一种采用高效液相色谱法同时检测乳中克拉维酸和他唑巴坦的方法与流程
本发明涉及一种采用高效液相色谱法同时检测乳中克拉维酸和他唑巴坦的方法,属于食品检测技术领域。
背景技术:
克拉维酸和他唑巴坦都是竞争性β-内酰胺酶抑制剂,可以与β-内酰胺酶结合后使其失活,具有抑制革兰氏阳性菌和阴性菌的作用。
抗生素的大量应用,导致其在乳中的残留问题受到广泛关注。为了躲避抗生素的检出,有些人在乳中添加β-内酰胺酶,用以降解抗生素,从而无法判断乳中是否含有抗生素。所以,针对这一现象,建立了关于乳中β-内酰胺酶的检测方法。但是,有些人又利用β-内酰胺酶抑制剂能不可逆地与β-内酰胺酶结合的性质,在乳中添加β-内酰胺酶抑制剂,使β-内酰胺酶失去活性,导致阳性奶不能被检测出来。
关于乳中β-内酰胺酶抑制剂的检测方法在国内外还没有现行的检测标准发布,国外由于其文化发展程度的差异,主要将目标放在临床医疗中的使用情况,并没有看到克拉维酸和他唑巴坦在乳中的报道;而国内对于克拉维酸和他唑巴坦的检测方法的报道很少,文献报道的方法又主要集中在微生物法、酶法、毛细管电泳法等。
由于目前尚无良好有效的检测手段,建立乳中β-内酰胺酶抑制剂克拉维酸和他唑巴坦的检测方法非常重要。
技术实现要素:
为解决现有技术的不足,本发明提供了一种采用高效液相色谱法同时检测乳中克拉维酸和他唑巴坦的方法,采用的技术方案如下:
本发明的目的在于提供一种采用高效液相色谱法同时检测乳中β-内酰胺酶抑制剂克拉维酸和他唑巴坦的方法,该方法步骤如下:
1)将样品进行前处理;
2)按照如下色谱条件利用高效液相色谱进行测定;所述色谱条件为:色谱柱为C18色谱柱;检测波长为210nm-220nm;流动相为磷酸溶液-甲醇;流速为0.7mL/min-1.2mL/min;进样量为10μL;洗脱时间为10min。
优选地,所述前处理,是称取1份牛奶与乙腈混合,震荡混匀后离心,取上清液与正己烷混合,震荡混匀后离心,弃去正己烷层,氮气吹干后,用流动相复溶,最后过微孔滤膜。
优选地,所述磷酸溶液浓度为0.01%-0.03%;pH为3.5-4.5。
更优选地,所述磷酸溶液浓度为0.02%;pH为4。
优选地,所述磷酸溶液-甲醇,体积比为95:5-90:10。
更优选地,所述磷酸溶液-甲醇,体积比为90:10。
优选地,所述C18色谱柱,规格为4.6mm×250mm,5μm。
优选地,具体步骤如下:
1)将样品进行前处理:称取1份牛奶与3份-5份乙腈混合于离心管中,震荡混匀后离心,取上清液与4份-6份正己烷混合于另一离心管中,震荡混匀后离心,弃去正己烷层,在35℃-45℃氮气吹干后,用流动相复溶,最后过微孔滤膜;
2)按照如下色谱条件利用高效液相色谱进行测定;所述色谱条件为:色谱柱为4.6mm×250mm,5μm的C18色谱柱;检测波长为210-220nm;流动相为体积比为95:5-90:10的磷酸溶液-甲醇;流速为0.7mL/min-1.2mL/min;进样量为10μL;洗脱时间为10min;所述磷酸溶液浓度为0.01%-0.03%,pH为3.5-4.5。
更优选地,具体步骤如下:
1)将样品进行前处理:称取1份牛奶与4份乙腈混合于离心管中,震荡混匀后离心,取上清液与5份正己烷混合于另一离心管中,震荡混匀后离心,弃去正己烷层,在40℃氮气吹干后,用流动相复溶,最后过微孔滤膜;
2)按照如下色谱条件利用高效液相色谱进行测定;所述色谱条件为:色谱柱为4.6mm×250mm,5μm的C18色谱柱;检测波长为215nm;流动相为体积比为90:10的磷酸溶液-甲醇;流速:1.0mL/min;进样量为10μL;洗脱时间为10min;所述磷酸溶液浓度为0.02%,pH为4。
以上所述任一方法在同时检测克拉维酸和他唑巴坦中的应用。
本发明对采用HPLC法同时检测乳中β-内酰胺酶抑制剂克拉维酸和他唑巴坦的色谱条件进行了优化,取得最佳色谱条件,然后针对此条件进行克拉维酸和他唑巴坦方法学验证。
本发明有益效果:
本发明采用高效液相色谱法建立了乳中β-内酰胺酶抑制剂克拉维酸的同时检测方法,样品采用乙腈除蛋白,正己烷脱脂的方法,最佳色谱条件为:色谱柱为Hypersil ODS-2C18(4.6mm×250mm,5μm),流动相为0.02%磷酸溶液(pH4)-甲醇(90:10),检测波长为215nm,流速为1mL/min。该方法的线性范围为12.5~200μg/mL,相关系数为0.9990和0.9992,平均回收率在81%~92%之间,相对标准偏差小于1.650%。本发明方法操作简便,结果可靠,对于保证产品的质量,保护消费者健康具有十分重要的作用,具有一定的现实和指导意义。适用于乳中β-内酰胺酶抑制剂克拉维酸和他唑巴坦的同时检测。
附图说明
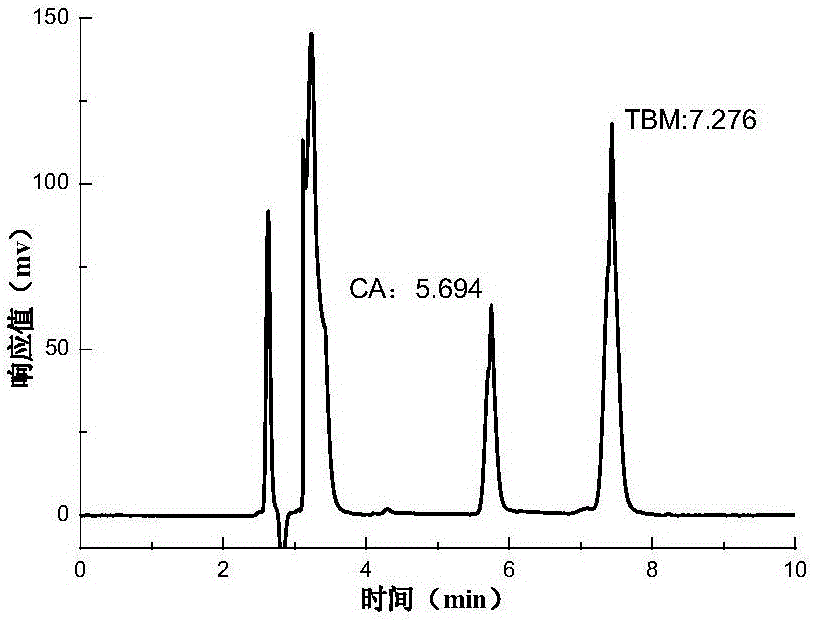
图1最佳色谱条件下克拉维酸和他唑巴坦标准溶液的色谱图。
图2最佳色谱条件下原料乳中添加克拉维酸和他唑巴坦的色谱图。
图3克拉维酸和他唑巴坦的标准曲线。
具体实施方式
下面结合附图对本发明的技术方案作进一步的说明,但并不局限于此,凡是对本发明技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,均应涵盖在本发明的保护范围中。
实施例1:
一、标准储备液的制备
精确称取克拉维酸和他唑巴坦标准品,将其配成浓度为1mg/mL的标准储备液,使用时用流动相稀释成所需的浓度,所有溶液使用前需过0.45μm的微孔滤膜。
二、液相色谱条件
色谱柱:Hypersil ODS-2C18(4.6mm×250mm,5μm);检测波长:210-220nm(优选为215nm);流动相:0.02%磷酸溶液(pH4)-甲醇(90:10);流速:0.7mL/min-1.2mL/min(优选为1mL/min);进样量:10μl;梯度洗脱时间:10min。
三、样品前处理方法
称取1份牛奶与3份-5份乙腈混合于离心管中,震荡混匀后离心,取上清液与4份-6份正己烷混合于另一离心管中,震荡混匀后离心,弃去正己烷层,在35℃-45℃氮气吹干后,用流动相复溶,最后过微孔滤膜。
经试验发现牛奶与乙腈的最佳体积比为1:4,牛奶与正己烷的最佳体积比为1:5时,氮气吹干最佳温度为40℃,即称取1份(如1mL)牛奶与4份(如4mL)乙腈混合于离心管中,震荡混匀(如1min)后离心(如4000r/min,15min),取上清液与5份(如5mL)正己烷混合于另一离心管中,震荡混匀(如1min)后离心(如4000r/min,8min),弃去正己烷层,重复除脂2次,在40℃氮气吹干后,用流动相复溶,最后过微孔滤膜(如0.45μm)。
四、色谱条件的优化
1、流动相选择及配比的优化
本试验首先考察了同等条件下甲醇-水(10:90)和乙腈-水(10:90)两种流动相对物质分离效果的影响。结果表明,当流动相为乙腈-水时,色谱峰拖尾;当流动相为甲醇-水时,峰型好,分离度高,待测物质可得到好的洗脱效果。因此,本试验确定甲醇-水为流动相。
确定甲醇-水为流动相后,考察甲醇与水以不同的比例存在时,对待测物质分离效果的影响。结果表明,甲醇比例从5%到35%的变化过程中,随着甲醇浓度的降低,保留时间逐渐延长(表-1)。但当甲醇浓度低于10%时,分离条件没有达到最佳状态,杂质和克拉维酸与他唑巴坦没有完全分开。因此甲醇-水最佳体积比=10:90。
表1流动相配比与保留时间的关系
2、缓冲溶液的选择及浓度的优化
在色谱分析中,向水相中添加缓冲溶液,达到防止样品解离,峰型拖尾,增加方法的可靠性等作用。本试验考察四丁基氢氧化铵和磷酸氢二钾两种缓冲溶液对分离效果的影响。结果表明,以四丁基氢氧化铵为缓冲溶液时,克拉维酸的色谱峰出现包峰现象,分离效果较差;而选择磷酸氢二钾时,克拉维酸得到较好的洗脱,且分离度好。因此,本试验选择以磷酸作为缓冲溶液。
在水相中添加磷酸,配制成不同浓度的磷酸溶液,按照前面确定的甲醇与水的最佳体积比进行混合,考察缓冲溶液的浓度对待测物质分离效果的影响。结果表明,在一定范围内,流动相的离子强度越高,则保留时间越长(表-2),但考虑液相色谱对浓度的限制,在保证有效分离的前提下,应选择离子强度相对较低的缓冲液体作为流动相。因此,本试验确定磷酸的最佳浓度为0.02%。
表2磷酸浓度与保留时间的关系
3、缓冲溶液pH值的优化
缓冲溶液pH值的选择对于试验的结果非常重要,若pH值的选择不恰当会导致分裂峰、不对称峰、肩峰或宽峰等。本试验考察了不同的pH值对保留时间及分离效果的影响。结果显示,pH值的改变对保留时间影响不大(表-3),但可以影响色谱峰的峰型以及实验的重现性。当pH值为4时,色谱峰峰型好,分离效果也好,并且与0.02%磷酸时的pH值最为接近。因此,本试验最佳pH为4。
表3 pH值与保留时间的关系
五、方法学的验证
1、系统适用性
由图1、图2可以看出,溶剂不干扰主峰和有关物质峰的测定,克拉维酸和他唑巴坦标准品溶液在最佳色谱条件下的保留时间与原料乳中克拉维酸和他唑巴坦的保留时间基本一致。综上表明,此最佳色谱条件适用于乳品中克拉维酸和他唑巴坦的同时检测应用。
2、标准曲线与检测限
分别精密称取克拉维酸和他唑巴坦标准品25mg置于25mL容量瓶中,加流动相稀释至刻度,摇匀;然后分别精密量取上述溶液,用流动性逐步稀释浓度为12.5、25、50、100、200μg/mL的标准溶液,按上述最佳色谱条件进样测定。每个浓度平行测定5次,按所得峰面积平均值(y)对克拉维酸和他唑巴坦的浓度(μg/mL)做标准曲线,得出克拉维酸和他唑巴坦的回归方程与相关系数(图3)。
3、回收率与精密度
本试验采用空白加标的方法进行回收率和精密度测定。将5、50、100μg/mL 3个浓度的标准品添加到空白样品中,按照1.3.2节的方法进行处理,每个浓度样品测定5次,记录峰面积,分别带入回归方程,并依据公式:回收率=样品测定值/标准加入量×100%进行测定,结果如表4。由表中结果可以看出,该方法的平均回收率在81%~92%之间,相对标准偏差小于1.650%。
表4乳中克拉维酸和他唑巴坦的加标回收率及精密度
4、实际样品的测定
按照本试验所建立的高效液相色谱的方法对市场销售的15种液态奶进行测定,暂未发现阳性样品。
虽然本发明已以较佳的实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可以做各种改动和修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!