一种含氟烷基共聚醚及其制备方法与流程
本发明涉及一种含氟烷基共聚醚及其制备方法,属于精细化学品合成技术和高分子材料领域。
背景技术:
聚氨酯具有优越的力学性能、良好的粘接性能,以及优良的耐候性、电气性能等优点。正因如此,聚氨酯在胶黏剂、油漆油墨、涂料、塑料和弹性体,以及纤维等领域获得了广泛应用。聚氨酯一般由聚合物多元醇与二异氰酸酯加成聚合而成,根据聚合物多元醇和二异氰酸酯种类的不同,可以获得性能丰富多彩、用途多变的聚氨酯品种。
合成含氟聚氨酯的通用方法是在聚氨酯主结构中嵌入氟烷基等疏水性链段。例如,使用聚(六氟五亚甲基己二酸酯)和聚(六氟五亚甲基丙二酸酯)与 TDI等反应可合成出聚酯型含氟聚氨酯。或者使用六氟戊二醇聚醚等全氟醚小分子二元醇为原料,其用于制备含氟聚氨酯使聚醚链段以软段进入聚氨酯,产物柔顺性好;然而,这些含氟聚氨酯由于采用的含氟基团氟含量低,提供的表面能不够低而不能起到防水防油的功能。
技术实现要素:
本发明的目的为提供一种侧基型含氟烷基共聚醚,其由两种含氟环氧单体共聚而成,氟含量高,疏水性强,且制备工艺简便、反应条件温和,适合工业化生产,在聚氨酯大分子中引入氟元素获得的含氟聚氨酯不但可以提供优越的耐水性,控制含氟聚氨酯的分子结构甚至可以提供强疏水性聚氨酯产品,可大大拓展聚氨酯在防水防油涂层、防污自洁涂料等领域的应用。
本发明公开了一种含氟烷基共聚醚,其化学结构式为:
其中,n为2~7;x为3~100;y为1~100。
本发明公开了一种含氟烷基共聚醚的制备方法,其制备步骤包括单体制备和共聚反应,包括以下步骤:
(1)混合乙酸烯丙基酯、碳酸氢钠、连二亚硫酸钠、有机溶剂、水;然后于-20~5℃下,滴加全氟烷基碘,进行合成反应;然后滴加碱液,进行环化反应,制备含氟环氧单体;
(2)混合乙酸烯丁基酯、碳酸氢钠、连二亚硫酸钠、有机溶剂以及水;然后于-20~5℃下,滴加全氟烷基碘,进行合成反应;然后加入碱液进行环化反应得到含氟氧杂环丁烷;
(3)在反应器中加入氯代烷烃类溶剂和起始剂;然后于0~10℃下加入三氟化硼乙醚;然后滴加混合单体,进行开环聚合反应,得到含氟烷基共聚醚;所述混合单体由含氟氧杂环丁烷与含氟环氧单体组成。
上述技术方案中,所述全氟烷基碘为七氟碘丙烷、九氟碘丁烷、十三氟碘己烷、十五氟碘庚烷、十七氟碘辛烷中的任意一种;所述有机溶剂为N,N-二甲基甲酰胺;所述氯代烷烃类溶剂为二氯甲烷、二氯乙烷、三氯甲烷、四氯化碳中的任意一种;所述的起始剂为乙二醇、1,4-丁二醇、1,6-己二醇中的任意一种;所述的碱液为NaOH水溶液、KOH水溶液中的任意一种;。
上述技术方案中,步骤(1)中,合成反应完成后,以芳香烃溶剂萃取反应液,然后在得到的萃取液中滴加碱液进行环化反应,所述碱液的质量浓度为10%~25%;步骤(2)中,合成反应完成后,以芳香烃溶剂萃取反应液,然后在得到的萃取液中加入碱液进行环化反应,所述碱液的质量浓度为5%~20%。
上述技术方案中,步骤(1)中,所述合成反应为于-20~5℃下反应1~10小时,再于室温反应1~10小时;所述环化反应为于5~40℃反应1~8小时;步骤(2)中,所述合成反应为于-20~5℃下反应1~12小时,再于室温反应1~12小时;所述环化反应为于5~30℃反应1~12小时;步骤(3)中,加入三氟化硼乙醚后反应10分钟~2小时然后滴加混合单体;开环聚合反应的温度为10~45℃,时间为1~48小时
上述技术方案中,步骤(1)中,乙酸烯丙基酯、碳酸氢钠、连二亚硫酸钠、有机溶剂、水、全氟烷基碘、碱液的质量比为(1~3)∶(1~3)∶(3~7)∶(10~30)∶(10~30)∶(2~10)∶(10~50);步骤(2)中,乙酸烯丁基酯、碳酸氢钠、连二亚硫酸钠、有机溶剂、水、全氟烷基碘、碱液的质量比为(1~3)∶(2~3)∶(4~7)∶(10~30)∶(10~30)∶(3~10)∶(10~60);步骤(3)中,卤代烷烃类溶剂、丁二醇、三氟化硼乙醚、含氟氧杂环丁烷与含氟环氧单体的质量比为(10~100)∶(0.1~3)∶(0.1~3)∶(10~50)∶(10~50)。
上述技术方案中,步骤(1)中,滴加全氟烷基碘的时间为0.5~2小时;步骤(2)中,滴加全氟烷基碘的时间为0.5~2小时;步骤(3)中,滴加混合单体的时间为10分钟~3小时。
上述技术方案中,步骤(1)中,环化反应结束后,反应液中的有机层经过水洗、干燥、减压蒸馏得到含氟环氧单体;步骤(2)中,环化反应结束后,反应液中的有机层经过水洗、干燥、减压蒸馏得到含氟氧杂环丁烷;步骤(3)中,开环聚合反应结束后,在聚合反应液中加入碱金属碳酸盐水溶液,搅拌0.5~6小时;然后用氯代烷烃类溶剂萃取,再将萃取液干燥、过滤、除溶剂得到含氟烷基共聚醚。
上述技术方案中,所述碱金属碳酸盐为NaHCO3、Na2CO3、K2CO3中的任意一种;所述碱金属碳酸盐水溶液的质量浓度为1.0~10%;所述芳香烃溶剂为甲苯、三氟甲苯、二(三氟甲基)苯中的任意一种;所述减压蒸馏条件为温度40~80℃、真空度10~20mmHg。
上述制备方法具体为:
(1)含氟环氧单体制备
a. 按重量计,在反应器中投入10~30份乙酸烯基酯,10~30份碳酸氢钠,30~70份连二亚硫酸钠,100~300份N,N-二甲基甲酰胺,100~300份水。滴加20~100份全氟烷基碘,于-20~ -5℃反应1~10小时后,缓慢升至室温,保温反应1~10小时;
b. 反应结束后,以50~200份芳香烃溶剂分3~5次萃取。合并萃取液,水洗3~5次,制得4-九氟丁基-3-碘丁基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应;
c. 将上述含氟烷基碘丁基乙酸酯溶液直接加入到反应器中,滴加100~500份质量浓度为10%~25%的氢氧化钠溶液,保温于5~40℃反应1~8小时;
d. 反应结束后分层。有机层水洗3~5次,干燥、减压蒸馏,得含氟环氧单体;
(2)含氟氧杂环丁烷合成
a. 加成:按重量计,在反应器中投入10~30份乙酸烯丁基酯,20~30份碳酸氢钠,40~70份连二亚硫酸钠,100~300份N,N-二甲基甲酰胺,100~300份水。控制温度为-20~-5℃,滴加30~100份全氟烷基碘,滴加时间为30分钟~2小时;加完后保温反应1~12小时,再缓慢升至室温反应1~12小时;
b. 萃取:以芳香烃溶剂分3~5次萃取反应液,每次使用芳香烃溶剂15~60份;合并,水洗萃取液3~5次;
c. 环化:在萃取液中加入100~600份碱液,于5~30℃反应1~12小时;
d. 后处理:取反应液中的有机层,水洗3~5次,加入0.5~3份干燥剂干燥1~12小时;减压蒸馏得含氟氧杂环丁烷单体;
(3)共聚反应
a. 混合单体
按重量计,将10~50份的含氟氧杂环丁烷和10~50份的含氟环氧丙烷混合均匀,得到混合单体;
b. 开环共聚
在反应器中加入10~100份氯代烷烃类溶剂和0.1~3份起始剂,于0~10℃下加入0.1~3份三氟化硼乙醚(BF3.Et2O)反应10分钟~2小时。滴加含氟混合单体,10分钟~3小时加完。加完后,升温至10~45℃继续反应1~48小时,停止反应;
c. 后处理
在反应液中加入2~30份碱金属碳酸盐的稀溶液,搅拌反应30分钟~6小时后,用40~200份氯代烷烃类溶剂萃取3~5次。合并有机层,加入干燥剂干燥1~12小时,过滤除去干燥剂,滤液蒸去溶剂得产物含氟烷基共聚醚。本发明提供的一种含氟烷基共聚醚的制备方法,采用全氟烷基碘分别与乙酸烯丙基酯和乙酸烯丁基酯通过单电子转移反应、环化反应制得含氟烷基环氧丙烷和含氟烷基氧杂环丁烷两种单体,再将两种环氧单体在路易斯酸催化下开环共聚,制得含氟烷基共聚醚。
反应式如下:
其中,m= 1 或2;n为2~7;x= 3~100;y= 1~100。
本发明在聚醚二元醇侧基上引入含氟烷基可以克服了现有技术含氟基团氟含量低缺点。聚醚侧基上含氟烷基由具有一定碳链长度的全氟烷基组成,当侧基氟烷基碳链长度大于等于4时,获得的含氟聚氨酯具有氟原子堆积密集、氟含量高,氟烷基可以在高分子表面聚集的特性;同时,通过调控侧基全氟烷基的链长等结构还可调空聚氨酯的表面性能,采用长碳链全氟烷基作为侧基甚至可以获得具有超疏水功能的聚氨酯材料,且具有良好的外观,稳定性、耐水性和综合力学性能较好。因此本发明还公开了上述含氟烷基共聚醚在制备含氟聚氨酯中的应用。
与现有技术相比,本发明的优点在于:
1、本发明合成的含氟烷基共聚醚侧基为全氟烷基,聚合物提供的氟含量高,用于合成聚氨酯可以得到低表面能和优良疏水性能的产品,聚氨酯表面性能易于调控;
2、本发明合成的含氟烷基共聚醚由两种不同链节交替形成主链,因此分子链柔顺性,产品不易结晶且具有低熔点,适合制备聚氨酯使用;
3、本发明合成的共聚醚引入了氧杂丁烷链节,该链节结构有利于共聚醚的疏水性;可以获得具有超疏水功能的聚氨酯材料,且具有良好的外观,稳定性、耐水性和综合力学性能较好;
4、本发明合成共聚醚的工艺简便,反应条件温和,仅需常规设备即可合成,适合工业化生产。
附图说明
图1是实施例1制备的含氟烷基共聚醚的1H NMR图;
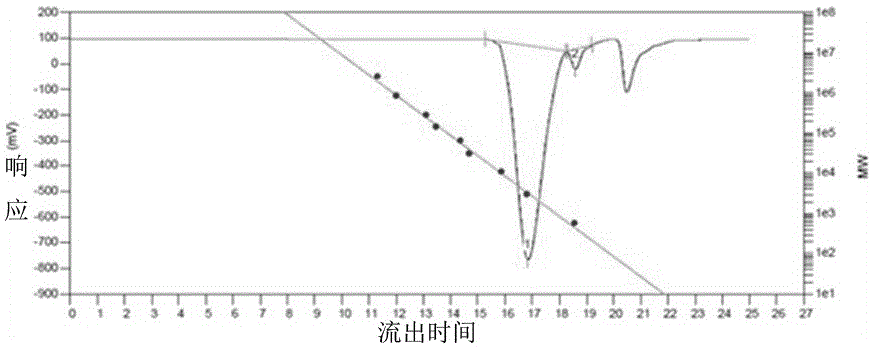
图2是实施例1制备的含氟烷基共聚醚的凝胶渗透色谱(GPC)测试图;
图3是实施例2制备的含氟烷基共聚醚的凝胶渗透色谱(GPC)测试图;
图4是实施例3制备的含氟烷基共聚醚的凝胶渗透色谱(GPC)测试图。
具体实施方式
下面结合实施例和附图对本发明技术方案作进一步描述。
实施例1
(1)一锅法制备含氟烷基环氧单体
a. 九氟氧杂环丁烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入16.5g乙酸烯丁基酯,26.6g碳酸氢钠(NaHCO3),55.0g连二亚硫酸钠,220g N,N-二甲基甲酰胺,210g水,滴加55.4g全氟丁基碘烷,-15℃反应3h,缓慢升至室温,保温反应2h。反应结束后,以150g甲苯分3次萃取。合并萃取液,水洗3次,制得4-九氟丁基-3-碘丁基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丁基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加305g质量浓度为20%的氢氧化钠溶液,保温于15℃反应3h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏,得3-(1H,1H-九氟戊基)氧杂环丁烷33.8g,收率为75.9%。
b. 九氟环氧丙烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入14.2g乙酸烯丙基酯,26.6g碳酸氢钠,55.0g连二亚硫酸钠,220g N,N-二甲基甲酰胺,210g水,滴加55.4g全氟丁基碘烷,-20℃反应4h,缓慢升至室温,保温反应4h。反应结束后,以180g三氟甲苯分3次萃取。合并萃取液,水洗4次,制得3-九氟丁基-2-碘丙基乙酸酯液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丙基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加533g质量浓度为10%的氢氧化钠溶液,保温于12℃反应1h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏除去溶剂,得1H,1H-九氟戊基环氧丙烷33.9g,收率为80.3%。
(2)共聚反应
a. 混合单体
将27.8g的3-(1H,1H-九氟戊基)氧杂环丁烷和26.4g的1H,1H-九氟戊基环氧丙烷混合均匀,得到混合单体。
b. 开环共聚
在配有温度计、搅拌器和恒压滴液漏斗的三口烧瓶中,在氮气保护下加入,加入50g二氯甲烷和0.32g1,4-丁二醇,冰水浴降温至2℃,慢慢加入0.11gBF3.Et2O,温度约上升3℃,反应30分钟后,滴加上述配制好的混合单体,35分钟加完。加完后,升温至30℃,继续反应24h。停止反应。
c. 后处理
上述开环聚合反应液加入10g2.5%NaHCO3水溶液,终止反应,搅拌反应1小时后,用120g二氯甲烷萃取3次,合并有机层,加入干燥剂无水硫酸镁干燥1小时,过滤除去干燥剂后,滤液蒸去溶剂得产物49.3g,收率91.2%。产物结构式如下。
其中,x为3~10;y为2~10。
参见附图1,是上述含氟烷基共聚醚的氢核磁共振图,其中,在化学位移δ4.25ppm处的峰可归属于聚醚主链-CH2-CH2-的氢质子峰,δ3.68ppm处的峰为-CH2-CH2-O-的氢质子峰,δ3.58ppm为-OCH2-CH2-O-的氢质子峰,δ1.87ppm为聚醚侧基上-CH2-的氢质子峰。
参见附图2,是上述含氟烷基共聚醚的凝胶渗透色谱图,其中,含氟烷基共聚醚产物数均分子量Mn为2640,重均分子量Mw为3430,分散系数为1.30。
实施例2:
(1)一锅法制备含氟烷基环氧单体
a. 九氟氧杂环丁烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入15.9g乙酸烯丁基酯,27.1g碳酸氢钠,55.3g连二亚硫酸钠,218g N,N-二甲基甲酰胺,209g水,滴加54.9g全氟丁基碘烷,-12℃反应3h,缓慢升至室温,保温反应3h。反应结束后,以165g三氟甲苯分3次萃取。合并萃取液,水洗3次,制得4-九氟丁基-3-碘丁基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丁基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加310g质量浓度为20%的氢氧化钠溶液,保温于14℃反应3h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏,得3-(1H,1H-九氟戊基)氧杂环丁烷32.3g,收率为72.3%。
b. 九氟环氧丙烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入14.6g乙酸烯丙基酯,26.9g碳酸氢钠,56.8g连二亚硫酸钠,208g N,N-二甲基甲酰胺,219g水,滴加56.4g全氟丁基碘烷,-18℃反应4h,缓慢升至室温,保温反应6h。反应结束后,以168g三氟甲苯分3次萃取。合并萃取液,水洗4次,制得3-九氟丁基-2-碘丙基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丙基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加535g质量浓度为10%的氢氧化钠溶液,保温于13℃反应3h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏除去溶剂,得1H,1H-九氟戊基环氧丙烷31.9g,收率为75.6%。
(2)共聚反应
a. 混合单体
将18.5g的3-(1H,1H-九氟戊基)氧杂环丁烷和35.9g的1H,1H-九氟戊基环氧丙烷混合均匀,得到混合单体。
b. 开环共聚
在配有温度计、搅拌器和恒压滴液漏斗的三口烧瓶中,在氮气保护下加入,加入45g二氯乙烷和0.32g1,6-己二醇,冰水浴降温至3℃,慢慢加入0.10gBF3.Et2O,温度约上升3℃,反应40分钟后,滴加上述配制好的混合单体,1小时加完。加完后,升温至25℃,继续反应24h。停止反应。
c. 后处理
上述开环聚合反应液加入11g2.5%NaHCO3水溶液,终止反应,搅拌反应1.5小时后,用130g二氯乙烷萃取3次,合并有机层,加入干燥剂无水硫酸钠干燥1小时,过滤除去干燥剂后,滤液蒸去溶剂得产物49.0g,收率90.1%。产物结构式如下。
其中,x为3~8;y为5~20。
参见附图3,是上述含氟烷基共聚醚的凝胶渗透色谱图,其中,含氟烷基共聚醚产物数均分子量Mn为4400,重均分子量Mw为5640,分散系数为1.28。
实施例3:
(1)一锅法制备含氟烷基环氧单体
a. 十三氟氧杂环丁烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入16.8g乙酸烯丁基酯,27.9g碳酸氢钠,54.8g连二亚硫酸钠,235g N,N-二甲基甲酰胺,230g水,滴加72.6g全氟己基碘烷,-14℃反应5h,缓慢升至室温,保温反应6h。反应结束后,以188g三氟甲苯分3次萃取。合并萃取液,水洗3次,制得4-十三氟己基-3-碘丁基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丁基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加511g质量浓度为10%的氢氧化钠溶液,保温于15℃反应6h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏,得3-(1H,1H-十三氟庚基)氧杂环丁烷39.4g,收率为65.8%。
b. 十三氟环氧丙烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入14.7g乙酸烯丙基酯,27.1g碳酸氢钠,55.4g连二亚硫酸钠,226g N,N-二甲基甲酰胺,215g水,滴加73.9g全氟己基碘烷,-16℃反应4h,缓慢升至室温,保温反应6h。反应结束后,以190g的三氟甲苯分3次萃取。合并萃取液,水洗3次,制得3-十三氟己基-2-碘丙基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丙基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加522g质量浓度为10%的氢氧化钠溶液,保温于20℃反应4h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏,得1H,1H-十三氟庚基环氧丙烷41.3g,收率为70.9%。
(2)共聚反应
a. 混合单体
将25.2g的3-(1H,1H-十三氟庚基)氧杂环丁烷和48.1g的1H,1H-十三氟庚基环氧丙烷混合均匀,得到混合单体。
b. 开环共聚
在配有温度计、搅拌器和恒压滴液漏斗的三口烧瓶中,在氮气保护下加入,加入60g二氯甲烷和0.52g1,4-丁二醇,冰水浴降温至2℃,慢慢加入0.8gBF3.Et2O,温度约上升5℃,反应50分钟后,滴加上述配制好的混合单体,1小时加完。加完后,升温至28℃,继续反应24h。停止反应。
c. 后处理
上述开环聚合反应液加入16g2.5%Na2CO3水溶液,终止反应,搅拌反应2小时后,用186g二氯甲烷萃取3次,合并有机层,加入干燥剂无水硫酸钠干燥1小时,过滤除去干燥剂后,滤液蒸去溶剂得产物65.2g,收率88.0%。产物结构式如下。
其中,x为3~10;y为3~20。
图4是上述含氟烷基共聚醚的凝胶渗透色谱(GPC)测试图,其中,含氟烷基共聚醚产物数均分子量Mn为4630,重均分子量Mw为5930,分散系数为1.28。
实施例4
(1)一锅法制备含氟烷基环氧单体
a. 十三氟氧杂环丁烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入16.5g乙酸烯丁基酯,26.8g碳酸氢钠,55.3g连二亚硫酸钠,233g N,N-二甲基甲酰胺,215g水,滴加72.6g全氟己基碘烷,-13℃反应5h,缓慢升至室温,保温反应4h。反应结束后,以165g甲苯分3次萃取。合并萃取液,水洗3次,制得4-十三氟己基-3-碘丁基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丁基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加520g质量浓度为10%的氢氧化钠溶液,保温于16℃反应3h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏,得3-(1H,1H-十三氟庚基)氧杂环丁烷44.3g,收率为73.3%。
b. 十三氟环氧丙烷合成
在配有温度计、冰水浴和磁力搅拌器保护的1000mL三口烧瓶中,加入14.2g乙酸烯丙基酯,26.8g碳酸氢钠,54.8g连二亚硫酸钠,218g N,N-二甲基甲酰胺,208g水,滴加73.0g全氟己基碘烷,-18℃反应5h,缓慢升至室温,保温反应3h。反应结束后,以165g的1,3-二(三氟甲基)甲苯分3次萃取。合并萃取液,水洗3次,制得3-十三氟己基-2-碘丙基乙酸酯溶液,溶液不经处理,直接投入下一步环化反应。
将上述含氟烷基碘丙基乙酸酯溶液直接加入到1000mL三口烧瓶中,滴加512g质量浓度为10%的氢氧化钠溶液,保温于19℃反应4h。反应结束后分层。有机层水洗3次,干燥、减压蒸馏,得1H,1H-十三氟庚基环氧丙烷46.3g,收率为76.6%。
(2)共聚反应
a. 混合单体
将37.9g的3-(1H,1H-十三氟庚基)氧杂环丁烷和36.1g的1H,1H-十三氟庚基环氧丙烷混合均匀,得到混合单体。
b. 开环共聚
在配有温度计、搅拌器和恒压滴液漏斗的三口烧瓶中,在氮气保护下加入,加入45g二氯甲烷和0.48g1,4-丁二醇,冰水浴降温至2℃,慢慢加入0.16gBF3.Et2O,温度约上升5℃,反应45分钟后,滴加上述配制好的混合单体,50分钟加完。加完后,升温至30℃,继续反应36h。停止反应。
c. 后处理
上述开环聚合反应液加入15g2.5%NaHCO3水溶液,终止反应,搅拌反应2小时后,用150g二氯甲烷萃取3次,合并有机层,加入干燥剂无水硫酸钠干燥1小时,过滤除去干燥剂后,滤液蒸去溶剂得产物60.1g,收率81.2%。产物结构式如下。
其中,x为3~10;y为2~10。
- 还没有人留言评论。精彩留言会获得点赞!