
在本发明的含义内,“微凝胶”被理解为在干燥状态(即含水量小于2wt%)下具有100nm至500nm(优选在350nm和450nm之间、更优选约400nm)的尺寸的球形颗粒形式的交联聚合物。本发明的微凝胶从能够通过若干单体的水相共聚合过程得到这个角度而言是微水凝胶。本发明的微凝胶不具有核/壳结构:形成该微凝胶的单体均匀地分布在整个颗粒体积中,这赋予它特定的性能。
本发明的微凝胶可同时具有单分散性、温度响应性、ph响应性和生物相容性这些优点。本发明的微凝胶可以同时具有温度响应性和生物相容性,这不同于现有技术中通常具有基于聚(烷基丙烯酰胺)的结构的温度响应性微凝胶。
本发明的微凝胶创新地包含支化环氧乙烷重复单元以及具有羧酸(-cooh)或羧酸根(-coo-)基团的单元的混合物,其含量可以根据目标应用而变化。这些基团赋予微凝胶ph响应性的性能。
因此,本发明的第一主题是能够在交联剂的存在下通过以下三种单体的水相沉淀聚合获得的微凝胶:
-二(乙二醇)甲基醚甲基丙烯酸酯(m(eo)2ma),
-寡聚(乙二醇)甲基醚甲基丙烯酸酯(m(eo)nma),n为3-12的整数,优选为8-10的整数,
-式cr1r2=cr3r4的单体,其中,r1、r2、r3和r4表示氢、卤素或烃基基团,四个基团中的至少一个包含-cooh基团或-coo-m+基团,m+表示阳离子。
在本文的其余部分中,为了简化,术语“-cooh”表示-cooh酸形式或-coo-m+盐化形式。
m(eo)2ma占例如单体摩尔总数的50mol%至90mol%,m(eo)nma优选占单体摩尔总数的10mol%至50mol%,式cr1r2=cr3r4的单体优选占单体摩尔总数的0.1mol%至20mol%,这三种单体含量的总和等于100%。
m(eo)2ma和m(eo)nma之间的摩尔比优选在1:1和20:1之间,例如在5:1和10:1之间。在本发明的含义内,表述“在……之间(between)”不包括所涉及的数值限制。另一方面,表述“……至……”包括所述限制。
式cr1r2=cr3r4的单体的摩尔数可以在三种单体的摩尔总数的0和20mol%之间,例如0.1mol%至5mol%。
根据一个实施方式,m(eo)2ma占例如三种单体的摩尔总数的80mol%至90mol%,m(eo)nma优选占单体摩尔总数的5mol%至15mol%,并且甲基丙烯酸优选占单体摩尔总数的0.1mol%至10mol%,这三种单体含量的总和等于100%。
式cr1r2=cr3r4的单体优选如下单体:r1和r2各自表示氢;r3表示h或任选被-oh或-cooh取代的烷基基团,优选任选被-oh或-cooh取代的c1-c6烷基基团;r4独立于r3,表示-cooh基团或任选被-oh或-cooh取代的烷基基团,优选任选被-oh或-cooh取代的c1-c6烷基基团。所述烷基基团可以为甲基、乙基或正丁基。根据一个具体实施方式,r1和r2各自表示氢,r3和r4独立地表示-h、-cooh或-ch2-cooh。
式cr1r2=cr3r4的单体例如可以选自于甲基丙烯酸(methylacrylicacid)、甲基甲基丙烯酸(methylmethacrylicacid)、乙基丙烯酸(ethylacrylicacid)、乙基甲基丙烯酸(ethylmethacrylicacid)、正丁基丙烯酸(n-butylacrylicacid)和正丁基甲基丙烯酸(n-butylmethacrylicacid)。
根据一个实施方式,式cr1r2=cr3r4的单体可以是甲基丙烯酸或衣康酸。在某些情况下,丙烯酸可以从式cr1r2=cr3r4的单体的定义中排除。
交联剂可以选自于由以下交联剂所组成的组:包含1至10个乙二醇单元的寡聚(乙二醇)二丙烯酸酯、1,3-丁二醇二丙烯酸酯、1,4-丁二醇二丙烯酸酯、1,6-己二醇二丙烯酸酯、季戊四醇二丙烯酸酯单硬脂酸酯、甘油1,3-二甘油化物二丙烯酸酯(glycerol1,3-diglycerolatediacrylate)、甘油二丙烯酸新戊酯、聚(丙二醇)二丙烯酸酯、1,6-己二醇乙氧基化物二丙烯酸酯、三羟甲基丙烷苯甲酸酯二丙烯酸酯、乙二醇二甲基丙烯酸酯、1,3-丁二醇二甲基丙烯酸酯、1,4-丁二醇二甲基丙烯酸酯、1,6-己二醇二甲基丙烯酸酯、二甲基丙烯酸甘油酯、n,n-二乙烯基苯、n,n-亚甲基双丙烯酰胺、n,n-(1,2-二羟基乙烯)双丙烯酰胺、聚(乙二醇)二丙烯酰胺、烯丙基二硫醚、双(2-甲基丙烯酰基)氧乙基二硫化物和n,n-双(丙烯酰)胱胺。
交联剂例如占三种单体的摩尔总数的1mol%至5mol%。
所使用的单体优选为二(乙二醇)甲基醚甲基丙烯酸酯(m(eo)2ma,mn250g·mol-1)、寡聚(乙二醇)甲基醚甲基丙烯酸酯(m(eo)9ma,mn475g·mol-1)、甲基丙烯酸(maa)。
交联剂例如是包含4至5个环氧乙烷单元的寡聚(乙二醇)二丙烯酸酯(oegda,mn250g·mol-1)。优选的单体和交联剂的化学结构示于图1中。
本发明的微凝胶的平均尺寸可以根据其是干燥的还是在水溶液中而变化:因此,当将干燥状态下的微凝胶置于20℃的水溶液中时,其尺寸可能达到初始尺寸的四倍。在干燥状态下,本发明的微凝胶的平均尺寸可以为100nm至1000nm。在20℃的温度下以60°的角度测量的微凝胶的流体动力学径向分布函数有利地小于1.1,这赋予微凝胶单分散性的品质。
本发明的微凝胶可以包含有机颗粒或无机颗粒:在这种情况下,它们通常被称为杂化微凝胶。引入的颗粒的尺寸优选在1nm和150nm之间,例如在5nm和50nm之间,并称为纳米颗粒。纳米颗粒可以是磁性的或可以不是磁性的。
本发明的第二个主题是含有磁性纳米颗粒的基于聚(寡聚(乙二醇)甲基丙烯酸酯)的具有单分散性、温度响应性和磁性的杂化微凝胶以及用于制备这些杂化微凝胶的方法。
本发明的某些基于聚(寡聚(乙二醇)甲基丙烯酸酯)的具有单分散性、温度响应性和磁性的杂化微凝胶任选在第三单体的存在下由至少两种单体获得:
-二(乙二醇)甲基醚甲基丙烯酸酯(m(eo)2ma),和
-寡聚(乙二醇)甲基醚甲基丙烯酸酯(m(eo)nma),n为3-12的整数,
所述第三单体为:
-式cr1r2=cr3r4的单体,其中,r1、r2、r3和r4表示氢、卤素或烃基基团,条件是四个基团中的至少一个包含-cooh或-coo-m+基团,m+表示阳离子。
用于制备本发明的基于聚(寡聚(乙二醇)甲基丙烯酸酯)的具有单分散性、温度响应性和磁性的杂化微凝胶的方法在于:
-制备在其表面带正电荷并置于水溶液中的纳米颗粒的胶体分散体,
-制备如权利要求1至4中任一项所述的微凝胶的水性胶体分散体,
-将两种所述胶体分散体混合并调节ph至高于所述纳米颗粒的等电点。
根据一个实施方式,当将纳米颗粒置于水溶液中时,纳米颗粒在其表面带正电荷,使得可以通过简单混合两种胶体分散体(微凝胶的第一胶体分散体和纳米颗粒的第二胶体分散体)来制备本发明的杂化微凝胶。使该方法成功的关键参数一方面在于在微凝胶内均匀分布的羧基或羧酸根基团的加入,另一方面在于纳米颗粒的正表面电荷。所有的一切使得能够以受控的方式将纳米颗粒包封在微凝胶内,同时保留最终材料的胶体性能和温度响应性性能。在本发明中证明了微凝胶的杂化结构及它们的温度敏感性性能。首先用大量且定量的包封的磁性纳米颗粒(测试的填料(filler)含量为各杂化微凝胶含0wt%至33wt%的纳米颗粒)证明了纳米颗粒被掺入微凝胶内。杂化微凝胶在水溶液中的温度响应性性能也被证明与磁性纳米颗粒填料含量无关。
根据一个实施方式,纳米颗粒是在光学、化妆品、农产品或药学领域中常用的色素、染料或遮光剂。
所述颗粒可以包含至少一种金属或一种金属氧化物。所述金属可以是金、银、锡、钛、铜或铝。所述金属氧化物可以选自于由氧化铁、氧化钛、氧化锌、氧化铬和氧化锡所组成的组。所述颗粒包含例如以下化合物中的至少一种:tio2、fe2o3、tife2o5、ti低价氧化物(ti-suboxides)、fe3o4、cr2o3、zro2、zno、sno2和sn(sb)o2。
根据一个实施方式,纳米颗粒是尺寸在1nm和150nm之间(例如在6nm和30nm之间)的氧化铁(fe2o3,磁赤铁矿)磁性纳米颗粒。
可以通过金属盐(fe2+和fe3+)在水相中的共沉淀来合成磁性纳米颗粒,然后进行氧化以藉由正电荷产生在溶液中稳定化的磁性纳米颗粒(γ-fe2o3)。用于合成磁赤铁矿纳米颗粒的方法可以是由massart,r.,preparationofaqueousmagneticliquidsinalkalineandacidicmedia.ieeetrans.magn.,1981,17(2):p.1247-1248研发的方法。
基于聚(寡聚(乙二醇)甲基丙烯酸酯)和纳米颗粒的温度响应性杂化微凝胶的制备可优选通过将处于胶体分散体形式的两种成分混合的方法而进行。当置于水溶液中的纳米颗粒与微凝胶接触时,纳米颗粒能够在表面带正电荷。通过提高ph至高于无机纳米颗粒的等电点而获得杂化微凝胶的稳定性,同时保持纳米颗粒的包封。
该方法具有实施简单的优点。羧酸或羧酸根基团在微凝胶内的均匀分布以及表面带正电荷的纳米颗粒的选择使得能够以受控的方式将纳米颗粒包封在微凝胶内,同时保持最终材料的胶体性能和温度响应性性能。
本发明的杂化微凝胶可以含有高达50wt%,特别是高达35wt%的纳米颗粒,而不损失其胶体性能、ph响应性性能和温度响应性性能。可以通过热重分析(tga)确定各杂化微凝胶的纳米颗粒含量。
上述任选包含纳米颗粒的微凝胶能够通过干燥或蒸发所述微凝胶的水悬浮液的方法而自组装,从而形成由一层或多层微凝胶组成的膜。
所形成的膜具有粘性和弹性。因此,本发明的微凝胶可用作化妆品组合物中的成膜剂,以改善这些组合物在角蛋白材料上的保持性。干燥后,当将膜浸入水中时,膜不会再分散。
微凝胶及其所形成的膜可以通过压缩效应(donnan效应)产生电位。该膜由具有衍生自羧酸根(coo-)官能团的离子位点的微凝胶制备而成。这些离子位点被限制在结构中并在微凝胶(例如聚电解质微凝胶)内形成极化。当膜受到压力时,抗衡离子(counterions)的移动有助于在膜内产生极化,这在膜的表面和体积(bulk)之间产生电位差。本发明人发现甲基丙烯酸的存在改善了膜的机电性能。
单分散性微凝胶的自组装还能够进行光衍射,从而产生颜色。光子性能可以通过微凝胶的特定组成来进行调节。
本发明的单分散性微凝胶可以以胶体晶体的形式周期性地(periodically)自组装。这种特殊的自组装能够实现入射光的衍射,从而产生根据视角可以观察到的颜色。该效应随微凝胶组成的变化而变化:a)不含纳米颗粒的微凝胶分散体的干燥导致形成不会衍射光的完全透明的无色干膜。而该相同的膜在湿态下衍射光,带有可以在溶液中观察到的颜色。b)含有磁性纳米颗粒的微凝胶(杂化微凝胶)分散体的干燥导致形成衍射光的透明、有色(棕色)干膜。因此,在90°的视角下该膜呈棕色(来自磁性纳米颗粒的颜色),并且在较小视角下在反射中改变了颜色。
微凝胶内磁性纳米颗粒的存在使得可以借助永磁体使微凝胶定向并改善膜的机械性能。在给定表面的干燥期间,具有温度响应性和磁性的微凝胶可以借助永磁体被引导并集中在精确的位点处。这具有改变膜的厚度和颜色(较深或较浅的棕色色调)的效果。此外,磁性纳米颗粒改善了膜在湿介质中的机械性能,从而可以对膜施加更大的压缩力。
所有这些性能使得可以想到本发明的微凝胶及其所形成的膜在用于制备化妆品产品或药物产品中的用途。这些产品可以通过产生电流并任选地通过经由压缩效应递送生物活性分子来刺激皮肤。生物活性分子可以被包封在微凝胶中或存在于产品中。
因此,本发明的另一主题是由如上所述的微凝胶以及任选的选自于由表面活性剂、油、生物活性产品、色素和染料所组成的组中的至少一种化合物组成的化妆品产品或药物产品,或者含有如上所述的微凝胶以及任选的选自于由表面活性剂、油、生物活性产品、色素和染料所组成的组中的至少一种化合物的化妆品产品或药物产品。
本发明的微凝胶可以包含在化妆品和药物领域中使用的各种成分或赋形剂,优选基于氧化铁的色素或生物活性物质。
本发明的微凝胶可以使用预溶解在水溶液中的单体通过沉淀聚合法合成。
本发明还涉及用于如上所述的聚(乙二醇)甲基丙烯酸酯微凝胶的沉淀聚合的方法,该方法包括在交联剂的存在下,在40℃和90℃之间的温度下,在水相中使上述三种单体接触的步骤。本发明的方法不需要诸如sds(十二烷基硫酸钠)的表面活性剂的存在。
可以通过在40℃和90℃之间,优选约70℃的温度下加入水溶性自由基引发剂(例如过硫酸钾(kps))来引发单体的聚合。
优选将式cr1r2=cr3r4的单体的水溶液逐渐加入到两种其它单体的水溶液中,以保证-cooh基团在沉淀的微凝胶中均匀分布。
在聚合温度下,形成的聚合物是疏水性的,并在水性反应介质中以球形颗粒的形式沉淀,聚合过程中交联剂oegda的存在使得可以通过产生交联点将颗粒固定于该球体形状中。
本发明的微凝胶具有结构均匀的优点,这是因为-cooh基团和交联点在其整个体积内均匀分布。
通过在20℃下干燥或蒸发溶剂的方法而有利地制备微凝胶膜,例如通过从在水中的重量浓度可以为1.4wt%至5wt%的单分散性微凝胶的胶体分散体起始。
根据该方法,溶液的至少第一体积可以保持进行干燥,直到水在环境温度下完全蒸发。该步骤可以重复若干次,以获得由若干层单分散性微凝胶组成的膜,并且其在干燥状态的厚度可以在350μm和450μm之间。
本发明还涉及:i)由如上所述的微凝胶以及任选的选自于由表面活性剂、油、生物活性产品、色素和染料所组成的组中的至少一种化合物组成的化妆品产品或药物产品,或者含有如上所述的微凝胶以及任选的选自于由表面活性剂、油、生物活性产品、色素和染料所组成的组中的至少一种化合物的化妆品产品或药物产品;ii)包含磁体(magnet)和化妆品产品的套装,所述化妆品产品含有如上所述且包含磁性纳米颗粒的微凝胶,所述磁体和所述产品被包装在一起;iii)美容化妆方法或护理方法,所述方法在于将上述微凝胶或化妆品产品施用至皮肤;iv)包含至少一层上述微凝胶或杂化微凝胶的薄膜;以及v)它们在诸如光学、电子学、传感器、化妆品、药学和医学诊断等各种应用领域中的用途。
附图说明
图1表示用于合成本发明的生物相容性微凝胶的单体的化学结构。
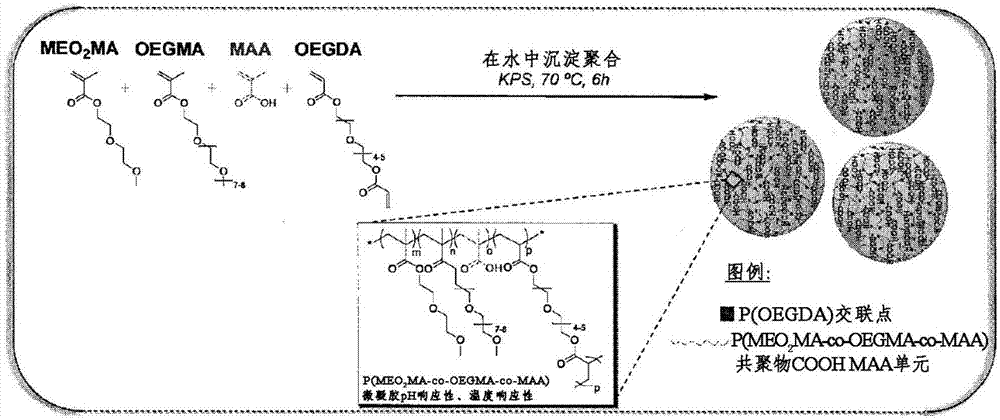
图2是本发明的基于聚(meo2ma-co-oegma-co-maa)的ph响应性和温度响应性的生物相容性微凝胶的合成方案。
图3是用于制备γ-fe2o3颗粒的方案。
图4和图5分别是用于制备含有γ-fe2o3颗粒的根据本发明的杂化微凝胶的方法的第一合成步骤和第二合成步骤的示意图。
图6表示用于形成根据本发明的微凝胶膜的方法的方案。
图7表示用于表征本发明的微凝胶膜的机电效应的组件。
图8是本发明的微凝胶膜的压缩和松弛(relaxation)时间的示意图。
图9表示本发明的微凝胶的压缩和松弛程序的图。
图10是处于干燥状态和在溶液中溶胀的本发明的微凝胶膜的照片。
图11是在各种视角下拍摄的含有γ-fe2o3纳米颗粒的本发明的杂化微凝胶的干膜的照片。
图12是在浅色表面和深色表面上拍摄的含有γ-fe2o3纳米颗粒的本发明的杂化微凝胶的干膜的照片。
图13是具有磁体和不具有磁体的含有γ-fe2o3纳米颗粒的本发明的杂化微凝胶的干膜的照片。
实施例
还通过以下实施例对本发明进行说明。
实施例1:本发明的基于聚(寡聚(乙二醇)甲基丙烯酸酯)的微凝胶的合成
使用以下单体:二(乙二醇)甲基醚甲基丙烯酸酯(m(eo)2ma,mn250g·mol-1)、寡聚(乙二醇)甲基醚甲基丙烯酸酯(m(eo)4-5ma,下文也称oegma,mn475g·mol-1)和甲基丙烯酸(maa)。交联剂是寡聚(乙二醇)二丙烯酸酯(oegda,mn250g·mol-1)。
实验方案:
将0.966gmeo2ma(5.14×10-3mol)、0.272goegma(5.73×10-4mol)和0.029goegda(1.17×10-4mol)引入体积为57.5ml的水中,并置于磁力搅拌下直到单体完全溶解。然后将混合物过滤,并引入装有机械搅拌器的体积为250ml的三颈烧瓶中,然后在机械搅拌(150rpm)下在氮气下脱气45分钟。然后将maa的水溶液(0.026g,3.05×10-4mol溶解在2ml水中)引入反应介质中。将混合物置于70℃下20分钟,然后引入预先在氮气下脱气的过硫酸钾的水溶液(kps,0.0143g溶解在2.5ml水中)中。加入kps使得可以引发聚合,并在70℃下将反应介质置于机械搅拌(50rpm)下6小时。
然后通过加入氧气终止聚合,并将其冷却至环境温度。然后通过离心(10000rpm,30分钟)将微凝胶与反应介质分离,并用纯水(milliq级别)替换反应介质;该步骤重复五次。
然后,最终溶液由水相中的p(meo2ma-co-oegma-co-maa)微凝胶的胶体分散体组成,将该分散体保持在环境温度下。
微凝胶的性能
通过使用质子核磁共振(1hnmr)光谱法对单体进行动力学监测来表征微凝胶的合成。观察到单体的完全转化,还观察到均质组成的微凝胶,交联点和甲基丙烯酸单元均匀分布。
通过反应介质的固含量对交联微凝胶的最终产率进行分析,可以确定交联微凝胶的产率为70wt%。
通过纯化微凝胶的酸碱滴定测定掺入的甲基丙烯酸的含量。可以证实微凝胶在水溶液中的ph响应性以及掺入了70mol%的初始maa单体。
通过透射电子显微术(tem)对微凝胶进行观察,并通过动态光散射测定其尺寸。观察到的微凝胶是单分散性的,在干燥状态下的尺寸为400nm,在湿态下可高达1000nm。
实施例2:基于聚(寡聚(乙二醇)甲基丙烯酸酯)的微凝胶的溶液的制备
将1.2ml根据实施例1制备的微凝胶溶液(含有15g·l-1微凝胶)分散在10ml纯水(milliq级别)溶液中。通过加入0.1mol·l-1盐酸或氢氧化钾溶液调节分散体的ph。将混合物置于磁力搅拌下直到ph稳定。通过动态光散射测量溶液中微凝胶的尺寸,并在分析过程中控制溶液温度。
通过利用动态光散射对溶液中微凝胶的尺寸进行研究,可以评估介质的ph和温度对微凝胶在水中溶胀或收缩的能力的影响。
微凝胶对介质的ph变化具有响应性,从ph<5.5时400nm的尺寸变化至ph>6.0时1000nm的尺寸。
微凝胶具有温度响应性,从20℃的溶胀状态变为高温下的收缩状态。收缩温度取决于ph(35℃,ph<6.0;55℃,ph>7.0)。最终,当温度超过收缩温度时,20℃下的溶胀微凝胶的体积相对于其初始体积减少高达3倍。实施例3:含有磁性纳米颗粒的基于聚(寡聚(乙二醇)甲基丙烯酸酯)的微凝胶的制备
磁赤铁矿γ-fe2o3纳米颗粒的合成
使用以下反应物:氯化亚铁四水合物(fecl2·4h2o)、氯化铁六水合物(fecl3·6h2o)、28%-30%w/w氢氧化铵(nh4oh)、硝酸铁(feiii(no3)3·9h2o)、36%v/v盐酸(hcl)和硝酸(hno3)。
本研究中使用的磁赤铁矿纳米颗粒是通过金属盐(feii和feiii)的共沉淀合成的。这种合成方法是通过加入氢氧化铵(nh4oh),在碱性介质中通过氯化亚铁(fecl2)和氯化铁(fecl3)的共沉淀来形成磁铁矿(fe3o4)纳米颗粒。然后将磁铁矿氧化形成磁赤铁矿(γ-fe2o3)品种。将磁铁矿氧化成磁赤铁矿使得可以在纳米颗粒表面建立ph响应性羟基官能团,这些官能团在中性ph(ph≈7.2)下具有零电荷点。因此,在酸性或碱性ph值下,这些纳米颗粒通过阴离子电荷(在碱性ph下)或阳离子电荷(在酸性ph下)的静电排斥而在水相中具有胶体状态。这是用于合成在水相中稳定化的磁性纳米颗粒的通用方法,根据稳定化的ph,通常被称为“阳离子铁磁流体”或“阴离子铁磁流体”。
实验方案
步骤1:磁铁矿的形成。
将12.2g氯化铁六水合物fecl3·6h2o(0.0451mol)引入含有500ml纯水的3l烧杯中。将4.49g氯化亚铁四水合物fecl2·4h2o(0.0226mol)溶解在24.3ml1.5mol·l-1盐酸(hcl)溶液中,加入到3l烧杯中,并将所有物质在温和的机械搅拌下进行混合(初始feii/feiii比=0.5)。然后将体积v=43ml的28/30%w/w氢氧化铵加入到烧杯中,并在环境温度下进行剧烈的机械搅拌。加入氢氧化铵导致在碱性水溶液(ph>10)中形成絮凝的磁铁矿(fe3o4),然后使磁铁矿絮凝物在由永磁体产生的磁吸引力的作用下沉降,然后将上清液移除并用纯水(milliq级别)替换。洗涤步骤重复两次,以除去过量的氢氧化铵。
步骤2:铵抗衡离子的解吸和表面氧化。
在洗涤磁铁矿的连续步骤之后,将体积v=28.6ml的2mol·l-1硝酸hno3水溶液加入到磁铁矿絮凝物中,并置于机械搅拌下30分钟以对磁铁矿颗粒的表面进行处理。
硝酸的添加使得可以使介质酸化并通过与硝酸根no3-离子的离子交换在纳米颗粒的表面诱导过量铵nh4+抗衡离子的解吸。在表面处的颗粒的氧化还使得可以溶解未沉淀并存在于纳米颗粒表面的亚铁离子。
使表面处理的磁铁矿絮凝物在永磁体下进行沉降,然后将上清液移除并用纯水替换,该步骤重复两次。
步骤3:纳米颗粒的核的氧化。
在洗涤表面处理的磁铁矿的连续步骤之后,将沸腾的体积v=85.7ml的新鲜制备的0.33mol·l-1硝酸铁feiii(no3)3·9h2o溶液加入到磁铁矿絮凝物中,并在机械搅拌下回流45分钟。
通过硝酸铁引入fe3+离子使得可以将颗粒的feii氧化,从而形成磁赤铁矿γ-fe2o3品种。在颗粒完全氧化后,使磁赤铁矿絮凝物在永磁体下进行沉降,将上清液移除,然后用纯水替换,该操作重复两次。
步骤4:磁铁矿纳米颗粒的“胶溶作用(peptization)”。
将体积v=28.6ml的2mol·l-1硝酸hno3溶液加入到磁赤铁矿絮凝物中,并在环境温度下置于机械搅拌下30分钟。加入硝酸使得可以在磁赤铁矿的表面引入水合氢h+离子。使阳离子磁赤铁矿絮凝物沉降,然后用丙酮洗涤三次。然后将体积v=70ml的水加入到纳米颗粒中,使得能够在水中进行纳米颗粒的“胶溶作用”,然后通过纳米颗粒表面的正电荷的静电斥力来使纳米颗粒的分散体稳定化。最后,通过在40℃下真空蒸发除去残留的丙酮。
p(meo2ma-co-oegma-co-maa)/γ-fe2o3杂化微凝胶的合成
通过将p(meo2ma-co-oegma-co-maa)微凝胶的水分散体与在ph2稳定化的磁赤铁矿纳米颗粒的分散体(带有阳离子电荷的纳米颗粒)简单混合而合成杂化微凝胶。纳米颗粒在微凝胶内的包封分两步进行:
·第一步是将阳离子纳米颗粒加入到在ph3和环境温度下分散的微凝胶溶液中。这些混合条件使得可以在γ-fe2o3纳米颗粒的表面保留阳离子电荷。由于由微凝胶中所含的甲基丙烯酸单元产生的羧酸基团,纳米颗粒将优先与微凝胶相互作用。具体而言,羧酸基团具有被吸附在诸如氧化铁等金属氧化物的颗粒表面的能力,进而,纳米颗粒表面的正电荷使得能够进行有利的相互作用。在这个意义上,加入γ-fe2o3纳米颗粒后,后者将优先位于微凝胶内(步骤概括在图4中)。
·第二步是将介质(微凝胶+纳米颗粒)的ph值从ph3开始提升至ph7.5。ph升高引起:1)混合物中阳离子γ-fe2o3纳米颗粒的去稳定化。具体而言,由于纳米颗粒在中性ph处具有零电荷点(等电点=7.2),后者在该ph下由于缺乏静电排斥而絮凝。2)由处于羧酸根(coo-)基团形式的羧酸(cooh)官能团衍生的微凝胶内的负电荷产生。由于羧酸根官能团的负电荷,这两种现象的共同作用使得可以将磁性纳米颗粒锚定在微凝胶内并提高杂化微凝胶的稳定性(步骤概括在图5中)。
实验方案
将体积为40ml的重量浓度为1.45g·l-1的p(meo2ma-co-oegma-co-maa)微凝胶的水分散体引入100ml圆底烧瓶中并置于磁力搅拌下,通过加入0.1mol·l-1硝酸(hno3)溶液将分散体的ph调节至3.0。接下来,在环境温度和磁力搅拌下,将体积为10ml的重量浓度为1.34g·l-1的ph3的阳离子磁铁矿纳米颗粒的分散体逐滴加到混合物中,这相当于各杂化微凝胶中纳米颗粒含量为约18.8%。将反应混合物在环境温度下置于搅拌下12小时。然后通过逐滴加入0.5mol·l-1氢氧化钾(koh)溶液来提高反应混合物的ph。最后,通过离心(5000rpm,20分钟)将杂化微凝胶从反应介质中分离,并将反应介质用纯水(milliq级别)替换。然后,最终溶液由水中的p(meo2ma-co-oegma-co-maa)微凝胶的胶体分散体组成,将该分散体保持在环境温度下。已经通过使每杂化微凝胶的纳米颗粒理论重量分数在0和33%之间变化,来进行各种合成。
γ-fe2o3/p(meo2ma-co-oegma-co-maa)杂化微凝胶的性能
通过透射电子显微术(tem)对干燥状态下的杂化微凝胶进行表征,并通过动态光散射对湿态下的杂化微凝胶进行表征。通过tem证实了微凝胶的杂化结构,在干燥状态下观察微凝胶揭示了磁性纳米颗粒在微凝胶内的良好包封,其在干燥处理期间不被排出(expelled)。通过热重分析测定包封的纳米颗粒填料的含量,该分析证实了纳米颗粒的定量包封和有效包封(所测试的填料含量范围为每杂化微凝胶含0至33wt%的纳米颗粒)。还通过杂化微凝胶的收缩证明了杂化微凝胶在中性ph下在水溶液中的温度响应性性能,其从20℃下的1000nm变化至450nm,收缩温度为37℃。中性ph下的这种收缩的发生与磁性纳米颗粒填料含量无关。
实施例4:微凝胶的膜
使用总结在表1中的微凝胶的组成来制备膜。
表1.微凝胶的单体组成
a微凝胶1含有0mol%maa单元;
b微凝胶2、3、4和5含有3.5mol%掺入至微凝胶中的maa单元(即初始maa单体的70%);
c通过热重分析(tga)测定的每杂化微凝胶的γ-fe2o3纳米颗粒含量。
从微凝胶在水中的重量浓度为1.4wt%至5wt%的单分散性微凝胶的胶体分散体(在溶液中的尺寸可在500μm和1000μm之间)(溶液1)开始,通过图6中所示的干燥方法制备膜。将恒定体积的溶液引入塑料模具中并保持进行干燥直到水完全蒸发(图6的步骤1)。然后,保留在模具底部的膜由若干层单分散性且完全干燥的微凝胶组成(在干燥状态下的尺寸可在350μm和450μm之间)。小心地回收膜并将其重新引入水溶液(溶液2)。多种参数发生变化:1)分散体的重量浓度从1.4wt%变化至5wt%,使得可以改变溶胀膜厚度(步骤3结束:厚度从200μm变化至1000μm)。2)溶液2的ph在5.5和7.5之间变化。
实验方案:基于聚(寡聚(乙二醇)甲基丙烯酸酯)的温度响应性微凝胶的膜的形成。
将体积为5ml的重量浓度为1.4wt%至5wt%的微凝胶的胶体分散体引入塑料模具中,并在32℃(±2℃)的温度下干燥。在溶剂完全蒸发后,小心地回收膜,然后将其引入水溶液中,并在环境温度下溶胀。
通过原子力显微镜在干燥状态下观察回收的膜,并在湿态下使用流变仪(rheometer)进行表征(图6的步骤3的结尾)。微凝胶形成由若干层微凝胶组成的弹性膜(步骤2),这与微凝胶(表1中的微凝胶1至5)的组成无关。相反,当微凝胶在水中溶胀而不在溶液中再分散时,它们的机械性能丧失。
微凝胶1和2:膜厚度从200μm变化至1000μm,微凝胶层的增加未改变“成膜”现象,并且膜在干燥状态下保持其弹性。
微凝胶3、4和5:在杂化微凝胶的情况下,对约300μm的厚度(步骤3中的溶胀膜)进行了研究。纳米颗粒的添加未改变微凝胶的成膜性能。相反,在湿态下,膜的机械性能大大提高。
膜的机电性能的评估
1.膜的机电性能的表征
对微凝胶膜的机电性能进行研究。这是涉及当在这些微凝胶上施加压力时证明微凝胶产生电流的能力的问题。更具体地,本发明的思想是通过在衬底表面上压缩材料来产生电流。该电位可以由具有共价键合的离子官能团的材料(或聚电解质材料)产生。具体地,由于离子基团连接在微凝胶中,所以只有各羧酸根基团的抗衡离子在微凝胶中具有移动性。当对这些聚电解质微凝胶施加单向压力/变形时,抗衡离子的移动性是有利的,由此在移动的抗衡离子的正电荷和所连接的羧酸根基团的负电荷之间产生极化。该离子梯度导致界面处的电位。因此,微凝胶中可离子化官能团的存在将使得能够在材料内产生极化并产生电位。
使用组件来证明膜的机电性能。为此,以平板-平板几何结构(plate-plategeometry)来使用antonpaarmcr301流变仪,其中,基于氧化铟锡或ito的两个平坦的导电电极(图7的实体1)在该几何结构的任一侧相连接。下电极固定,上电极可拆卸。将微凝胶的湿膜(图7的实体3)沉积在下电极表面上并降低上电极,以在膜上施加压缩力fn。通过控制两个电极之间的距离,可以控制施加在膜上的挤压力(crushingforce,fn)。
进行压缩/松弛程序以改变施加在膜上的力。首先,测定湿膜的初始厚度(表示为l0),并通过降低上电极来使两个电极之间的距离逐渐减小距离δl。该程序的特点是以最终距离l进行短时间(τ=2秒)的挤压,然后进行长时间(τ=20秒)的松弛,返回初始状态l0,所有这些使得可以在膜上模拟“触碰敏感”型动作(图8)。
在挤压期间,记录法向力fn(以牛顿计)。该力fn与挤压厚度(δl)成比例。将程序设置为:将膜压缩到距离l=l0-δl,δl=(3x)10%l0,然后(3x)20%l0,然后(3x)25%l0等。所用程序的实例如图9所示。
最后,将上电极和下电极连接到转换器/放大器,以记录在整个程序中在两个电极之间产生的电位差(表示为e)。
2.微凝胶膜的研究
p(meo2ma-co-oegma-co-maa)微凝胶膜
通过改变3个参数来对p(meo2ma-co-oegma-co-maa)生物相容性微凝胶的膜进行表征:
1.)压缩倍增(multiplication)的作用:对于各膜,将相同力的压缩连续重复(如图9所示的3次),并对各压缩的电位进行分析。
2.)膜厚度:对两种膜厚度进行测试,以确定厚度对膜在界面处产生电位的能力的影响(约200μm和约900μm-1000μm)。
3.)羧酸官能团的组成:为了评估maa单元对电位的影响,将maa单元的组成从0变化至3.5mol%maa(微凝胶1和2)。
4.)膜在其中溶胀的溶液的ph:ph使得可以改变微凝胶内离子官能团(coo-)的量。具体而言,羧酸官能团以质子化(cooh)物类和去质子化或电离(coo-)物类这两种形式存在。这两种物类的比例取决于溶液的ph值,当ph升高时,电离物类coo-增加(ph5.5→coo-%=0;ph6.5→coo-%=50%;ph7.5→coo-%=75%)。
表2.所研究样品的概述
结果:
通过观察微凝胶膜压缩期间电信号的变化,在所有表征的膜上都证实了机电效应。这种机电效应是施加在膜上的力fn的函数,电位随压缩力而增加。此外,似乎显现出作为分析参数的函数的趋势:
·重复压缩的影响:在第一次压缩期间记录的电位非常高。然而,在第二次和第三次压缩期间,产生的电位较低。该首要观察可能是由于在膜的第一次压缩中离子的显著移动产生高瞬时电位(约12mv)。在20秒的松弛时间之后,接下来以相同力进行的压缩似乎不足以产生这一相同的离子运动,所产生的电位减小。
·膜厚度的影响:当膜厚度过厚时,由对不同厚度的膜进行压缩而产生的电位显示出弱机电效应(对于fn,max=0.35n,e=2-5.6mv)。相反,当膜厚度较小时,看到更大的机电效应,对于力fn为0.38n至0.4n而言为11mv至5mv。因此,太大的厚度将使得无法对离子的移动性产生足够的影响。(参见表2.膜厚度的影响)
·羧酸官能团的组成:甲基丙烯酸的存在似乎改善了膜的机电性能。具体而言,这些膜似乎对压缩效应更为敏感,对于弱力测得电位(在0.4n有maa时为11-5mv;而在0.4n无maa时为1-0.5mv)。此外,在ph6.5进行的测量突出了衍生自maa单元的电离羧酸根基团(电离maa基团的50%)对微凝胶膜敏感性的重要性。(参见表2.maa组成的影响)
·对于相同组成的微凝胶膜,5.5至7.5的溶液ph似乎不会改变膜的电位值,但是当溶液ph值增加时,面对压缩重复,电位损失似乎减少。具体而言,在ph5.5时,压缩重复使电位降至2mv,而在较高ph下,电位降至5.4mv。这可能是由于电离羧酸根官能团的比例(cooph5.5%=0%;cooph6.5%=50%;cooph7.5%=75%)增加,增加了形成膜的微凝胶的极化能力。从而,当ph增加时,膜更敏感。(参见表2.ph的影响)
p(meo2ma-co-oegma-co-maa)/γ-fe2o3杂化微凝胶的膜
在ph7.5下对杂化微凝胶的膜进行表征,并将其与不含磁性纳米颗粒(np)的微凝胶膜进行比较。不含纳米颗粒的微凝胶膜对于压缩力fn=0.4n而言最大电位为6mv。对于具有纳米颗粒的微凝胶的膜,产生的电位取决于掺入的纳米颗粒的量:
·对于掺入~5wt%的磁性纳米颗粒而言,纳米颗粒对产生的电位无影响,膜对压缩的响应性为6mv-7mv。
·对于掺入9wt%和17wt%的磁性纳米颗粒而言,观察到无论压缩力如何,电位均有所降低,低至2.5mv。这种电位的损失可能归因于在ph7.5下衍生自羧酸单元的电荷减少(由于它们已经与np相互作用)。具体而言,通过吸附包含于微凝胶中的离子位点(coo-)处的后者而发生纳米颗粒的掺入。这种吸附似乎降低了微凝胶内仍然可利用的离子位点的分数(fraction),从而降低了微凝胶的极化能力。掺入5wt%的量的纳米颗粒不影响杂化膜的聚电解质行为(参见表2.γ-fe2o3的影响)。
微凝胶的光学性能
除了机电性能之外,微凝胶膜的特征还在于其光学性能,这与这些膜衍射光的能力有关。观察到取决于膜组成的差异(disparity):
p(meo2ma-co-oegma-co-maa)微凝胶的膜
在不含纳米颗粒的微凝胶的胶体分散体的干燥期间,所形成的膜在干燥状态下是透明的,而在湿态下呈虹彩色(iridescent)(图10)。看起来,在微凝胶溶胀期间,颗粒之间的距离和直径有利于可见光区域中的光的衍射,该衍射通过观察光子晶体而得以证明。
p(meo2ma-co-oegma-co-maa)/γ-fe2o3杂化微凝胶的膜
在微凝胶的胶体分散体的干燥期间,获得在干燥状态下透明且有色的膜,其在非常小的视角下具有反射的虹彩性质。当在90°观察时该材料则呈现棕色(由于磁性纳米颗粒而产生的颜色),并且当以更小的角度观察时呈虹彩色(图11)。
如图12所示,光子性能特别是在反射(在深色背景上)中可见,而在透射(在浅色背景上)中几乎不可见。
杂化膜的机械性能和磁性能
除了杂化微凝胶的成膜性能及其光学性能之外,磁性纳米颗粒的添加还使得可以在干燥期间使微凝胶定向。图13清楚地示出了这些性能,因为可以通过施加磁场(在我们的情况下将磁体放置在分散体下方)在干燥期间使微凝胶集中在精确位点。干燥一方面使得可以将所有物质设置在目标点上,另一方面使得可以通过杂化微凝胶的局部浓度来修饰最终膜的色调,同时保持反射中的虹彩性质(有磁体时干燥的溶液,图13)。
- 还没有人留言评论。精彩留言会获得点赞!