一种利用原位碳化模板合成有序介孔ZSM‑5的方法与流程
本发明属于zsm-5分子筛的合成方法技术领域,具体涉及一种利用原位碳化模板合成有序介孔zsm-5的方法。
背景技术:
分子筛具有尺寸和形状筛分作用,被广泛应用于催化、炼油、吸附、分离等多个领域,并被做成各种产品如离子交换剂、挥发性有机化合物(voc)的吸附剂、光电器件、传感器、药物胶囊、护肤品等等。在催化反应中,以分子筛为主要催化剂的甲醇制烯烃、催化裂解、加氢等反应,实现了由煤炭或天然气经甲醇生产基本有机化工原料,为解决能源危机提供了有效途径。
影响分子筛催化性能的主要因素有分子筛的拓扑结构、酸性、扩散速率和催化测试条件等。拓扑结构指的是孔口或笼的大小、有无边带,酸性指的是酸量和酸位点分布,扩散速率指的是晶体尺寸、有无介孔,催化测试条件指的是温度和空速。因此探讨如何提高低碳烯烃的选择性和减少积炭的生成以延长催化剂的使用寿命,将对于实际工业生产具有很大的指导意义。合成同时具有微孔-介孔甚至大孔多级结构的层状分子筛,将会提高催化活性,减少空间/扩散限制。此外,二级孔隙为活跃成分的沉积和有机基团的功能化提供了充足的空间。然而,二级表面的出现阻断了晶体骨架的连续性,骨架连通性变低,si-oh浓度增加。最终,层状分子筛与传统分子筛相比,在酸性、亲疏水性、空间限制效应、形状选择性和水热稳定性方面显示了并不那么“沸石化”的行为。但是,二级表面并不是无定型的,这赋予了介孔分子筛一系列新颖的特征。目前已经开发了很多合成介孔分子筛的方法。
与传统的沸石相比,在微孔沸石中引入二级孔道可以缩短扩散通道并暴露更多的酸性位点,从而有望改善催化剂的催化性能,如活性和选择性等。介孔分子筛的合成策略有软模板法(如表面活性剂、大分子聚合物等)、硬模板法(碳材料等固体)、蒸汽辅助干凝胶转化法(sac)、酸/碱刻蚀脱al/si等,也可以将多种合成方法相结合。在这些合成方法中,在硬模板中进行限域合成能够获得均匀的介孔和可控的晶体尺寸,这在其他合成方法中不太容易得到。无定形前驱体在由硬模板围成的有限空间内形成沸石晶体,固体模板在沸石晶化后通过煅烧或酸洗可以很容易地除去。
尽管近些年来在寻求新颖的合成多级孔道沸石的方法上取得了巨大进展,但是仍然面临很大的挑战。新的合成方法必须满足以下要求:可以用于合成具有多种结构和组成的分子筛类型、层状分子筛的特性可调、成本低廉、简单易行。未来的发展方向有可能是多种方法相结合,比如酸和碱双步骤脱铝、脱硅与表面活性剂组装相结合等。此外,在二级孔内掺杂其他的活性物种还没有被充分研究,比如可以开发活性物种高度分散的多功能层状分子筛。此外,尽管很多工作将层状分子筛进行放大合成,并做成适用于工业反应器的形状和尺寸,但是经济成本仍然是目前绝大多数合成策略的瓶颈。例如,脱除骨架原子会造成分子筛质量的巨大损失,硬模板合成在分子筛结晶之后被完全破坏,而双模板合成所使用的介孔模板剂价格高昂。因此,有必要优化合成策略以达到尽量降低成本和获取最优异的催化性能之间的平衡。
碳纳米粒子和介孔碳是合成介孔分子筛广泛使用的硬模板。采用多孔碳做硬模板可以做成介孔分子筛晶体、纳米分子筛或者碳支持的分子筛等。此外采用有序介孔碳模板如cics、cmks、3dom-i等,并通过控制合成条件,可以合成高度有序的介孔分子筛。chen等人使用三维有序介孔碳(3dom),通过水热合成得到了三维有序的介孔bea、lta、fau和ltl型分子筛,这种方法需要大量的水,合成的分子筛介孔有序但较大。cho等人使用具有10nm介孔的cmk-l合成了高度有序的介孔silicalite-1,这种分子筛的比表面积较小。hu等人通过晶化cmk-5原位碳化填充的sba-15,得到了介孔壁被完全晶化的有序介孔沸石,然而,以cmk-3介孔碳为直接模板或者晶化cmk-3原位碳化填充的sba-15均不容易得到有序介孔分子筛,而是无序的沸石纳米晶体的团聚体,或者未完全结晶的sba-15与沸石碎片的混合物。
对于原位碳化法合成介孔分子筛,控制碳化方法以确保得到高度有序的介孔碳以及选用合适的合成条件,是获得有序介孔沸石的关键。kim等人通过控制碳化条件,成功地将sab-15合成初期的介孔内的p123模板碳化成了有序的介孔cmk-3,其中的关键步骤是对sba-15/p123复合物仅用h2so4进行预碳化。此外,传统的水热方法可能对cmk-3外表面的沸石生长不利,沸石前驱体可能从cmk-3的外表面迁移出来,团聚成无序的介孔结构或形成大尺寸沸石晶体。
技术实现要素:
基于背景技术中提出的技术问题,本发明提供了一种利用原位碳化模板合成有序介孔zsm-5的方法,该合成方法能够充分利用最初的p123模板剂,在少量水的条件下可以合成出有序介孔zsm-5,且该有序介孔zsm-5的比表面积较大,介孔体积较大。
为实现上述技术目的,本发明采用的技术方案如下:
一种利用原位碳化模板合成有序介孔zsm-5的方法,所述的合成方法包括以下步骤:
步骤一,制备sba-15/p123复合物和mcf-p123复合物:
sba-15/p123复合物的制备,称取3.5g~6g的p123加入由15ml~20ml质量百分数为36.5wt%的hcl和100ml~150ml的h2o配制的hcl溶液中,剧烈搅拌直至完全溶解,再加入5ml~10ml分析纯的teos混合,静置老化,过滤并室温干燥,即得到sba-15/p123复合物;mcf-p123复合物的制备,称取3.5g~6g的p123加入由15ml~20ml质量百分数为36.5wt%的hcl和100ml~150ml的h2o组成的hcl溶液中,再加入2,4,6-三甲基苯,剧烈搅拌直至完全溶解,然后加入5ml~10ml分析纯的teos混合,静置老化,过滤并室温干燥,即得到mcf-p123复合物;
步骤二,分别对sba-15/p123复合物和mcf-p123复合物进行碳化:
称取0.8g~1.2g的sba-15/p123复合物或0.8g~1.2g的mcf-p123复合物加入h2so4溶液中,搅拌30min,干燥后,得到的棕色固体放置于管式炉中,在高纯ar的保护下,以1℃/min升温到900℃并保持3h,得到黑色固体为碳化的sba-15/p123复合物或mcf-p123复合物。
步骤三,蒸汽辅助结晶化法合成有序介孔zsm-5:
称取1.0g~1.2g质量百分数为25wt%的tpaoh、3.1g~3.4g的h2o和0.04g~0.05g的异丙醇混合,充分搅拌5h,直至得到澄清溶液,再加入0.8g~1.1g步骤二碳化得到的sba-15/p123复合物或mcf-p123复合物,混合搅拌1h,于烘箱中第一次干燥后,压碎,并悬置于内设有不锈钢支架的反应釜内衬中的支架上,所述反应釜内于支架的下方装有水用于形成饱和蒸汽压,封釜,并将反应釜置于160℃的烘箱中加热0h~48h后,将固体于烘箱中第二次干燥后,转移至马弗炉中,以2℃/min升温至550℃并保持6h,然后在60℃的条件下加入到nh4no3溶液中缓慢搅拌2h进行离子交换后,煅烧得到h+形式两种不同的白色固体粉末,即两种介孔zsm-5。
采用上述技术方案的发明,利用原位碳化介孔模板法合成了两种有序介孔zsm-5分子筛。将未除p123模板的sba-15进行原位两步碳化,即硫酸预碳化和氩气900度高温碳化,使内部的p123软模板最大程度地碳化成介孔有序的硬模板,经浸渍tpaoh溶液和蒸汽辅助晶化后,最后空气煅烧除碳,得到了两种具有不同介孔大小的结晶有序介孔zsm-5分子筛。利用蒸汽辅助晶化法在少量水的条件下可以合成出介孔zsm-5,比水热法或足量水晶化法更节约溶剂,且后处理更简单,合成后从反应釜中取出直接干燥煅烧即可,省去了煅烧前的洗涤等繁琐的后处理步骤。
作为本发明的一种优选方案,所述步骤一中的2,4,6-三甲基苯的加入量为4.0ml~4.5ml,这样的设计,2,4,6-三甲基苯作为扩孔剂,有利于增大由mcf-p123复合物碳化得到的介孔zsm-5的比表面积和孔容。
作为本发明的一种优选方案,所述步骤一中的静置老化均为两级老化,先放置于40℃的环境下静置老化20h,再置于100℃的环境下静置老化24h后,这样的设计,有利于sba-15/p123复合物或mcf-p123复合物反应前驱体的形成,为后续的碳化做准备。
作为本发明的一种优选方案,所述步骤二中的干燥分两级干燥,先在100℃的烘箱中干燥12h,然后在160℃的条件下继续干燥12h,这样的设计,干燥充分彻底,为后续的高纯氩气高温干燥做准备,使内部的p123软模板最大程度地碳化成介孔有序的硬模板。
作为本发明的一种优选方案,所述步骤二中的h2so4溶液由4ml~10mlh2o和0.05ml~0.1ml质量百分数为98wt%的h2so4配制而成,这样的设计,该浓度的h2so4溶液可以将内部的p123软模板完全碳化。
作为本发明的一种优选方案,所述步骤三中的反应釜内水的体积为0.4ml~1ml,这样的设计,0.4ml~1ml水足够在密封的160℃反应釜中形成饱和蒸汽压用于辅助晶化,比水热法或足量水晶化法更节约溶剂。
作为本发明的一种优选方案,所述步骤三中第一次干燥和第二次干燥的温度均为60℃,这样的设计,既能进行充分干燥,又能保持干燥物质的宏观结构不被破坏。
作为本发明的一种优选方案,所述步骤三中向澄清溶液中加入sba-15/p123复合物或mcf-p123复合物后,得到的溶液中的al2o3、sio2、tpaoh、h2o的重量比值为1:100:12:1920,这样的设计,有利于合成较理想的有序介孔zsm-5。
作为本发明的一种优选方案,所述步骤三中加入的nh4no3溶液的体积为0.8ml~1ml,这样的设计,有利于充分地进行离子交换。
作为本发明的一种优选方案,所述步骤三中的煅烧温度为560℃~600℃,煅烧时间为6h这样的设计,该煅烧温度和煅烧时间有利于除去碳支撑体,同时不破坏有序介孔的形貌,有利于得到理想的有序介孔zsm-5。
相对于现有技术,本发明利用原位碳化介孔模板法合成了两种有序介孔zsm-5分子筛,将未除p123模板的sba-15进行原位两步碳化,即硫酸预碳化和氩气900度高温碳化,使内部的p123软模板最大程度地碳化成介孔有序的硬模板,经浸渍tpaoh溶液和蒸汽辅助晶化后,最后空气煅烧除碳,得到了两种具有不同介孔大小的结晶度较高的有序介孔zsm-5分子筛,由sba-15/p123碳化得到的有序介孔zsm-5标记为mesozsm-5a,由mcf-p123碳化得到的有序介孔zsm-5标记为mesozsm-5b。
蒸汽辅助晶化法是一种有效解决相分离的方法,在此方法中,位于介孔模板剂空隙内的沸石前驱体被浓缩成了干凝胶,相比于传统模板,此方法只需要消耗少量的水和小分子有机模板。在晶化过程中,保证充足的饱和蒸气压对凝胶的转化非常重要,本发明只需用0.4ml~1ml水来保证充足的饱和蒸气压。在蒸汽处理过程中,干凝胶和水是分开的,所以模板剂和固体sio2之间的相分离可以被成功避免。在原位碳化的碳材料作为碳模板,并运用蒸汽辅助晶化的双合成方法中,介孔内碳材料不仅作为硬模板从而保留介孔结构,还可以作为限域空间内干凝胶转化过程中的支架。利用蒸汽辅助晶化法在少量水的条件下可以合成出介孔zsm-5,比水热法或足量水晶化法更节约溶剂,且后处理更简单,合成后从反应釜中取出直接干燥煅烧即可,省去了煅烧前的洗涤等繁琐的后处理步骤。
mesozsm-5a和mesozsm-5b均为有序的六角相介孔分子筛,采用本发明的方法所合成的mesozsm-5a具有4nm~5nm的均一介孔尺寸,比sba-15的介孔尺寸(6.6nm)小很多,mesozsm-5b具有9.5nm~9.8nm的均一介孔尺寸,明显比mesozsm-5a的介孔尺寸大,mesozsm-5a的bet比表面积(sbet=375m2/g~396m2/g)、mesozsm-5b的bet比表面积(sbet=372m2/g~396m2/g)相较于商业zsm-5(sbet=312m2/g)更大;由mesozsm-5a和mesozsm-5b的29si和27almasnmr固体核磁谱图、xrd衍射谱图和ft-ir红外谱图可以看出mesozsm-5a和mesozsm-5b的结晶度很高。
附图说明
本发明可以通过附图给出的非限定性实施例进一步说明;
图1为本发明一种利用原位碳化模板合成有序介孔zsm-5的方法实施例的合成过程示意图;
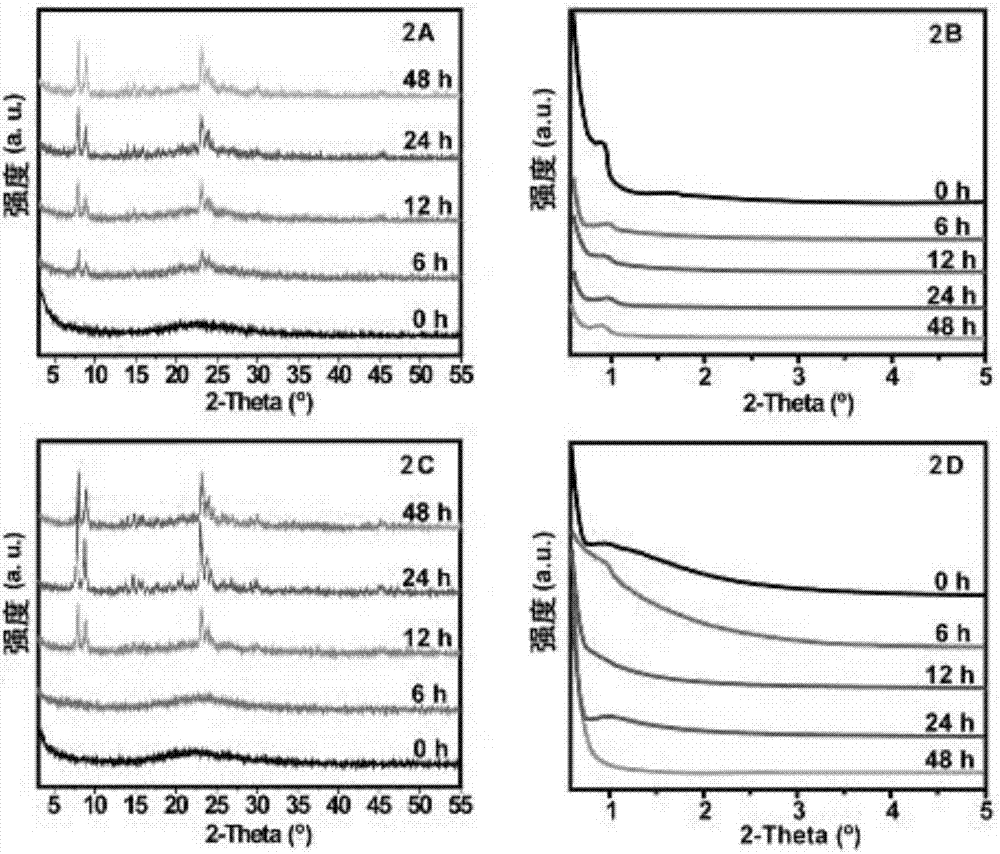
图2为本发明一种利用原位碳化模板合成有序介孔zsm-5的方法实施例得到的两种介孔zsm-5的xrd图像,其中,2a为mesozsm-5a的广角xrd图像,2b为mesozsm-5a的小角xrd图像,2c为mesozsm-5b的广角xrd图像,2d为mesozsm-5b的小角xrd图像;
图3为本发明一种利用原位碳化模板合成有序介孔zsm-5的方法实施例得到的两种介孔zsm-5的n2吸脱附曲线图,其中,3a为mesozsm-5a的n2吸脱附曲线图,3b为mesozsm-5b的n2吸脱附曲线图;
图4为本发明一种利用原位碳化模板合成有序介孔zsm-5的方法实施例得到的两种介孔zsm-5的tem图像,其中,4a、4b、4c、4d、4e分别代表蒸汽晶化时间为0h、6h、12h、24h、48h得到的mesozsm-5a的tem图像,4f、4g、4h、4i、4j分别代表蒸汽晶化时间为0h、6h、12h、24h、48h得到的mesozsm-5b的tem图像;
图5为本发明一种利用原位碳化模板合成有序介孔zsm-5的方法实施例得到的两种介孔zsm-5的sem图像,其中,5a、5b、5c、5d、5e分别代表蒸汽晶化时间为0h、6h、12h、24h、48h得到的mesozsm-5a的sem图像,5f、5g、5h、5i、5j分别代表蒸汽晶化时间为0h、6h、12h、24h、48h得到的mesozsm-5b的sem图像;
图6为本发明一种利用原位碳化模板合成有序介孔zsm-5的方法实施例得到的两种介孔zsm-5的ftir光谱与商业zsm-5的对比示意图,其中,6a为mesozsm-5a的ftir光谱与商业zsm-5的对比示意图,6b为mesozsm-5b的ftir光谱与商业zsm-5的对比示意图;
图7为本发明一种利用原位碳化模板合成有序介孔zsm-5的方法实施例得到的两种介孔zsm-5的29si和27almasnmr固体核磁谱与商业zsm-5的对比示意图,其中a代表商业zsm-5,b代表mesozsm-5a,c代表mesozsm-5b。
具体实施方式
为了使本领域的技术人员可以更好地理解本发明,下面结合附图和实施例对本发明技术方案进一步说明。
以下实施例中所用到的tpaoh即四丙基氢氧化铵,质量百分数为25wt%,来源于acros公司;采用p123即聚环氧乙烷-聚环氧丙烷-聚环氧乙烷三嵌段共聚物作为表面活性剂,p123的分子量为5800,来源于aldrich公司;teos(原硅酸乙酯)、hcl和浓h2so4均来源于北京化学试剂公司。以上所有的化学试剂不经任何纯化处理直接使用。以下实施例中由sba-15/p123碳化得到的有序介孔zsm-5标记为mesozsm-5a,由mcf-p123碳化得到的有序介孔zsm-5标记为mesozsm-5b。
实施例一
如图1所示,首先,分别制备sba-15/p123复合物和mcf-p123复合物。
称取4gp123加入由16.8ml质量百分数为36.5wt%的hcl和104ml的h2o配制的hcl溶液中,剧烈搅拌直至完全溶解,再加入7.36ml分析纯的teos混合,先放置于40℃的环境下静置老化20h,再置于100℃的环境下静置老化24h后,过滤,得到的滤渣室温干燥,即得到sba-15/p123复合物;称取4gp123加入由16.8ml质量百分数为36.5wt%的hcl和104ml的h2o配制的hcl溶液中,再加入4.3ml的2,4,6-三甲基苯,剧烈搅拌直至完全溶解,然后加入7.36ml分析纯的teos混合,过滤,得到的滤渣室温干燥,即得到mcf-p123复合物。
其次,分别对sba-15/p123复合物和mcf-p123复合物进行碳化。
sba-15/p123复合物和mcf-p123复合物的碳化过程和工艺条件均相同,分别量取4mlh2o和0.08ml质量百分数为98wt%的h2so4混合得到h2so4溶液,称取1g的sba-15/p123复合物加入h2so4溶液中,搅拌30min,在100℃的烘箱中干燥12h,然后在160℃的条件下继续干燥12h后,将得到的棕色固体放置于管式炉中,在高纯ar的保护下,以1℃/min升温到900℃并保持3h,得到黑色固体为碳化的sba-15/p123复合物;称取1gmcf-p123复合物按照上述碳化方法进行碳化得到碳化的mcf-p123复合物。
最后,分别由sba-15/p123复合物和mcf-p123复合物合成不同的有序介孔zsm-5。
分别称取1.024gtpaoh、3.2422gh2o和0.0428g异丙醇混合,充分搅拌5h,直至得到澄清溶液,再加入1g碳化得到的sba-15/p123复合物于澄清溶液中,该澄清溶液中的al2o3、sio2、tpaoh、h2o的重量比值均为1:100:12:1920,混合搅拌1h,转移到60℃的烘箱中第一次干燥后,压碎,又将固体于60℃的烘箱中第二次干燥后,转移至马沸炉中,以2℃/min升温至550℃并保持6h,然后在60℃的条件下加入到1mlnh4no3溶液中缓慢搅拌2h进行离子交换后,于560℃的温度下煅烧6h,得到h+形式的白色固体,即由sba-15/p123碳化得到的有序介孔zsm-5标记为mesozsm-5a1。称取1g碳化的mcf-p123复合物按照同样的方法得到有序介孔zsm-5标记为mesozsm-5b1。
实施例二
首先,分别制备sba-15/p123复合物和mcf-p123复合物。
称取4gp123加入由16.8ml质量百分数为36.5wt%的hcl和104ml的h2o配制的hcl溶液中,剧烈搅拌直至完全溶解,再加入7.36ml分析纯的teos混合,先放置于40℃的环境下静置老化20h,再置于100℃的环境下静置老化24h后,过滤,得到的滤渣室温干燥,即得到sba-15/p123复合物;称取4gp123加入由16.8ml质量百分数为36.5wt%的hcl和104ml的h2o配制的hcl溶液中,再加入4.3ml的2,4,6-三甲基苯,剧烈搅拌直至完全溶解,然后加入7.36ml分析纯的teos混合,过滤,得到的滤渣室温干燥,即得到mcf-p123复合物。
其次,分别对sba-15/p123复合物和mcf-p123复合物进行碳化。
sba-15/p123复合物和mcf-p123复合物的碳化过程和工艺条件均相同,分别量取4mlh2o和0.08ml质量百分数为98wt%的h2so4混合得到h2so4溶液,称取1g的sba-15/p123复合物加入h2so4溶液中,搅拌30min,在100℃的烘箱中干燥12h,然后在160℃的条件下继续干燥12h后,将得到的棕色固体放置于管式炉中,在高纯ar的保护下,以1℃/min升温到900℃并保持3h,得到黑色固体为碳化的sba-15/p123复合物;称取1gmcf-p123复合物按照上述碳化方法进行碳化得到碳化的mcf-p123复合物。
最后,分别对sba-15/p123复合物和mcf-p123复合物采用蒸汽辅助晶化法合成不同的有序介孔zsm-5。
分别称取1.024gtpaoh、3.2422gh2o和0.0428g异丙醇混合,充分搅拌5h,直至得到澄清溶液,再加入1g碳化得到的sba-15/p123复合物于澄清溶液中,该澄清溶液中的al2o3、sio2、tpaoh、h2o的重量比值均为1:100:12:1920,混合搅拌1h,转移到60℃的烘箱中第一次干燥后,压碎,并悬至于内设有不锈钢支架的反应釜内衬中的支架上,该反应釜内于支架的下方装有0.4ml的水用于形成饱和蒸汽压,小心封釜,并将反应釜置于160℃的烘箱中加热6h后,将固体于60℃的烘箱中第二次干燥后,转移至马沸炉中,以2℃/min升温至550℃并保持6h,然后在60℃的条件下加入到1mlnh4no3溶液中缓慢搅拌2h进行离子交换后,于560℃的温度下煅烧6h,得到h+形式的白色固体,即由sba-15/p123碳化得到的有序介孔zsm-5标记为mesozsm-5a2。称取1g碳化的mcf-p123复合物按照同样的方法得到有序介孔zsm-5标记为mesozsm-5b2。
实施例三
首先,分别制备sba-15/p123复合物和mcf-p123复合物。
称取3.5gp123加入由15ml质量百分数为36.5wt%的hcl和100ml的h2o配制的hcl溶液中,剧烈搅拌直至完全溶解,再加入5ml分析纯的teos混合,先放置于40℃的环境下静置老化20h,再置于100℃的环境下静置老化24h后,过滤,得到的滤渣室温干燥,即得到sba-15/p123复合物;称取3.5gp123加入由15ml质量百分数为36.5wt%的hcl和100ml的h2o配制的hcl溶液中,再加入4ml的2,4,6-三甲基苯,剧烈搅拌直至完全溶解,然后加入5ml分析纯的teos混合,过滤,得到的滤渣室温干燥,即得到mcf-p123复合物。
其次,分别对sba-15/p123复合物和mcf-p123复合物进行碳化。
sba-15/p123复合物和mcf-p123复合物的碳化过程和工艺条件均相同,分别量取5mlh2o和0.05ml质量百分数为98wt%的h2so4混合得到h2so4溶液,称取0.8g的sba-15/p123复合物加入h2so4溶液中,搅拌30min,在100℃的烘箱中干燥12h,然后在160℃的条件下继续干燥12h后,将得到的棕色固体放置于管式炉中,在高纯ar的保护下,以1℃/min升温到900℃并保持3h,得到黑色固体为碳化的sba-15/p123复合物;称取0.8gmcf-p123复合物按照上述碳化方法进行碳化得到碳化的mcf-p123复合物。
最后,分别对sba-15/p123复合物和mcf-p123复合物采用蒸汽辅助晶化法合成不同的有序介孔zsm-5。
分别称取1.0gtpaoh、3.1gh2o和0.04g异丙醇混合,充分搅拌5h,直至得到澄清溶液,再加入0.8g碳化得到的sba-15/p123复合物于澄清溶液中,该澄清溶液中的al2o3、sio2、tpaoh、h2o的重量比值均为1:100:12:1920,混合搅拌1h,转移到60℃的烘箱中第一次干燥后,压碎,并悬至于内设有不锈钢支架的反应釜内衬中的支架上,该反应釜内于支架的下方装有0.5ml的水用于形成饱和蒸汽压,小心封釜,并将反应釜置于160℃的烘箱中加热12h后,将固体于60℃的烘箱中第二次干燥后,转移至马沸炉中,以2℃/min升温至550℃并保持6h,然后在60℃的条件下加入到0.8mlnh4no3溶液中缓慢搅拌2h进行离子交换后,于580℃的温度下煅烧6h,得到h+形式的白色固体,即由sba-15/p123碳化得到的有序介孔zsm-5标记为mesozsm-5a3。称取0.8g碳化的mcf-p123复合物按照同样的方法得到有序介孔zsm-5标记为mesozsm-5b3。
实施例四
实施例四所采用的化学试剂量和工艺条件均与实施例三相同,唯一不同的是将反应釜置于160℃的烘箱中加热24h,最后得到由sba-15/p123碳化得到的有序介孔zsm-5标记为mesozsm-5a4,由mcf-p123复合物按照同样的方法得到有序介孔zsm-5标记为mesozsm-5b4。
实施例五
首先,分别制备sba-15/p123复合物和mcf-p123复合物。
称取6gp123加入由20ml质量百分数为36.5wt%的hcl和150ml的h2o配制的hcl溶液中,剧烈搅拌直至完全溶解,再加入10ml分析纯的teos混合,先放置于40℃的环境下静置老化20h,再置于100℃的环境下静置老化24h后,过滤,得到的滤渣室温干燥,即得到sba-15/p123复合物;称取6gp123加入由20ml质量百分数为36.5wt%的hcl和150ml的h2o配制的hcl溶液中,再加入4.5ml的2,4,6-三甲基苯,剧烈搅拌直至完全溶解,然后加入10ml分析纯的teos混合,过滤,得到的滤渣室温干燥,即得到mcf-p123复合物。
其次,分别对sba-15/p123复合物和mcf-p123复合物进行碳化。
sba-15/p123复合物和mcf-p123复合物的碳化过程和工艺条件均相同,分别量取10mlh2o和0.1ml质量百分数为98wt%的h2so4混合得到h2so4溶液,称取1.2g的sba-15/p123复合物加入h2so4溶液中,搅拌30min,在100℃的烘箱中干燥12h,然后在160℃的条件下继续干燥12h后,将得到的棕色固体放置于管式炉中,在高纯ar的保护下,以1℃/min升温到900℃并保持3h,得到黑色固体为碳化的sba-15/p123复合物;称取1.2gmcf-p123复合物按照上述碳化方法进行碳化得到碳化的mcf-p123复合物。
最后,分别对sba-15/p123复合物和mcf-p123复合物采用蒸汽辅助晶化法合成不同的有序介孔zsm-5。
分别称取1.2gtpaoh、3.4gh2o和0.05g异丙醇混合,充分搅拌5h,直至得到澄清溶液,再加入1.2g碳化得到的sba-15/p123复合物于澄清溶液中,该澄清溶液中的al2o3、sio2、tpaoh、h2o的重量比值均为0.9:100:11.5:1900,混合搅拌1h,转移到60℃的烘箱中第一次干燥后,压碎,并悬至于内设有不锈钢支架的反应釜内衬中的支架上,该反应釜内于支架的下方装有1ml的水用于形成饱和蒸汽压,小心封釜,并将反应釜置于160℃的烘箱中加热48h后,将固体于60℃的烘箱中第二次干燥后,转移至马沸炉中,以2℃/min升温至550℃并保持6h,然后在60℃的条件下加入到0.9mlnh4no3溶液中缓慢搅拌2h进行离子交换后,于600℃的温度下煅烧6h,得到h+形式的白色固体,即由sba-15/p123碳化得到的有序介孔zsm-5标记为mesozsm-5a5。称取1.2g碳化的mcf-p123复合物按照同样的方法得到有序介孔zsm-5标记为mesozsm-5b5。
分别取实施例一、实施例二、实施例三、实施例四和实施例五最后得到的两种产品作为待测样品进行分析检测,即采用广角xrd物相表征、小角x射线衍射(xrd)物相分析、比表面积和孔体积测定、透射电子显微镜(tem)表征、扫描电子显微镜(sem)表征、ftir红外光谱表征进行分析检测,具体结果和分析如下:
一、广角xrd物相表征和小角x射线衍射(xrd)物相分析
对干燥后的待测样品进行广角x射线物相分析,该测试在shimadzu/xrd-7000上进行,x射线源为石墨单色器滤波的cukα辐射(λ=0.15406nm),束流大小30ma,束流电压为20kv,数据采集步长为8°/min,扫描角度范围为5-55°;对干燥后的待测样品进行小角x射线物相分析,x射线衍射物相分析(smallanglex-raydiffraction,saxrd)在rigakud/max2500衍射仪上进行,x射线源为石墨单色器滤波的cu-kα辐射(λ=0.15406nm),束流大小200ma,束流电压为40kv,数据采集步长为0.5°/min,扫描角度范围为0.5-6°。分析结果如图2所示。
图2显示了分别由普通的sba-15/p123和加2,4,6-三甲基苯扩充介孔后得到的两种介孔材料的xrd图像,由图2可以看出蒸汽晶化时间对介孔材料的结晶度有比较明显的影响。如图2a所示,随晶化时间延长,mesozsm-5a的xrd峰强度逐渐增加,并在晶化24h后达到完全结晶,显示了典型的纯相mfi的沸石结构;由图2b可知,所有的样品均在2θ=1°左右显示了尖锐的衍射峰,说明蒸汽晶化过程并没有破坏初始sba-15的有序介孔;由图2c可知,由2,4,6-三甲基苯扩充介孔的mcf-p123碳化得到的mesozsm-5b的结晶过程显示了与mesozsm-5a相同的结晶度变化趋势,并在晶化24h后达到了完全结晶;但是从图2d可以看到,高温蒸汽对介孔的有序度产生了不利影响,在结晶48h时其有序度基本丧失,可能的解释是mcf-p123碳化后形成的介孔碳具有超大笼的介孔胶束,并且笼与笼之间由开放的窗口彼此连接,这将导致窗口旁边的sio2没有固体支架支撑,导致局部介孔壁塌陷。而普通的s15-p123碳化后形成的介孔碳具有独立的六方胶束,在合适的碳化控制下,所环绕的sio2被六方碳胶束支撑,所以介孔的有序度得到保持。所以说,蒸汽晶化时间对介孔材料的结晶度有比较明显的影响,而介孔碳的结构对最终的介孔分子筛产物的形貌影响很大。
在碳化过程中,如果要得到有序的cmk-3,初期的h2so4预碳化过程几乎是必需的,如果省略这一步,用ar直接碳化,在sba-15介孔内只能得到不连续的纳米碳粒子,最终导致介孔沸石的介孔孔道也不是连续有序的。经过反复实验证明三嵌段共聚物在惰性气氛下热处理很容易裂解成气相产物,但是经h2so4处理后,p123可以转化为富碳材料。h2so4作为脱水催化剂从p123中除去h2o,使得剩余的聚合物链发生交联,这些交联聚合物在惰性气氛下即高纯氩气热处理而转化为硬质碳材料。在900℃的ar气处理下,交联的聚合物开始碳化,并形成局部高密度的石墨化结构,导致疏松的碳发生收缩,最终形成中空的碳胶束cmk-3。碳材料在介孔内外收缩的空间恰恰为tpaoh前驱体溶液渗入复合物内部提供了充足的运输通道。
二、比表面积和孔体积测定
将各待测样品于120℃下真空脱气过夜,在77k下使用quantachromeautosorbas-1仪器测定比表面积,结果如图3所示,比表面积采用bet(brunauer-emmett-teller)方法计算测定,孔径分布使用bjh(barret‐joyner‐halenda)的吸附支得到,微孔体积通过dft(nonlocaldensityfunctionaltheory)计算方法得到。测试得到的比表面积(sbet)、介孔体积(vmeso)、介孔尺寸(dmeso)汇总如表1所示:
表1
由图3可以看出,结晶之前的样品(0h)具有与典型的sba-15相似的吸附等温线和比表面积,说明浸渍、碳化与煅烧的整个处理过程对介孔结构基本没有影响。从bjh模型计算结果可知,所合成的mesozsm-5a具有4nm~5nm的均一介孔尺寸,比sba-15的介孔尺寸(6.6nm)小很多,这是由于介孔壁中的无定形sio2结晶成沸石,导致介孔孔道收缩。此外,结晶也导致介孔体积的缩小。然而,即使在结晶48h后,介孔体积依然维持在一个较大数值(0.36cm3/g)。经tmb扩孔的sba-15前驱体制备得到的mesozsm-5b的介孔体积明显比mesozsm-5a的介孔体积大很多。所有的介孔分子筛均比商业zsm-5(sbet=312m2/g)具有更大的bet比表面积。与传统的沸石相比,所有的介孔材料在相对压力p/p0=0.4~0.9之间出现了明显的回滞环,随高温蒸汽晶化时间延长,回滞环有变小的趋势,并且向高压区偏移,说明高温蒸汽导致部分介孔纳米沸石晶体发生塌缩,可能出现了更大的介孔。
三、透射电子显微镜(tem)表征
将各待测样品分别超声分散于乙醇中,滴于喷有碳膜的铜网上,自然干燥后进行透射电子显微镜(transmissionelectronmicroscope,tem)表征。tem表征在jeoljem-2100f上进行,工作电压为200kv,电流为180ma。结果如图4所示。
对两种介孔zsm-5分子筛来说,晶化时间小于24h时,介孔的有序度均可以得到很大程度的保持,而延长晶化时间则对介孔均有一定程度的破坏。结晶48h后,两种沸石的介孔结构均开始变为无序,这与小角xrd的结果相吻合。从图4a和4f可知,浸渍tpaoh溶液不会对介孔结构造成破坏。此外,在晶化过程中,介孔碳模板的结构塌陷的可能性也比较小,因此无序介孔结构最可能是在最终除去碳模板的煅烧过程中形成的。随着晶化时间的延长,介孔壁上的无定形sio2逐渐进行周期性有序排列,聚合成纳米沸石颗粒,该过程中从低密度的无定形氧化硅生成高密度的沸石晶体会导致介孔壁上的沸石晶体之间出现空隙,因此在煅烧除去碳支撑体后介孔沸石壁会在微观上发生轻微塌陷,表现在tem图像上即为介孔无序。
四、扫描电子显微镜(sem)表征
将割成小块的单晶硅片用导电铜胶固定在sem样品台上,将乙醇超声分散的各待测样品滴在硅片上,待干燥后溅射一薄层金属pt以增加导电性后进行扫描电镜(scanningelectronmicroscope,sem)观察。sem表征在jeol-6701f上进行,加速电压为10kv。结果如图5所示。
对所有样品的sem图像进行测试,发现在宏观上并没有出现大面积塌陷的现象。mesozsm-5a仍然保持类似前驱体sba-15的蠕虫状结构,而mesozsm-5b则保持前驱体的球状形貌,说明此原位碳化模板的合成方法能够原位形成介孔分子筛而不会引起前驱体重组。
五、ftir红外光谱表征
将含有3wt%的kbr粉末压片,在thermonicoletin10-iz10上对各待测样品进行ftir谱图扫描,扫描范围为4000-400cm-1。结果如图6所示。
ftir谱图可间接反映前驱体sba-15的晶化过程,550cm-1左右出现的红外强吸收峰代表了si-o-si组成的双五元环,该峰被广泛用于指示mfi型分子筛的结晶程度。大量实验证明,随着晶化时间的延长,两种介孔材料在550cm–1与455cm–1处的峰强度比逐渐增加,说明在介孔zsm-5的介孔壁中含有mfi沸石的初级结构单元。对mesozsm-5a来说,当晶化时间为24h时,550cm–1与455cm–1处的峰强度比为0.51,略低于商业zsm-5(0.62),说明大于80%的sba-15介孔壁均结晶生成了mfi结构;对于mesozsm-5b当晶化时间为24h时,550cm–1与455cm–1处的峰强度比为0.55,说明大约90%的sba-15介孔壁均结晶生成了mfi结构。对比结晶6h后的样品,可以发现在相同的结晶时间内,具有更大介孔的前驱体的沸石结晶速度更快,说明在更大限域空间内,结晶所需的介质蒸汽传输更快。以上结果表明,经足够的晶化时间后,介孔壁几乎可以完全结晶成沸石。
六、29si和27almasnmr固体核磁表征
我们对晶化24h的两种介孔zsm-5与商业zsm-5的29si和27almasnmr光谱进行了表征,以研究介孔材料中硅和铝物种的存在形式。在brukeravanceiii400mhz波谱仪上进行各待测样品的29si和27almasnmr固体核磁表征,同时本发明还对商业zsm-5按照相同的方法进行表征,方便对比。29simasnmr的测试条件为:探针长度4mm,脉冲频率79.3mhz,接触时间2ms,脉冲间隔2s,扫描速度12khz。27almasnmr的测试条件为:探针长度4mm,脉冲频率104mhz,接触时间2ms,脉冲间隔1s,扫描速度5khz。结果如图7所示。
27almasnmr表明所合成的介孔zsm-5与商业zsm-5均含有大量的mfi骨架内四面体al。两种介孔zsm-5的八面体al峰强度均比商业zsm-5稍高,说明它们含有较多的外骨架al,这是由于介孔孔道的引入导致外表面积增大,或者残余的少量未结晶的无定形sio2所致。从29simasnmr谱图中可明显看出,三种材料均没有出现q2的峰,说明几乎不存在无定形的si-oh。三种材料均呈现了比较弱的q3峰,代表少量的外骨架si-oh,这与27almasnmr谱图吻合。q4的相对强度越高,说明结晶的沸石成分越多。所合成的两种介孔材料与商业zsm-5均具有很大的q4/q3比值,说明两种介孔材料的结晶度很高。
本发明利用原位碳化介孔模板法合成了两种有序介孔zsm-5分子筛。将未除p123模板的sba-15进行原位两步碳化,即硫酸预碳化和氩气900度高温碳化,使内部的p123软模板最大程度地碳化成介孔有序的硬模板,经浸渍tpaoh溶液和蒸汽辅助晶化后,最后空气煅烧除碳,得到了两种具有不同介孔大小的结晶有序介孔zsm-5材料。此硬模板原位转化方法比以介孔sio2为原料浸渍葡萄糖或糠醇等方法更简单,且能充分利用最初的p123模板剂,所得到的比表面积和介孔体积大小均在现有相关产品之上。在少量水(0.4ml~1ml)的条件下可以合成出介孔zsm-5,比水热法或足量水晶化法更节约溶剂,且后处理更简单,合成后从反应釜中取出直接干燥煅烧即可,省去了煅烧前的洗涤等繁琐的后处理步骤。
以上对本发明提供的一种利用原位碳化模板合成有序介孔zsm-5的方法进行了详细介绍。具体实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!