试剂盒及其用途的制作方法
本发明涉及生物医学领域,具体的,涉及试剂盒及其用途,更具体的,本发明涉及一种试剂盒、试剂盒的用途、一种构建目标区域测序文库的方法、一种测序方法、一种检测目标区域变异的方法和装置。
背景技术:
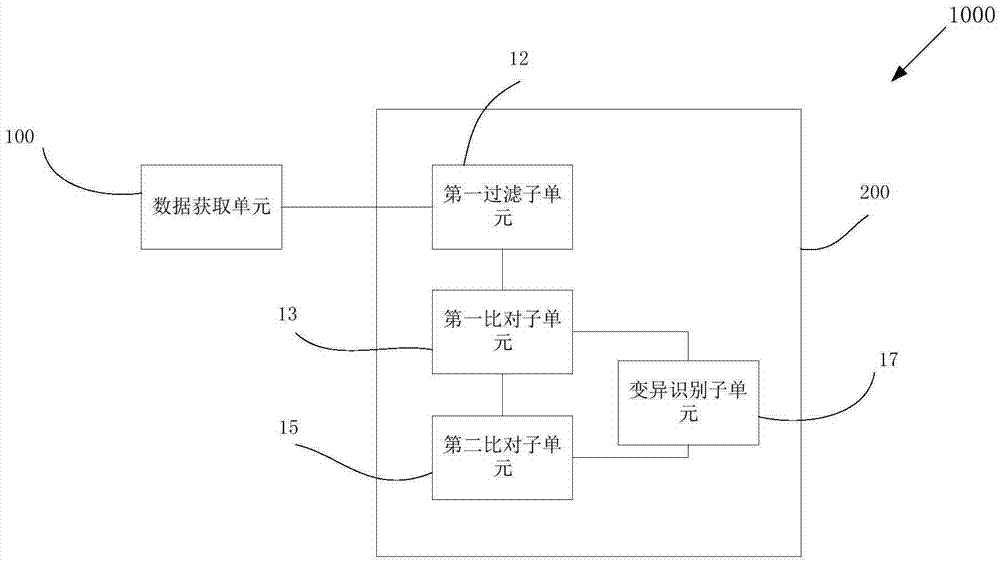
:胃癌是世界范围内最常见的恶性肿瘤之一,在全球肿瘤致死原因中排名第二位,2012年全球胃癌的新患者人数约95万,胃癌死亡人数约70万,胃癌在东亚的发病率最高。我国是胃癌的高发国家,据国家癌症中心的数据显示:2011年胃癌在我国恶性肿瘤发病率和死亡率均居第三位[陈万青,郑荣寿,曾红梅等.2011年中国恶性肿瘤发病和死亡分析.中国肿瘤.2015;24(1):2.]。由于缺乏有效的治疗手段,胃癌整体治疗效果并不理想,患者5年生存率在5%-20%左右,总体平均生存时间小于1年。胃癌的发生是一个多因素共同作用的复杂过程,目前认为幽门螺杆菌慢性感染是最重要的致病因素,约90%的新发非贲门胃癌的发生与之相关。幽门螺杆菌产生的细菌毒力因子(如caga)导致炎症发生和激活致癌信号通路,在这一持续的慢性炎症环境中,过度的氧化应激导致胃上皮细胞dna损伤发生,诱发癌基因、抑癌基因、细胞周期调控因子、细胞粘附分子、dna损伤修复基因的遗传改变及表观遗传改变,促进胃癌的发生发展。胃癌是一种异质性很高的肿瘤,有显著的表型多样性,包括不同的分子亚型。早在1965年,lauren将胃癌分为肠型和弥漫型,后者侵袭性强、患者预后差。2013年lei等基于248例胃癌标本的基因分析,将胃癌分为增殖型、代谢型和间质型。其中增殖型胃癌细胞存在高度的遗传不稳定性、tp53突变和dna的低甲基化;代谢型胃癌对5-fu敏感,患者术后是否接受5-fu辅助治疗的生存差异明显;间质型胃癌具有肿瘤干细胞的特性,对pi3k/akt/mtor抑制剂敏感。根据分子分类系统,胃癌分为四种类型:eb病毒感染型胃癌、微卫星不稳定型胃癌、基因稳定型胃癌和染色体不稳定型胃癌。肿瘤治疗已进入分子靶向药物和个体化治疗时代。大量的研究显示,患者对抗肿瘤药物(化疗药物、靶向药物)是否敏感与其癌细胞的基因组突变直接相关。2012年10月,中国第一个能明显降低her2阳性晚期胃癌患者死亡风险、提高总生存期的靶向药物赫赛 汀(曲妥珠单抗)正式上市,意味着胃癌靶向治疗时代的到来。其他几种靶向her2、c-met和vegf等受体的胃癌新药已经取得了可观的研究成果。进行胃癌个体化治疗,首先需要对胃癌患者的靶药基因进行检测。随着癌症认知领域的发展和科学技术的进步,基因组测序技术逐渐进入到肿瘤临床应用中。在科研及临床转化领域的现有技术中,对于体细胞基因突变的检测方法主要有pcr-sanger法、pcr-mass法和arms-pcr法等。这几种方法所应用的样本均为肿瘤组织样本。pcr-sanger法准确性好,能检测新的点突变类型及indel,但灵敏度很低,仅能检测突变率大于20%的高频突变,且难以胜任多基因多位点检测。pcr-mass法不能检测未知突变,也无法检测indel,灵敏度在5%左右,该技术局限性较大。arms-pcr法特异性好,灵敏度高,能检测1%-5%的低频突变,但不能检测未知突变,且同样难以胜任多基因多位点的同时检测。技术实现要素:本发明旨在至少解决上述问题至少之一或者提供至少一种可选择的商业手段。依据本发明的一方面,本发明提供一种试剂盒,其包含探针,所述探针固定在固相基质上或者游离于溶液中,所述探针能够特异性识别表1里的17个基因中的每个基因的至少一部分区域。本发明的试剂盒中的探针能够特异性识别的基因区域组合,是发明人经过信息收集、多次筛选、针对性地设计探针和试验获得的,这些基因区域组合与结胃癌的发生发展相关。该试剂盒能够用于获取胃癌相关基因序列、和/或在检测胃癌相关基因序列中的突变、和/或在辅助指导胃癌用药、和/或在辅助预测胃癌风险。从而能够用于预测个体的患胃癌风险、预测胃癌患者使用相应抗肿瘤靶向药物的疗效,辅助指导临床用药,辅助用于胃癌的个体化治疗。表1根据本发明的实施例,本发明的这一方面的试剂盒还可以具有以下附加技术特征至少之一:根据本发明的一个较佳实施例,所述探针能够特异性识别表2中的区域。这些区域包含表1中17个基因的332个外显子,是发明人结合cosmic、ncbi等数据库的记载数据,利用迭代算法对已选入表1中的17个基因的外显子进行分析并确定各外显子的目标区域的 序列,使目标区域能对记载病例进行最大化的覆盖,即表2中的区域。表2根据本发明的实施例,试剂盒包含的探针的长度为25~300nt,较佳的为100nt左右。为获得能够在同一反应体系中同时特异性捕获所说的区域组合,根据本发明的一个实施例,探针是通过先获得初始探针集,再筛选所述初始探针集来确定的。获取所述初始探针集包括:确定所述区域的参考序列,从所述基因的参考序列的一端开始,在所述参考序 列上依次获取dna片段直至所述参考序列的另一端,其中,一条dna片段为一条初始探针,全部所述dna片段构成所述初始探针集,所述dna片段之间完全重叠、部分重叠或完全不重叠,所述初始探针集能够覆盖所述基因区域至少一次。所说的区域的参考序列可以从参考基因组上获取,例如从人参考基因组hg19上获得对应的基因区域,所有的hg19上的对应区域构成所说的区域的参考序列,hg19可以从ncbi数据库下载。根据本发明的一个较佳实施例,利用迭代算法设计获取所述初始探针集,包括:确定所述区域在参考基因组上的位置,获取所述基因区域的参考序列,从所述参考序列的第一个核苷酸开始拷贝所述参考序列获取第一条dna片段,从所述参考序列的第二个核苷酸开始拷贝所述参考序列获取第二条dna片段,从所述参考序列的第三个核苷酸开始拷贝所述参考序列获取第三条dna片段,这样依次获取后续dna片段直至第n条dna片段的一端超出所述参考序列,其中,一条dna片段为一条初始探针,全部所述dna片段构成所述初始探针集,n为所述初始探针集中包含的初始探针的总数,以获得能够全面覆盖目标基因区域的初始探针集。而且为使最终的探针具高特异性,根据本发明的一个较佳实施例,进一步对所述初始探针集进行筛选,包括:将所述dna片段(初始探针集)与所述参考序列比对,获得每一条dna片段在参考序列上的比对次数,过滤掉比对次数超过1的dna片段。为使最终的探针能在同一反应体系中捕获所说的基因区域,和/或使捕获的基因区域在同一反应条件下被一起洗脱下来,进一步对所述初始探针集进行筛选,包括:去除掉gc含量不在35-70%的dna片段。最终,获得的芯片/探针最终覆盖的区域包含了17个基因的332个外显子,总大小为79kb。依据本发明另一方面,提供上述任一实施例中的试剂盒在获取胃癌相关基因序列、和/或在检测胃癌相关基因序列中的突变、和/或在辅助指导胃癌用药、和/或在辅助预测胃癌风险中的用途。利用本发明上述一方面或者任一实施例中的试剂盒,能够一次性、简单方便且高特异性的获取胃癌的相关基因序列,检测分析这些相关基因序列,检测分析结果可以用于或者辅助用于胃癌的风险预测和用药指导。上述对本发明一方面的或者任一实施例中的试剂盒的优点和技术特征的描述,同样适用本发明这一方面的试剂盒的用途,在此不再赘述。依据本发明的再一方面,提供一种构建目标区域测序文库的方法,所述方法包括:(a)获取待测样本中的核酸,所述核酸由多个核酸片段组成,所述核酸片段来自断裂的基因组dna和/或游离的dna;(b)末端修复所述核酸片段,获得末端修复片段;(c)加碱基a 至所述末端修复片段的两端,获得粘性末端片段;(d)连接接头于所述粘性末端片段的两端,获得接头连接片段;(e)对所述接头连接片段进行第一扩增,获得第一扩增产物;(f)利用上述本发明一方面或者任一实施例中的试剂盒对所述第一扩增产物进行捕获,获得所述目标区域;以及(g)对所述目标区域进行第二扩增,获得第二扩增产物,所述第二扩增产物构成所述目标区域测序文库;任选地,所述接头末端为t-粘性末端。本发明的这一方面的测序文库构建方法,特别适用于样本含微量核酸的测序文库的构建。根据本发明的一个实施例,样本为含微量游离dna片段的血浆样本,包含极其微量的目标游离dna片段,第一扩增使得核酸的量能满足芯片/探针杂交捕获的需求,而因芯片杂交捕获会损耗一定量的核酸,第二扩增能使捕获下的目标片段获得再次扩增以满足上机测序和质控检测的要求。本发明的这一文库构建方法特别适用于总游离核酸不低于10ng或者常规组织基因组dna不低于1μg的样本的测序文库构建,利用本发明的这一方面的方法构建的目标区域文库,测序后的下机数据质量高,基于高质量的下机数据利于后续的准确检测分析。依据本发明的又一方面,提供一种测序方法,所述方法包括:根据本发明一方面或者任一实施例中的测序文库构建方法构建目标区域测序文库;对所述目标区域测序文库进行测序,获得测序数据,所述测序数据由多个读段组成。测序可以利用已知平台进行,包括但不限于illumina的hiseq2000/2500平台、lifetechnologies的iontorrent平台和单分子测序平台。测序方式可以选择单端测序,也可以是双末端测序,在本发明的一个实施例中利用双末端测序,所得的测序数据由多对读段对组成。本发明这一方面的方法,通过结合上述任一实施例中的试剂盒和高通量捕获测序技术,包括上述发明人优化了的建库方法,使芯片捕获目标区域的过程更高效率,能充分发挥高通量捕获测序技术通量高、灵敏度高等优势。上述对任一本发明一方面或者任一实施例中的试剂盒和测序文库构建方法的优点和技术特征的描述,同样适用本发明的这一方面的测序方法,在此不再赘述。依据本发明的一方面,本发明提供一种检测目标区域变异的方法,所述方法包括:(1)利用前述本发明任一实施例中的测序方法,获得目标样本的目标区域测序数据;(2)基于所述测序数据,检测所述目标区域变异,获得变异位点信息,所述变异包括snp和indel中的至少之一。根据本发明的一个实施例,步骤(2)包括:将所述测序数据与参考序列进行第一比对, 获得第一比对结果;将所述第一比对结果与所述参考序列的一部分进行第二比对,获得第二比对结果;基于所述第一比对结果和所述第二比对结果,同时检测所述目标区域中的snp和indel。为使变异检测结果更准确可信,根据本发明的一个实施例,在所述第一比对之前,对所述测序数据进行过滤,所述过滤包括去除掉不确定碱基比例超过10%的读段和/或碱基质量值不大于5的碱基数的比例不小于50%的读段。并且较佳的,在所述第二比对之前,去除掉第一比对结果中的一个读段对中的两个读段相同的读段对。所称的参考序列的一部分包括目标区域参考序列中的每个已知indel位点,以及所述每个已知indel位点上下游各1000bp的参考序列。这里,所称的第二比对为局部比对,第一比对为常规全局比对,可利用但不限于soap或bwa等软件依照其默认设置进行,获得第一比对结果,第一比对结果包括读段在参考序列上的匹配位置及匹配情况信息。根据本发明的一个实施例,进行第二比对即基于第一比对结果,对与所捕获的基因区域对应的参考序列中的所有已知indel附近的所有序列信息(reads)进行局部重新比对,能够消除第一比对中的错误,提高后续变异检测的准确性,第二比对可利用gatk重比对软件(https://www.broadinstitute.org/gatk/)进行。根据本发明的一个实施例,测序数据来自lifetechnologies公司的ionproton测序平台,通过tvc工具使用其默认参数同时检测snp和indel变异。利用本发明的这一方面的变异检测方法,能够准确检测出突变频率为1%的低频突变。通过上述发明人优化了的生物信息学分析的方法,能够准确地检测到更低频率的突变(1%)。综上,前述提供的采用高通量捕获的测序法,该方法是利用上述设计的芯片探针,对样本dna中感兴趣的目标基因区域进行杂交捕获,并进行高通量测序,对测序结果进行生物信息学分析,从而得到变异位点的信息。该方法较之前述方法存在的灵敏度不高、通量小、检测内容有限或只能检测已知突变等缺点,有着多方面的优势:首先,通量高,可以同时检测几十甚至上百的基因,在得到更丰富信息的同时也大大降低了检测成本;其次,灵敏度高,检测灵敏度能够达到1%-5%,且可以根据研究需要调整检测阈值,如需更高灵敏度可以加大测序深度从而得到1%以下的超低频突变,操作灵活;再次,特异性高,该方法能够保证较低的假阳性率、假阴性率,确保得到的检测结果能够准确的反映受检者的突变情况;最后,利用生物信息学分析可以挖掘到更多的数据,不仅能够提供已知突变位点的信息,还可以挖掘未知突变位点、indel、cnv等多方面数据,为科学研究提供更多有用 的信息。并且,与目前几种方法相比,上述本发明一方面的方法能避免已知方法存在的诸多不足。该发明提供的试剂盒探针结合高通量捕获测序,将有望为临床胃癌个体化治疗提供科学指导。为了能充分发挥高通量捕获测序技术通量高、灵敏度高等优势,发明人对测序的建库流程进行了优化,使芯片捕获目标区域的过程效率更高;同时,发明人也优化了生物信息学分析的流程,从而能够准确地检测到更低频率的突变,包括突变频率1%的突变。上述本发明一方面的芯片捕获结合高通量捕获测序,有助于胃癌个体用药疗效和复发风险预测的研究,并有望将研究成果转化并辅助用于临床。依据本发明的一方面,还提供一种检测目标区域变异的装置,用以实现或执行上述本发明一方面的或者任一实施例的目标区域变异检测方法,所述装置包括:数据获取单元,用于实现上述本发明一方面的测序方法,获取目标区域的测序数据,所述测序数据由多个读段组成;检测单元,用于基于来自数据获取单元的测序数据,检测所述目标区域变异,获得变异位点信息,所述变异包括snp和indel中的至少之一。本领域人员可以理解,本发明的装置中的全部或部分单元,可选择的、可拆卸的包含一个或多个子单元以执行或实现前述本发明方法的各个实施例。例如,根据本发明的一个实施例,如图1所示,装置1000中的检测单元200包括第一比对子单元13、第二比对子单元15和变异识别子单元17,所述第一比对子单元13用以将来自数据获取单元100的测序数据与参考序列进行第一比对,获得第一比对结果,所述第二比对子单元15用以将来自所述第一比对子单元13的第一比对结果与所述参考序列的一部分进行第二比对,获得第二比对结果,所述变异识别子单元17用以基于来自所述第一比对子单元13的第一比对结果和来自所述第二比对子单元15的第二比对结果,同时检测所述目标区域中的snv和indel,获得变异位点信息,其中,所述参考序列的一部分包括目标区域参考序列中的每个已知indel位点,以及所述每个已知indel位点上下游各1000bp的参考序列。再例如,根据本发明的一个实施例,如图2所示,所述装置1000的检测单元200还包括第一过滤子单元12,所述第一过滤子单元12与所述第一比对子单元13连接,用于在所述测序数据进入所述第一比对子单13元之前,对所述测序数据进行过滤,所述过滤包括去除掉不确定碱基比例超过10%的读段和/或碱基质量值不大于5的碱基数的比例不小于50%的读段。任选的,如图3所示,所述检测单元200还包括第二过滤子单元14,所述第二过滤子单元分别14与所述第一比对子单元13和所述第二比对子单元15连接,用于在所述第 一比对结果进入所述第二比对子单元15之前,去除掉来自所述第一比对子单元13的第一比对结果中的一个读段对中的两个读段相同的读段对。上述参考序列可以为hg19,所述第一比对单元中进行的第一比对为全局比对,所述第二比对子单元中进行的第二比对为局部比对。本发明的方法/装置,基于新一代测序技术的目标区域捕获测序,能够在很短的时间内同时进行多例样本检测,并且在目标区域捕获芯片的使用下,相同数据量可进行更高深度的数据挖掘。这样,在相同成本下,可以研究更大的样本量及获得更高的测序深度,从而更有可能获得所需的用药及预后相关基因突变的信息。而相比于目前科研界对于胃癌体细胞突变运用较多的arms-pcr等方法,利用实施例中的试剂盒包含的芯片/探针的多基因同时检测的优势则颇为明显。芯片/探针包含的17个基因的332个外显子,代表了目前已有报道的在胃癌用药及发生发展研究中的突变基因位点,这就可以在一次实验中获得多个基因突变的信息,较之单个或少数几个突变的检测方法,通量上有优势,更容易对胃癌预后进行全面、综合的研究。附图说明本发明的上述和/或附加的方面和优点从结合下面附图对实施方式的描述中将变得明显和容易理解,其中:图1是本发明的一个实施例中的目标区域变异检测装置的结构示意图;图2是本发明的一个实施例中的目标区域变异检测装置的结构示意图;图3是本发明的一个实施例中的目标区域变异检测装置的结构示意图;图4是本发明的一个实施例中的变异位点的sanger测序验证的测序峰图。具体实施方式下面结合附图和具体实施方式对本发明进行详细说明。所述实施方式在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。本发明中的“变异”、“核酸变异”、“基因变异”可通用,本发明中的“snp”(snv)、“插入缺失”(indel)和“结构变异”(sv)同通常定义,但本发明中对各种变异的大小不作特别限定,这样这几种变异之间有的有交叉。这些类型变异的大小交叉并不妨碍本领域人员通过上述描述执行实现本发明的方法和/或装置并且达到所描述的结果。本发明中的“参考序列”为已知基因组序列或者已知基因组序列的至少一部分。在本文中,所使用的术语“第一”、“第二”等仅用于描述目的,而不能理解为指示或暗示相对重要性、隐含指明所指示的技术特征的数量或者具有顺序关系。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者多个该特征。在本发明的描述中,除非另有说明,“多个”的含义是两个或两个以上。在本文中,除非另有明确的规定和限定,术语“顺序连接”、“相连”、“连接”等术语应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通。对于本领域的普通技术人员而言,可以根据具体情况理解上述术语在本发明中的具体含义。下文的公开了不同的具体实施方式或实施例用来实现本发明的不同方法步骤或装置结构。为了简化本发明的公开,下文中对特定例子的步骤和设置进行描述。当然,它们仅仅为示例,并且目的不在于限制本发明。此外,本发明可以在不同例子中重复参考数字和/或参考字母,这种重复是为了简化和清楚的目的,其本身不指示所讨论各种实施方式和/或设置之间的关系。以下结合附图和具体实施例对本发明的方法、装置和/或系统进行详细的描述。下面示例,仅用于解释本发明,而不能理解为对本发明的限制。除另有交待,以下实施例中涉及的未特别交待的试剂、序列(接头、标签和引物)、软件及仪器,都是常规市售产品或者开源的,比如购自lifetechnologies等。实施例一获得本发明一方面的试剂盒、实现本发明一方面的方法和/或装置,一般包括目标区域捕获探针/芯片的设计、微量样本建库及杂交上机测序、下机数据的生物信息分析和变异数据解读。(一)芯片入选基因的确定、序列设计及生产。首先,对cosmic数据库中胃癌分类中突变率在前20位的基因进行调研,筛选出与胃癌用药相关的基因、胃癌中发现的重要癌基因和抑癌基因,并加入了胃癌的中高频突变的基因,得到芯片的最终基因列表,共计17个,如表1所示。芯片的目标区域由华大自主编写并优化的程序计算得来。该算法结合了cosmic、ncbi等数据库的记载数据,利用迭代算法对已入选17个基因的外显子进行分析并得到捕获目标区域的序列。芯片最终目标区域覆盖了17个基因的332个外显子,总大小为79kb。 详细目标区域如表2所示。利用迭代算法设计获取所述初始探针集,包括:确定所述区域在参考基因组上的位置,获取所述基因区域的参考序列,从所述参考序列的第一个核苷酸开始拷贝所述参考序列获取第一条dna片段,从所述参考序列的第二个核苷酸开始拷贝所述参考序列获取第二条dna片段,从所述参考序列的第三个核苷酸开始拷贝所述参考序列获取第三条dna片段,这样依次获取后续dna片段直至第n条dna片段的一端超出所述参考序列,其中,一条dna片段为一条初始探针,全部所述dna片段构成所述初始探针集,n为所述初始探针集中包含的初始探针的总数,以获得能够全面覆盖目标基因区域的初始探针集,而且为使最终的探针具高特异性,进一步对所述筛选初始探针集,包括:将所述dna片段(初始探针集)与所述参考序列比对,获得每一条dna片段在参考序列上的比对次数,过滤掉比对次数超过1的dna片段。进一步的,为使最终的探针能在同一反应体系中捕获所说的基因区域,和/或使捕获的基因区域在同一反应条件下被一起洗脱下来,进一步对所述初始探针集进行筛选,包括:去除掉gc含量不在35-70%的dna片段。最终,获得的芯片/探针最终覆盖的区域包含了17个基因的332个外显子,总大小为79kb。确定了的芯片探针的制备由华大基因自主芯片生产部门负责。(二)测序文库的制备2.1对样本提取dna片段,并检测浓度;2.2对提取的dna片段进行片段化处理;2.3对dna片段进行末端修复;2.4连接adapter文库接头:文库接头(adapter)是指经过设计的一段碱基序列,在dna文库扩增时与引物相结合,使dna扩增进行,并且在上机测序时与standardanchor相结合,利于探针与待测序位点结合辅助dna测序进行;2.5文库进行第一轮pcr扩增;2.6纯化,质量控制,并进行胃癌芯片杂交捕获目的区域;2.7杂交后的文库进行第二轮pcr扩增;2.8文库定量及质量控制;2.9life公司的ionproton测序仪上机测序。(三)测序下机数据生物信息分析获得下机数据后需进行如下的生物信息分析,并得到最终的突变位点注释结果:3.1去除低质量reads;3.2与reference序列比对,产生bam文件;3.3标记重复序列;3.4比对结果不好的序列重新比对,并校正质量值;3.5去除错配序列;3.4和3.5中,为使变异检测结果更准确可信,在比对之前,对所述测序数据进行过滤,所述过滤包括去除掉不确定碱基比例超过10%的读段和/或碱基质量值不大于5的碱基数的比例不小于50%的读段。并且,在进行重新比对(第二比对)之前,去除掉前一次比对结果中的一个读段对中的两个读段相同的读段对。所说的第二比对为局部比对,第一比对为常规全局比对,可利用但不限于soap或bwa等软件依照其默认设置进行,获得第一比对结果,第一比对结果包括读段在参考序列上的匹配位置及匹配情况信息,进行第二比对即基于第一比对结果,对与所捕获的基因区域对应的参考序列中的所有已知indel附近的所有序列信息(reads)进行局部重新比对,能够消除第一比对中的错误,提高后续变异检测的准确性,第二比对可利用gatk重比对软件(https://www.broadinstitute.org/gatk/)进行。3.6分析下机数据qc;3.7寻找变异;3.8对变异结果进行注释,得到最终数据结果。(四)靶向药物数据库构建及肿瘤变异解读靶向药物在肿瘤治疗中具有药效显著、毒副作用小的特点,但其对靶点(包括蛋白、基因等)有特异性依赖,必须先对患者做靶点分析,才能确定患者是否适合用药。整合目前fda批准用于治疗胃癌的靶向药物、用于其他癌症治疗的靶向药物、以及处于临床ii、iii期药物,依据nccn临床治疗指南,最新的临床药物基因研究,整理药物靶点基因与靶向药物疗效关系,形成肿瘤个体化靶向药物解读数据库。并将靶向药物数据库整合入肿瘤个体化生物信息分析流程,完成靶向药物的数据库构建及自动化解读。对生物信息分析后的变异数据进行个体化解读,参考构建的肿瘤数据库及相关文献,对患者检出的变异进行分析,判断最适合的获益靶向药物及耐药性靶向药物,辅助让临床医生对于肿瘤患者的用药治疗更有针对性,免去无效用药所耽误的宝贵时间以及毒副作用给患者带去的治疗痛苦。在肿瘤个体化用药指导领域,目标区域捕获高通量测序技术是目前基因组学研究中的一个热点技术。通过对受检者治疗过程中切除的病灶组织进行高通量捕获测序,可有以下 临床应用:(1)胃癌靶向治疗关键基因变异情况分析;(2)胃癌患者术后靶向治疗指导建议。相比于前述的其他技术手段,上述应用具有如下优势:1、高通量。基于新一代测序技术的目标区域捕获测序,能够在很短的时间内同时进行多例样本检测,并且在目标区域捕获芯片的使用下,相同数据量可进行更高深度的数据挖掘。2、高灵敏度:通过高深度的目标区域捕获测序,可以检测出突变频率为1%的低频变异,对于胃癌患者个体的体细胞变异能够准确地检出。3、高特异性:该应用能够保证较低的假阳性率、假阴性率,确保得到的检测结果能够准确的反映受检者的突变情况。4、应用广:对于每个经手术切除病灶组织的胃癌患者,均可运用该项技术。样本采集过程不会额外给受检者造成负担。实施例21、样本选择与dna提取。在本发明的实施例中选择1个带有已知突变tp53基因i195f突变的福尔马林固定石蜡包埋(ffpe)的胃癌组织样本,具体样本信息见表3。表3样本编号组织类型来源突变位点获得途径sc1ffpe低分化胃腺癌患者tp53-i195f临床样本sc1样本所带的tp53基因i195f突变经过sanger测序验证,测序峰图见图4。采用dneasyblood&tissuekit基因组提取试剂盒(qiagen,货号:69504)提取其中的基因组dna。利用qubit2.0荧光分光光度计(lifetechnologies,货号:q32866)检测提取所获得的dna浓度。2、基因组dna片段化样品打断采用ab公司的covarissystem可将样品dna片段控制在100~500bp范围。样品打断结束后,取少量(约总量的1/30)打断后样品在2%琼脂糖凝胶上进行1*tae电泳检测,并保存好胶图。打断效果一般以所要求制备的文库片段大小在所打断dnasmea的主带位置较为理想。例如,若要求制备插入100bp的pairedend文库,则打断后dnasmear主带在100bp处即可,若打断效果不理想则需要进行重新打断。样品片段化步骤也 可使用其他打断系统,具体参数可根据仪器的要求进行调整。covarissystem打断具体操作为:1)每天使用前须更换covariss2仪器有机玻璃水槽中的水,水面须达到“waterline”的界线。使用的水须是milli-q的水或超纯水,每天仪器使用完毕后需将水槽中的水及时倒掉,每个月须用超纯水清理有机玻璃水槽至少一次。2)开启covaris专用的笔记本电脑,检查其与各仪器终端是否连接。打开covariss2仪器和制冷仪开关并检查制冷仪的温度设置是否为4℃。3)双击打开covaris主程序“sonolabs-seriesv2.54”后点击“start[enter]”,待仪器排气30min,且水温降为10℃左右后方可使用。打开主程序之前须先确定打碎仪和冷却仪的电源开关是否打开,否则主程序会提示错误并重试。4)将样品加入待用的100μl的covarismicrotube中,样品加入量5μg,最后将终体积用te补至100μl,用移液器将样品溶液吹打混匀。点击“configure”,按下表设置参数:选择模式为“frequencysweeping”。所有设置完成后点击“save”或“saveas...”保存好程序,再点击“returntomainpanel”回到主界面。将打断管放入covaris装置中,选择好程序,开始打碎。按所设置的程序打碎完成后便可以进行下一个样品的打碎,若无其他样品须打碎可以关闭打碎仪和制冷仪的电源然后关闭打碎主程序和电脑。5)将打断后的样品从玻璃管中吸出,放入1.5mlep管中。取出3μl的样品用于2%琼脂糖凝胶进行1*tae电泳检测,并用1.8倍beckmancoulter公司的agencourtampurexp磁珠(a63881)进行回收,产物溶于32μl的elutionbuffer(eb)中。3、末端修复预先从-20℃保存的试剂盒中取出10xpolynucleotidekinasebuffer和10mmdntpsmix,将其置于冰上融解并充分混匀10xpolynucleotidekinasebuffer。在1.5ml的离心管中配制末100μl端修复反应体系:30μl的片段化回收产物、12μl超纯水、5μl10xpolynucleotidekinasebuffer(b904)、0.4μldntpsolutionset(自己稀释混合为25mmeach)、1.2μlt4dnapolymerase、0.2μlklenowfragment与1.2μlt4polynucleotidekinase。20℃ 温浴30min后,用1.8倍agencourtampurexp磁珠回收纯化反应体系中的dna,产物dna溶于33.5μl的eb中。4、primeradapter连接1)在产物离心管中按照如下体系配制接头连接反应体系,充分混匀后轻微离心;试剂体积(μl)dna31.52×ligationbuffer37.5p1_adapters(10um,自合成)1.5a_adapters(10um,自合成)1.5dnaligase3totalvolume752)将离心管放置在thermomixer中20℃孵育30min;3)用1.5×体积agencourtampurebeads纯化反应产物,最后用52μlelutionbuffer回溶产物;4)用1.5×体积agencourtampurebeads纯化第一次纯化的回溶产物,最后用33μlelutionbuffer回溶产物;5)用qubit2.0检测接头连接之后的产物浓度。5、杂交前pcr扩增pre-pcr杂交体系如下:试剂体积(μl)dna15nuclease-freewater312×phusionhfpcrmastermix50p1primer(10μm自合成)2aprimer(10μm自合成)2totalvolume100将试剂完全转入一个新的pcr管中,按照如下程序进行pcr反应:反应后先加入0.9倍agencourtampurexp磁珠进行大片段选择,然后转移上清加入0.15倍agencourtampurexp磁珠进行纯化,用33μlnucleas-free水回溶,取上清后用qubit2.0质控并进行芯片杂交。6、目标区域捕获芯片杂交1)杂交体积计算:用qubit定量后将不同pcr产物等量混合(pooling),共750ng。2)取750ngpooling产物转入一个新的离心管中,使用真空浓缩仪60℃约40min将产物蒸干。3)将所需的试剂置于冰上融化,充分混匀后轻微离心;4)在离心管中按照下表配制用于富集目的片段的样品文库体系;试剂体积(μl)pre-pcrlibrary750ngnuclease-freewater补水至2.8μlbgiblock#12.5bgiblock#22.5ionxpress_p1block(500μm)0.6ionxpress_a_xblock(500μm)0.6totalvolume95)将反应体系全部转入一个新的pcr管中,按照如下反应条件进行预杂交;温度时间95℃5min65℃holding6)在一个新的1.5ml离心管中按照下表配制杂交缓冲液反应体系;试剂体积(μl)hybridizationbuffer125hybridizationbuffer21hybridizationbuffer310hybridizationbuffer414totalvolume507)根据样品反应个数,按每个反应13μl的量分别加入到pcr管中,盖好管盖,然后将其放入pcr仪中65℃孵育至少5min,确认体系中没有晶体沉淀才能使用;8)在一个新的96孔pcr管中按照下表配制oligolibrarymix;试剂体积(μl)nuclease-freewater1.5rnaseblock0.5oligocapturelibrary5totalvolume7注意事项:此操作在冰上进行。9)将反应体系放入pcr仪中65℃孵育2min;10)保持各反应体系于65℃,揭开样品文库管和杂交buffer管的管盖,迅速把13μl的杂交buffer转移到样品文库管中;11)保持各反应体系于65℃,揭开96孔板上的oligolibrarymix管盖,迅速将样品文库管中的所有反应体系全部转移到oligolibrarymix管中,用移液器吹打混匀;12)保持pcr板于65℃,杂交24h。7、芯片洗脱及第二轮pcr扩增1)每一个杂交反应取50μldynabeadsm-280streptavidinbeads于一新的1.5ml离心管;2)将200μlbindingbuffer加入磁珠中,用漩涡混合仪剧烈振荡5s重悬磁珠;3)将离心管置于磁力架上2min,待液体完全澄清;小心吸取并弃去上清;4)重复步骤2)到步骤3)2次;5)加入200μlbindingbuffer重悬磁珠(重悬磁珠和杂交体系一起在垂直混匀仪上旋转混匀);6)从pcr仪中直接将全部杂交混合液转移到准备好的磁珠中,上下颠倒离心管3到5次直到混匀;7)将装有杂交混合液和磁珠的离心管在垂直混匀仪上旋转混匀,室温下混匀30min;8)将样品从混匀装置取下,瞬时离心5s确保管盖上没有液体残留;9)将1.5ml离心管转移放置在磁力架上,静置3-5min待液体完全澄清,小心吸取并弃去上清;10)用500μlwashbuffer#1重悬磁珠,并于漩涡混合仪上振荡5s混匀样品,室温下孵育样品15min;11)将离心管瞬时离心3s,然后放置在磁力架上,静置3-5min待液体完全澄清,小心吸取并弃去上清;12)用500μlwashbuffer#2重悬磁珠,并于漩涡混合仪上振荡5s以混匀样品,将其置于thermomixer中65℃孵育10min;13)颠倒离心管以混匀样品,瞬时离心3s,然后将其放置在磁力架上,静置3-5min待液体完全澄清,小心吸取并弃去上清;14)重复步骤12)到步骤13)2次;15)用28μlnuclease-freewater重悬磁珠。16)在离心管中按照下表配制反应体系,充分混匀后轻微离心:试剂体积(μl)dna30nuclease-freewater162×phusionhfpcrmastermix50p1primer(10μm自合成)2aprimer(10μm自合成)2totalvolume10017)将试剂完全转入一个新的pcr管中,按照如下程序进行pcr反应:反应后加入1.5倍agencourtampurexpreagent,磁珠纯化后,使用52μlnuclease-freewater回溶,再次加入1.5倍agencourtampurexpreagent磁珠纯化,最后用33μlnuclease-freewater回溶,qubit2.0和安捷伦2100质控后上机测序。8、测序及结果分析。运用iontorrent公司的proton测序仪(lifetechnologies)对构建好的文库进行高通量测序。测序实验操作按照制造商提供的操作说明书操作。得到下机的数据后,对得到的测序结果进行数据过滤和质量控制,然后按照下列流程进行生物信息学分析:1)tmap:与reference序列比对,产生bam文件;2)bamduplicates:对pcr过程中生成的重复reads进行标记;3)qc:统计芯片的捕获效率、有效reads数、平均深度、重复率、覆盖度及未被覆盖的区间等信息;4)callsnp/indel:用tvc工具默认参数检测snp/indel变异;5)注释注释变异的功能、reads支持数、变异频率、氨基酸变异及cosmic中的变异等;本实例得到的下机数据结果如表4所示。表4实例测序下机数据结果样本测序数据量(bp)≥q20reads数量平均长度sc1465,768,497391,700,8613,102,543132bp数据质量结果如表5所示。表5实例测序结果数据质量分析样本sc1总有效数据量(mb)69.25目标区域捕获效率26.00%目标区域平均测序深度166.55目标区域覆盖度99.70%重复率81.61%同时,发明人在测序结果中验证了样本所携带的已知基因突变是否能被检测到,结果如表6所示。表6已知突变的检测结果样本阳性突变位点是否检出突变测序深度突变频率sc1tp53-i195f是16441%根据以上的数据分析结果,发明人得到以下结论:①根据表4的数据,此次测序的测序数据量、q20数据量、reads平均长度等指标均在正常范围内,说明此次测序结果数据可靠;②根据表5的数据,目标区域捕获效率均在26%左右,目标区域平均测序深度在166x左右,数值均在正常范围内,说明该芯片的捕获结果正常。同时,该芯片对于目标区域的覆盖度为99.7%,能较好地覆盖芯片设计的目标区域;③根据表6的结果,此次测试的ffpe样本所包含的已知突变成功被检测到,说明该芯 片可以正确检测出样本中所包含的基因突变。综上所述,本发明所涉及的芯片在实例中实现了对样本dna目标区域的捕获以及样本所包含基因突变的检测。本发明可以应用于胃癌个体用药指导的研究之中。在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1