改进的分子育种方法与流程
背景技术:
分子育种技术涉及将遗传特征(基因型)与表型性状(表型)联系起来。因为基因型可以比表型更快地确定,所以可以加速育种周期。在植物和动物育种中使用基因组预测来预测用于选择目的的育种值,并且在人类遗传学上使用基因组预测来预测疾病风险。基因组预测方法取决于一组个体(“训练个体”)的表型的数据集(“训练数据集”)和相关联的基因型数据(典型地在许多遗传标记上)。将统计方法与训练数据集和选择候选物基因型的组合一起用来预测该选择候选物的育种值或疾病风险。然而,通常,常规的基因组预测方法例如流行的gblup方法在非线性遗传效应的影响下不能对复杂性状进行准确预测,该非线性遗传效应例如是由在输出性状与环境中的潜在组分性状之间的非线性关系以及基因型与环境的相互作用(其进一步扩大与组分性状的非线性关系的复杂性)传达。因此,当该复杂性状与组分性状具有非线性关系并且具有基因型与环境的相互作用时,在基因组预测领域中需要改进的基因组预测,特别是对候选物的复杂性状的选择。
发明概述
一个实施例包括在育种程序中选择个体的方法,所述方法包括:种植和生长训练个体的遗传多样性群体(geneticallydiversepopulation)或遗传狭窄群体(geneticallynarrowpopulation);对该训练个体的遗传多样性群体或遗传狭窄群体进行表型分型以产生表型训练数据集;使用生物模型例如作物生长模型将该表型训练数据集与包含跨每个训练个体基因组的遗传信息的基因型训练数据集相关联,所述作物生长模型是用于评估基因型标记的效应的方法和用于将该基因型标记的效应的估计与该生物模型联系的方法;对育种个体的遗传多样性群体进行基因分型;使用该关联训练数据集、生物模型例如作物生长模型来预测育种个体的性状表现,所述作物生长模型是一种用于评估基因型标记的效应的方法和用于将该基因型标记的效应的估计与该生物模型联系的方法:使用关联训练数据集和生长模型、基于植物基因型从该育种个体的遗传多样性群体中选择育种对,以选择可能产生具有一种或多种所希望的性状的后代的育种对;将该育种对杂交以产生后代;并且使该具有一种或多种所希望的性状的后代生长。
在另一个实施例中,该方法可以用于对数量性状基因座(qtl)作图,该数量性状基因座然后可以用于标记辅助选择策略。
附图说明
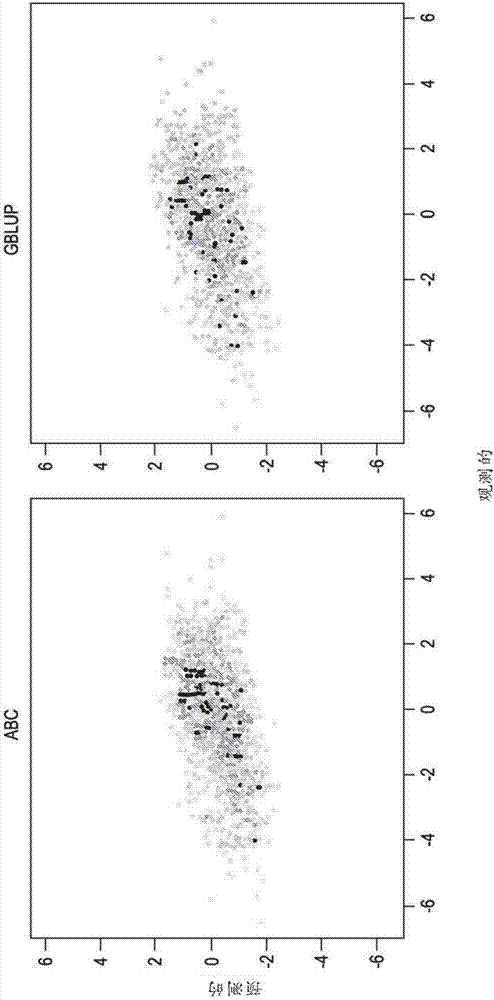
图1是abc(相关性0.78)和gblup(相关性0.52)的验证集中的预测值与观测值的图。
图2是abc(相关性0.57)和gblup(相关性0.51)的验证集中的预测值与观测值的图。
图3是总叶片数(tln)和最终生物量产量(bm)之间关系的图。
图4是abc和gblup的预测的和观测的最终生物量产量(bm)的图。
图5是由abc预测的总叶片数(tln)与在验证集中dh系的“观测的”tln值的图。注意,当拟合模型时,tln是未观测的。
图6是在环境e1(干旱)和e2(非干旱)中25个代表性基因型的生物量产量的相互作用图。
图7是在环境e1和e2中验证集dh系的观测和预测的生物量产量的图。使用的方法是abc。将来自e2的表型数据用于估计(但为不同dh系)。
图8是在环境e1和e2中验证集dh系的观测和预测的生物量产量的图。使用的方法是gblup。将来自e2的表型数据用于估计(但为不同dh系)。
图9是在环境e1和e2中25个代表性dh系的最终生物量产量的相互作用图。最终生物量产量值在每个环境中被标准化以改善可视化。
图10分别是代表性实例中对于2,000个dh系的在环境e2中的最终生物量产量(bm)和总叶片数(tln)和最大叶片面积(am)的的散点图。
图11是在方法abc-cgm的验证集中dh系的预测的与观测的tln和am值的图。在这个特定的实例中,在预测值和观测值之间的相关性为0.86(tln)和0.88(am)。对于比较,与gblup的相关性为-0.28(tln)和0.63(am)。
图12是使用abc-cgm和gblup方法获得的在环境e2和e1中在验证集中dh系的预测与观测的最终生物量产量(bm)的图。在这个特定的实例中,对于abc-cgm,在预测值和观测值之间的相关性为0.75(e2)和0.79(e1),并且对于gblup,为0.28(e2)和0.15(e1)。
图13是在环境e2中观测的最终生物量产量(bm)与对于不同的am值范围来自模型bm~tln+am的拟合值的图。
图14是在验证集中生理性状的预测值与观测值的图。使用具有最佳参数设置的abc-cgm获得预测。
图15是在验证集中生理性状的预测值与观测值的图。使用abc-cgm获得预测。(代表性实例)。
图16是对于环境e1和e2在验证集中预测的与观测的最终生物量(bm)值的图。使用abc-cgm(第1行)和gblup(第2行)获得预测。
图17是在验证集中生理性状的预测值与观测值的图(参见图15)。钟形曲线示出了在预测误差方面参数估计的不确定性。
图18是对于两种干旱环境模拟的水分供应/需求比率的时间模式。在种植后大约80天,对于两种环境通过水平条指示条目开花的时期(测量为花粉脱落的时间)。
图19显示了在两种干旱环境中评估的dh条目的全集的谷物产量blup。
图20显示了在两种干旱环境中评估的单个玉米杂交的20个重复的在cgm-wgp和gblup之间获得的测交谷物产量的预测准确度的比较:20a是观测环境中的估计条目,20b是新环境中的估计条目,20c是观测环境中的测试条目,20d是新环境中的测试条目。图例鉴定了对遗传模型参数进行估计的环境。
图21显示了基于使用50个dh条目的估计集在sfs估计环境中的模型选择,在fs预测环境中对于56个dh条目的测试集基于cgm-wgp和gblup的谷物产量预测的一次复制。
图22显示了基于gblup和cgm-wgp、对于一个重复的谷物产量预测的比较。
图23显示了作为平均动脉血压(map,mmhg)、醛固酮浓度(ald,ng/1)和血清钠(snameq/l)的函数的尿钠排泄量(una,meq/l)。
图24显示了与以摩根(m)计的遗传距离相关的成对连锁不平衡(ld,测量为r2)。
图25显示了在基于生物模型的全基因组预测方法(肾脏-wgp)和基准方法gblup的测试集中1,450个个体的预测与观测的尿钠排泄量(una,meq/l)。
说明书
对于复杂性状建模的先验基因组预测方法使用基于纯统计学的方法来明确地对统计模型条目之间的非线性关系进行建模。以前将生物信息纳入基因组预测方法以重建和预测来自组分性状的最终目标复杂性状的尝试,将组分性状与目标复杂性状分开建模,并且与其他组分性状分开建模。这些尝试未能明确地纳入参数估计过程中的性状之间的非线性关系。这些尝试也未能开发出将基于生物学知识的模型整合到估计过程中的框架,因此不允许同时对所有性状建模并且要求待观测的组分性状。无论目标复杂性状与组分性状是观测的或未观测的组分性状,并且不考虑它们之间的关系的性质,通过对它们之间的关系同时建模,本发明提供了任意复杂性状的广义定量预测框架。组分性状包括但不限于在作物生长模型中包括的生理性状、基因网络内的个体基因、天然和转基因dna多态性。
当观测的复杂性状和组分性状之间的关系是非线性的时,数值算法,例如包括近似贝叶斯计算(abc)的拒绝取样算法允许对任意参数集的同时估计,而不需要测量所有组分性状。通过在取样算法中将基于生物知识的模型整合、将观测的复杂性状与(可能)未观测的组分性状之间的关系明确地作图,可以促进预测参数的估计。基于生物学知识的模型的实例包括生理作物生长模型、基因网络和生物化学途径。然后可以使用预测参数来预测复杂性状和组分性状(当任一者或两者未被观测时)。
本发明的一个实施例包括用于增强的全基因组预测的方法,以选择具有干旱耐受性的近交种和杂交种,从而在干旱条件下改善作物产量和在更有利的环境条件下改善相等产量表现。本发明的另一个实施例包括增强的多性状全基因组预测,用于选择对于特定目标环境具有提高的产量和农艺性状表现的近交种和杂交种。本发明的另一个实施例包括增强的全基因组预测,用于选择对于目标地理具有提高的产量和农艺性状表现的近交种和杂交种,其中基因型与环境的相互作用是重要的。本发明的另一个实施例是对于上述每种方法转基因和天然遗传变异对近交种和杂交种产量和农艺性状表现的联合效应的增强的全基因组预测。
实例
实例1:一般性状模拟。
在这个实例中,模拟了两性状模型,然后使用近似贝叶斯计算方法进行建模,并且使用基因组最佳线性无偏预测(gblup)方法将这些结果进行比较。在两个性状模型中,第一个性状t1控制基因型是否跨越某一生理阈值,例如从营养发育阶段到生殖发育阶段的过渡,其反过来确定在水分有限的环境中在干燥的天气开始之前或之后开花是否发生。例如,t1可以是开花时间本身,或者它可以是作物生长模型(cgm)中的基因型依赖参数,例如可以计算开花时间的发育过渡的基础温度需求。第二个性状t2对于是否跨越特定阈值敏感。一个实例可以是产量形成,其在当水分不受限时的环境中与在水分有限的环境中的作用不同,因为生殖发育阶段的发生太晚。为了表现这种机制,第二个性状可以被指定为t2+或t2-,以分别表示在水分不受限和有限的条件下的产量形成。
为了模拟这种性状层次,t1由25个snp控制、t2+由25个snp控制并且t2-由25个snp控制。这些snp中的一些对几种性状(例如对t1和t2+)具有影响。由于这个原因,主效snp(causativesnp)的平均数为65。主效snp对于t1、t2+、和t2-的影响u=[u1,u2+,u2-]分别从标准正态分布绘制,并且总是性状特异的(即使snp对多个性状有影响)。将主效snp随机置于3摩根长度的单个染色体上。除了被假设是未观测的主效snp之外,将100个观测的snp标记置于染色体上。
模拟从双亲杂交创建了2,000个双单倍体(dh)系,其中染色体的减数分裂根据霍尔丹(haldane)作图函数进行模拟。所有未观测和观测的snp在杂交中分离。t2的表型,表示为y2,其被计算为
其中y1=z1u1和z1、z2+和z2-表示在三个性状的因果snp9(causalsnp)处2,000个dh的基因型矩阵。t1表型y1为中心,0用作生理关键阈值。从2,000个dh中,随机选择50个作为估计集。
近似贝叶斯计算(abc)。标记基因型与表型相关的模型对应于用于从模拟的主效snp产生dh表型数据的模型,但仅在方程式(1)中体现的原则方面。将关键阈值0视为已知的并且将t1是确定基因型是否跨越阈值的性状这一事实视为已知的。进一步假设t2对是否跨越阈值有生理敏感为已知,即遗传控制可能不同并且上下文取决于是否跨越阈值。然而,假设t1和t2性状的未观测的遗传结构的任何细节为未知的,例如哪些snp控制哪个性状(因果snp无论如何都是未观测的)。因此,我们对所有100个观测的snp拟合了三个标记效应,t1、t2+和t2-各一个。拟合的模型是
其中x1=za1。这里z是100个观测的snp的基因型矩阵,并且a=[a1、a2+、a2-]是估计的标记效应的向量。对于标记效应a的先验,我们使用了平均值为0以及方差为0.05的正态分布。
取样。abc拒绝取样算法进行如下:
1.从先验抽取候选物a′。
2.根据方程式(2)生成新数据y′2。
3.计算向量y′2和y2之间的欧几里德(euclidean)距离d。
4.如果d低于公差水平,则a′被接受为后验分布p(a|y2,h)的样本。
5.对于足够数量的样品,重复1至4。
接受候选物的公差水平是在“初始化运行”中确定的,以实现
作为比较,对数据进行标准基因组blup模型拟合。将通过使用50个dh的估计集获得的结果用于预测剩余的1,950个dh个体的y2。图1显示了abc实现了0.78的准确度(预测值和观测值的相关性),而gblup实现了0.52的较低值的一个实例。模拟重复了几次,并且abc总是比gblup实现更高的预测准确度,尽管在某些情况下具有更小的差异(图2)。
实例2:结合作物生长模型预测最终生物量产量。
该实例表明,abc可以与通过作物生长模型提供的信息一起使用。马乔(muchow)等人(1990)作物生长模型将玉米生物量(bm)生长作为温度和太阳辐射以及植物的几种生理性状(pt)的函数进行建模。所有pt可以是基因型特异的,这意味着应该对这些的标记效应进行估计。然而,作为第一步,所有pt被设置为有意义的常数,并且仅总叶片数(tln)作为特异的基因型建模。tln是直接或间接地包含cgm的大多数方程式中的关键pt和因素。在tln和最终bm之间的非线性关系图解地显示于图3中。
基因型:tln被模拟为由具有相似幅度的加性效应的10个因果snp控制。将这些因果snp随机置于3摩根长度的单个染色体上,并且将其假设是未知的。另外的110个观测的snp标记也随机地置于染色体上。根据霍尔丹(haldane)作图函数,通过模拟染色体的减数分裂产生来自双亲杂交的2,000个dh系。所有未观测和观测的snp在杂交中分离。在确定所有dh的tln后,根据cgm计算其bm值。在2,000个dh系中,100个被用作估计集,剩余的用于验证。
abc:假设产生表型数据的cgm是已知的,包括所有pt的值,除了tln之外,其被建模为
tlni=μtln+ziutln,(1)
其中μtln是截距,z1是dh系i的110个snp标记的基因型向量并且utln是标记效应的向量。唯一观测的参数是zi和最终生物量bm,此后表示为y。没有观测tln。
abc拒绝取样算法进行如下:
1.从先验抽取μtln和utln的候选物。
2.根据方程式(1)计算预测值tln′
3.从cgm模拟新的bm数据y′
4.计算向量y′和y之间的欧几里德(euclidean)距离d
5.如果d低于公差水平,则μtln和utln的候选物被接受为来自后验分布p(μtln,utln|y,h)的样本。
6.重复1至5直到抽取足够数量的样品。
与μtln一样,使用具有平均值等于9.5(其为tln的模拟平均值)和标准偏差为2的高斯分布(gaussiandistribution)。因此,关于μtln相当多的先验知识被假设是可用的,这是一个合理的假设。utln的先验是具有平均值为零以及标准偏差为0.25的高斯分布。选择公差使得接受率是≈10-6,,抽取的样本数是200。作为基准,也对数据进行标准gblup模型拟合。
结果
预测bm:使用abc实现的bm预测准确度(在验证集中dh系之间的预测值和观测值之间的相关性)总是远高于用gblup实现的预测准确度。取该模拟的10个重复的平均值,abc实现了0.85的准确度,gblup只有0.15。代表性实例显示于图4中。
预测tln:使用abc,基于cgm中体现的定量关系和cgm在abc算法中的形式结合,可能获得对未观测的性状tln的预测。预测准确度非常高,在验证集中平均值为0.95。代表性实例显示于图5中。
即使tln是未观测的,abc成功地以高准确度预测了dh系的tln值。由于在tln和bm之间的非线性关系,前者只能通过cgm从后者推导出来。这表明abc实际上利用了cgm。预测bm的高准确度显示了该方法可以比标准线性全基因组回归方法(如gblup)具有巨大的潜在优势。
主效snp对tln的效应是线性和加性的。然而,在tln和bm之间的关系是强非线性的(图3)。其结果是主效snp对bm的效应也是非线性的。实质上,bm是所有可能命令的上位效应的函数。使用与bm和标记基因型直接相关的线性模型不能很好地捕获这些。因此,gblup的预测准确度非常低。
拟合非常复杂的上位模型的替代方法是将复杂性状bm分解成更简单的生理性状(pt)例如tln,这些生理性状更可能具有简单的、加性遗传结构。由于通常没有观测pt,模型参数只能通过pt与观测的性状(本实例中的ot、bm)之间的关系进行估计。然而,在这种情况下,给出模型参数的ot的可能性是未知的或甚至不存在的。在这种情况下,abc允许使用上面详述的算法从参数的近似后验分布中进行取样。abc要求的是一个模拟给定参数的数据的机制。cgm提供这种机制。
实例3:在新环境中的预测表现。
通过修改日常太阳辐射、温度和植物群体(植物m-2)来模拟两种不同的环境。环境1(e1)的植物群体为2个,日常温度为28℃,并且日常太阳辐射为36mjm-2。环境2(e2)的植物群体为10个,日常温度为18℃,并且日常太阳辐射为20mjm-2。因此,e1是一个非常干燥和热胁迫的环境,其中由于水分的限制只能使用非常低的植物密度。相比之下,e2具有低可能性的水分限制或热胁迫,对于植物生长更有利并且可以使用更高的植物密度。这里使用的cgm可以在e1和e2之间产生交叉的基因型与环境与管理(gxexm)的相互作用,秩相关性仅为0.62(图6)。
为了测试abc拒绝取样算法是否可以用于预测新环境中的表现,将来自100个dh系的随机子集的e2表型数据用于评估标记效应,然后在e1和e2中预测剩余的1,900个dh系的表现,如上所述。在e1和e2中的表现的平均预测准确度(超过10个重复)为0.87(图7)。因此,只要可以预测pt性状并且在cgm中的关系适用于新环境,就可以预测新环境中的表现。
gblup再次被用作基准。因为它不能解释在e1和e2中的不同环境条件,所以对于两者的预测值是相同的。对于在e2中的预测,gblup实现了0.36的平均准确度。然而,在新环境e1中,准确度为-0.27,可能是因为不能被解释的交叉相互作用(图8)。
两种基因型特异的性状:以前,只有tln被假设为是基因型特异的。现在,将影响平均叶片尺寸的pt‘最大叶片的面积’(am)也被模拟为基因型特异的。如上面针对tln所述对am进行模拟,并且线性内插到在450-650cm2之间的范围。将am建模为ami=μam+ziuam,,并且扩展了上文解释的abc拒绝取样算法以允许估计参数(tln和am)的两个向量。
实例4:增加作物生长模型的复杂性以预测最终生物量产量。
马乔(muchow)等人(1990)作物生长模型(cgm)将玉米生物量(bm)生长模型作为植物群体(植物密度)、温度和太阳辐射以及植物的几种生理性状(pt)的函数进行建模。pt是总叶片数(tln)、最大叶片面积(am)、太阳辐射利用效率(sr)和生理成熟度的热单位(mtu)。将sr设定为1.6gmj-1的恒定值,并且mtu设定为1150,为马乔(muchow)等人(1990)使用的值。如下所述,将tln和am模拟为基因型特异的。
通过修改日常太阳辐射、温度和植物群体(植物m-2)来模拟两种不同的环境。环境1(e1)的植物群体为2个,日常温度为28℃,并且日常太阳辐射为36mjm-2。环境2(e2)的植物群体为10个,日常温度为18℃,并且日常太阳辐射为20mjm-2。因此,e1被认为是一个非常干燥和热胁迫的环境,其中由于水分的限制只能使用非常低的植物密度。相比之下,e2被认为具有低可能性的水分限制或热胁迫,对植物生长更有利并且可以使用更高的植物密度。植物群体被认为是环境的农艺管理(m)组分。因此,这里使用的cgm可以在e1和e2之间产生交叉的基因型与环境与管理(gxexm)的相互作用,相关性为0.69(图8)。仅使用来自e2的表型数据进行估计。
数据模拟:tln和am由具有相似幅度的加性效应的10个因果snp的单独集控制。将这些因果snp随机置于3摩根长度的单个染色体上,并且将其假设是未知的。另外的100个观测的snp标记也随机地置于染色体上。根据霍尔丹(haldane)作图函数,通过模拟染色体的减数分裂产生来自双亲杂交的2,000个dh系。所有未观测和观测的snp在杂交中分离。对初始获得的tln和am值应用线性内插,使得tln具有平均值为9.5的[3,16]的范围,并且am具有平均值为550的[500,600]的范围。
在确定所有dh系的tln和am后,根据cgm计算环境e1和e2的bm值。在2,000个dh系中,100个被用作估计集,剩余的用于验证。为了模拟残余变异,我们在e2中估计集的系的bm值中添加了一个正态分布的噪音变量,该变量用于拟合该模型。选择噪音变量的方差,使得遗传性(h2)=0.85。通过复制整个模拟获得十个独立的数据集。
abc-cgm:假设产生数据的cgm是已知的,包括所有pt的值,除了tln和am之外,其被建模为
tlni=μtln+ziutln(1)
ami=μam+ziuam(2)
其中μtln和μam为截距,zi是dh系i的100个snp标记的基因型向量,并且utln和uam是标记效应的向量。唯一观测的特性是zi和最终生物量bm,此后表示为y。没有观测tln和am。
为了适应在cgm不能解释的估计数据集内的环境噪音的存在,使用以下似然函数作为数据的模型:
yi~n(cgmi,σ).(3)
因此,使用具有平均值等于从cgm获得的bm产量值和标准偏差σ的高斯分布,其取决于h2并且被假设为已知的。注意,由于(3)是一个已知的似然函数,所以abc的使用不会严格地要求,并且可以被更常规的拒绝取样器替代。
abc拒绝取样算法进行如下:
1.从它们的先验抽取μtln,μam、utln和uam的候选物。
2.根据方程式(1)和(2)计算tln′和am′的预测值
3.使用cgm来计算cgmi
4.从(3)模拟新的bm数据y′
5.计算向量y′和y之间的欧几里德(euclidean)距离d
6.如果d低于公差水平,则μtln,μam、utln和uam的候选物被接受为来自后验分布p(μtln,μam,utln和uam|y,h)的样本。
7.重复1至6直到抽取足够数量的样品。
如μtln和μam的先验一样,我们分别使用平均值等于9.5和550以及标准偏差为2.5和50的高斯分布。关于μtln和μam的相当多的先验知识被假设是可用的,这是一个合理的假设。utln的先验是平均值为零并且标准偏差为0.5的高斯分布。uam的先验是平均值为零并且标准偏差为5的高斯分布。选择公差使得接受率是5·10-7,抽取的样本数是200。作为基准,我们对数据进行标准的gblup模型拟合。
结果
增加复杂性:随着复杂性的增加,相比线性方法gblup,abc-cgm能够保留预测优势。然而,与先前调查的情景相比(其中只有单个变化的pt与bm具有强非线性关系),线性方法gblup也实现了大体上相当好的准确度。一个可能的解释是,个体pt与bm的非线性关系被几个pt的组合作用‘掩蔽’了。变化的pt的数量及其值范围会影响非线性关系被掩蔽的程度。图13显示了对于越来越窄的am范围,来自简单线性回归模型bm~tln+am的观测值与拟合值。当允许am在更宽的350和750cm2的范围之间变化时,即使当在pt之间存在潜在的非线性线性关系时,这种简单的线性模型也可以对数据提供合理的拟合。然而,当am的范围减小到[500,600](如在本研究中)时或者如果am实际上是固定的(如在实例2中),则揭示在tln和bm之间的确定的非线性关系。
除了掩蔽非线性关系之外,应该预期必须解释的pt越多,对每个关系的预测将越不准确。
环境噪音:abc-cgm可以适应环境噪音的存在,并且仍然比gblup实现更高的准确度。
实例5:三个潜在的生理性状。
在这个实例中,三个生理性状被模拟为基因型特异的并被纳入估计程序。在这种情景下,随着复杂性进一步增加,abc-cgm仍然胜过常规线性方法,并且仍然可以解释基因型与环境的相互作用。
作物生长模型:使用由马乔(muchow)等人(1990)开发的作物生长模型(cgm)。这个cgm将玉米生物量(bm)生长模型作为植物群体(植物密度)、温度和太阳辐射以及植物的几种生理性状(pt)的函数进行建模。pt是总叶片数(tln)、最大叶片面积(am)、太阳辐射利用效率(sre)和生理成熟度的热单位(mtu)。将mtu设定为1150,为马乔(muchow)等人(1990)使用的值。如下所述,将tln、am和sre模拟为基因型特异的。
通过修改日常太阳辐射、温度和植物群体(植物m-2)来模拟两种不同的环境。环境1(e1)的植物群体为10个,日常温度为18℃,并且日常太阳辐射为20mjm-2。环境2(e2)的植物群体为2个,日常温度为28℃,并且日常太阳辐射为36mjm-2。因此,e2是一个非常干燥和热胁迫的环境,其中由于水分的限制只能使用非常低的植物密度。相比之下,e1被认为具有低可能性的水分限制或热胁迫,对植物生长更有利并且可以使用更高的植物密度。这里使用的cgm可以在e1和e2之间产生交叉的基因型与环境与管理(gxexm)的相互作用(实例参见图9)。仅使用来自e1的表型数据进行估计。
数据模拟:tln、am和sre由具有相似幅度的加性效应的10个因果snp的单独集控制。将这些因果snp随机置于1.5摩根长度的单个染色体上,并且将其假设是未知的。另外的100个观测的snp标记也随机地置于染色体上。
根据霍尔丹(haldane)作图函数,通过模拟染色体的减数分裂产生来自双亲杂交的1,000个dh系。所有未观测和观测的snp在杂交中分离。对初始获得的tln和am值应用线性内插,使得tln具有平均值为9.5的[3,16]的范围,am具有平均值为550的[500,600]的范围,并且sre具有平均值为1.60的[1.55,1.65]的范围。
在确定所有dh系的tln、am和sre后,根据cgm计算环境e1和e2的bm值。在1,000个dh系中,200个被用作估计集,剩余的用于验证。为了模拟残余变异,在e2中估计集的系的bm值中添加了一个正态分布的噪音变量,该变量用于拟合该模型。选择噪音变量的变异使得h2=0.85。通过复制整个模拟获得了二十个独立的数据集。在e1中的bm和tln之间、在am和sre之间的关系分别显示于图14中。
abc-cgm:我们假设产生数据的cgm是已知的,然而,pt(除了mtu之外)不是已知的并且被建模为
tlni=μtln+ziutln(1)
ami=μan+ziuam(2)
srei=μsre+ziusre(3)
其中μtln,μam和μsre为截距,zi是dh系i和utln的100个snp标记的基因型向量,并且uam和usre是标记效应的向量。仅观测了zi和最终生物量bm,此后表示为y。没有观测tln、am和sre。
为了适应在cgm不能解释的估计数据集内的环境噪音的存在,使用以下似然函数作为数据的模型
yi~n(cgmi,σ).(4)
因此,使用具有平均值等于从cgm获得的bm产量值和标准偏差σ的高斯分布,其取决于h2并且被假设为已知的。
abc拒绝取样算法进行如下:
1.从它们的先验抽取μtln,μam、μsre,utln·uam和usre的候选物。
2.根据方程式(1-3)计算tln′、am′和sre’的预测值
3.使用cgm来计算cgmi
4.从方程式(4)模拟新的bm数据yi
5.计算向量yi和y之间的欧几里德(euclidean)距离d
6.如果d低于公差水平τ,则μtln,μam、μsre,utln,uam和usre的候选物被接受为来自后验分布p(参数|y,h)的样本。
7.重复1至6直到抽取足够数量的样品。
如μtln、μam和μsre的先验一样,我们分别使用平均值等于9.5、550和1.6以及标准偏差为0.75、25和0.1的高斯分布。这假设关于μtln,、μam和μsre的大量的先验知识是可用的,这是典型的情况。
第j个标记对性状x的效应的先验是
结果
常规的线性方法gblup。对20个相同的数据集进行gblup模型拟合以获得基准结果。对于e1和e2,gblup分别实现了0.51的0.05的bm的平均预测准确度。pt性状的预测准确度对于tln、am和sre分别为-0.49、0.19和0.42。
abc-cgm。在验证集中生理性状tln、am和sre的平均预测准确度分别为0.85、0.28和0.44(图15)。最终生物量产量bm的平均预测准确度在e1中为0.80,且在e2中为0.70(图16)。
abc-cgm的优越性。主效snp对生理性状tln、am和sre的效应是线性和加性的。然而,在这些性状和bm之间的关系是非线性的,尤其是在tln和bm之间(图14)。其结果是这些主效snp对bm的效应也是非线性的。bm是所有可能命令的上位效应的函数。使用与bm和标记基因型直接相关的线性模型不能很好地捕获这些。因此,gblup的预测准确度更低,即使在环境e1(估计数据来自的相同的环境)中。
abc-cgm对生物量产量和潜在的生理性状之间的函数关系进行建模并且纳入了环境特异的天气和管理信息(太阳辐射、温度和植物密度)。这使得abc-cgm甚至能够预测交叉的gxexm相互作用,并且甚至在根本不同的环境中提供高的预测准确度。常规的线性方法像gblup没有明确地对性状之间的非线性关系或gxexm的相互作用进行建模,因此未觉察具体环境的特征以及它们如何决定作物生长。因此,它们无法预测在强的gxexm下的环境中的表现。
因果推理。因为abc-cgm对功能关系进行建模,它还可以深入gxexm的生理和遗传决定因子内。在复杂系统中的参数(例如在cgm中的生理性状)的重要性越低,关于该参数的后验不确定性越高,因为被数据报告和约束的参数的后验越少。如果参数对确定环境中的bm根本没有起作用,那么它的后验不确定性应该等于先验不确定性。因此,后验不确定性可以被用作生理性状对于驱动在目标环境中的表现的相对重要性的指示器。因此,在这个实例中,在e1中确定bm表现的最重要的性状是tln,接着是am,然后是sre(图17)。
实例6:先验分布和取样参数。
在这个实例中,研究了先验分布和abc取样参数对预测准确度的影响。数据模拟和作物生长模型与实例3相同。
标记效应的先验定义:第j个标记对性状x的效应的先验是
参数σx被计算为
其中m是标记的数量,var(x)是性状x的表型变异,其被假设是已知的,并且s是用于模拟先验错误设定的比例因子。对于s的值,我们考虑了1.0、2.0、0.1和10.0。
对于先验模型列入概率(inclusionprobability),考虑了(1-π)值等于1.0、0.9、0.7、0.5、0.3和0.1。对于(1-π)<1.0,,先验(5)对应于贝叶斯cπ先验,而对于(1-π)=1.0,它对应于贝叶斯c。
取样参数:减小公差τ降低后验的近似程度,这将导致更准确的预测。然而,这与计算时间的增加有关联,因为减少τ需要降低接受率ρ。在实践中,通过设置ρ的目标值(这在计算上是可行的)来确定τ。在这项研究中,我们使用了ρ等于10-4,10-5,2·10-6,10-6和10-7的值。
增加从后验分布抽取的样品的数量t也预期增加预测准确度,因为数量像后验平均数或分位数可以更准确地估计。然而,增加样本的数量t也增加计算时间。在这里,我们使用了t等于25、50、100、150、200和500的值。
使用了步进式(step-wise)方法,其中一次仅研究了一个因素,所有其他因素固定在合理的值或在先前的步骤中发现为最佳的值。使用以下顺序:
1.s是变化的,而保持(1-π)=1.0且ρ=2·10-6并且t=100
2.ρ是变化的,而保持s在1.0不变、(1-π)=1.0并且t=100
3.t是变化的,而保持s在1.0不变、(1-π)=1.0并且ρ=2·10-6
4.(1-π)是变化的,而保持s在1.0不变、t=100并且ρ=2·10-6
结果
改变s:当方差比例因子等于1.0时,在两种环境中的bm以及tln、am和sre的预测准确度最高(表1)。对于低于或高于1.0的比例因子,准确度降低。
表1:不同的先验方差比例因子s的平均预测准确度
改变π:所有性状的预测准确度随着π增加而强烈下降。当标记效应不等于零时,可能需要更高的样本大小t来准确估计标记效应。
改变t:在两种环境中bm的预测准确度在t=100时达到最大值(表2)。tln、am和sre的预测准确度随着t增加也趋于增加。然而,pt的t水平之间的巨大波动在标准误差之内(细节未显示)。
表2:不同样本大小t的平均预测准确度
改变ρ:在两种环境中bm的预测准确度随着接受率ρ降低而增加。然而,在百万分之二的中等接受率下,已经观测到良好的准确度,对其的计算在合理的努力下是可行的。生理性状的预测准确度也增加了。在1/1,000,000的接受率下,它们似乎达到峰值,之后上下波动。然而,这些波动是在标准误之内(未显示)。
表3:不同接受率rhoρ(的平均预测准确度(在每百万的接受的样本中)。列tau显示公差τ的平均值(表示为均方根差)。
实例7:cgm-wgp在多环境试验中的应用
cgm-wgp方法被应用于经验玉米干旱多环境试验(met)数据集,以评估减少实践所涉及的步骤。对于来自杂交的双单倍体的样品,在两种干旱环境中对于杂交谷物产量实现了正的预测准确度。这通过将五个性状的变异包括在cgm中使得cgm-wgp方法成为可能来实现。在目标干旱环境中,这五个性状被先验地认为对在玉米杂交种之间的产量变异很重要。
选择用于评估cgm-wgp方法的经验实现的met数据集是基于在单个地点的两种干旱环境(处理)中测试的单个双亲杂交获得的谷物产量结果。选择双亲杂交的亲本以对比在干旱下它们的谷物产量育种值;一个亲本以前的特征是具有高的育种值,另一个具有低的育种值。将亲本杂交以产生f1代,并且将f1自花授粉以产生f2代。双亲杂交由源自于f2代个体的随机样品的106个双单倍体(dh)系代表。对106个dh系进行基因分型,其中总共86个单核苷酸多态性(snp)标记分布在10条染色体上。先前将snp鉴定为在两个亲本之间是多态的。将106个dh系与测交近交系杂交以产生测交杂交种子。测交系选自互补杂种优势群,并且被认为在干旱下具有高的谷物产量育种值。在106个dh系的测交杂交种子上产生所有的谷物产量数据。
在两种干旱环境下,在实验地块中评估了106个dh系的谷物产量。通过在相邻田间进行的两个实验中产生两个干旱处理来产生两种干旱环境。将灌溉的量和时间用于产生不同的干旱处理。通过在种植时在实验地块中安装的滴灌带系统来管理灌溉。每个实验地块有两行,长4.5m,行之间的间距为0.75m。在实验的每个地块中,在每行旁边的种植土壤中插入滴灌带。干旱处理是通过调节灌溉水的量来实施的,该灌溉水是通过实验中安装的滴灌带系统来供应的。在两个干旱实验之间,对水的供应的管理不同,以产生两种不同水平的水供应,因此产生两个不同的干旱处理。管理灌溉时间表,使最大缺水的时间与106个dh系的开花期相一致。在两个干旱处理中实现的缺水时间模式的表征显示于图1中。
两个环境的实验设计都是基于两个重复的。以行列配置以及许多其他dh系和一组商业杂交检查来评估106个dh系。出于本文的目的,这些另外的dh系和商业杂交检查将不被进一步考虑,除了认识到它们是获得106个dh系的数据集的一部分。使用两地块组合收获系统获得谷物产量数据,该系统测量从该地块获得的谷物重量和谷物含水量。每个地块的谷物收获重量被调整为在15%的含水量下每单位面积的谷物产量。使用混合模型分析谷物产量数据,该混合模型包括地块的行和列位置的条目和估计的地块残差的空间相关性。106个dh系被认为代表可能已经从双亲杂交获得的可能的dh的随机样本。因此,对于两个干旱处理,106个dh系中的性状变异的基因型术语被视为随机的,并且获得了106个dh系中的每一个的谷物产量的最佳线性无偏预测(blup)。
作物生长模型
在本研究中使用的cgm是基于马乔(muchow)等人(1990)开发的机械模型。cgm使用资源利用、资源利用效率和资源分配的概念针对谷物来模拟产量。光截获是基于叶片出现率、最大叶片尺寸(am)、总节数(tln)、种植密度和灭绝系数来建模的。以总质量计的日增加量的模拟是由给定日子的光截获和辐射利用效率产生的。开始于旗叶扩展结束后三天,结束于生理成熟度,从以收获指数计的日增加量模拟产量。
由于本研究的动机是为了证明cgm-gwp方法用于在干旱胁迫条件下评估玉米群体,该模型被修改以模拟土壤水分平衡、蒸腾和对缺水的生长反应。如里奇(ritchie)(1999)所描述的,土壤水分平衡采用多层方法进行建模。如马乔(muchow)和辛克莱(sinclair)(1991)所描述的,模拟了组分土壤水分平衡、渗透、径流和蒸发。蒸发使用两阶段模型进行建模。蒸腾基于质量生长和等于9pa的蒸腾系数进行建模。有限的蒸腾性状如墨西拿(messina)等人(出版中)描述的来实施,区别在于本研究中,对在蒸气压差(vpd)断点(vpdb)以上的vpd的蒸腾反应被建模为连续线性函数而不是恒定的最大值。使用具有描述根占有率和水力传导率的函数指数的一阶动力学模拟根吸水量;此参数设置为0.08。土壤层潜在吸水量的总和决定了土壤供水量,而蒸腾计算决定了需水量术语。这两个组分的比例定义了用于影响质量生长和叶片扩展的胁迫指数。
因为该模型的目的是模拟在开花时间经历缺水的玉米产量(图1),并且收获指数方法不足以模拟这些类型的胁迫环境中的玉米产量,该模型被修改为纳入对描述花丝出现和果穗生长(对缺水敏感的过程)的动力学重要的元素。可获得的收获指数被模拟为潜在收获指数(其对应于在没有缺水的情况下获得的收获指数)、由果穗中的最大行数和环数引起的潜在花丝数、吐丝后三天花丝的外露数、和当来源超过库容量时核重量的潜在增加的函数。在这些条件下,核重量可以增加约20%。使用负指数函数对外露的花丝数量进行建模。参数性状最小果穗生物量(meb)对应于果穗质量生长的阈值,低于该阈值花丝不会从壳中显露。花丝的潜在数量定义了产量潜力。该函数的指数定义了花丝出现/单位果穗生长的速率,其使用热时间的指数函数和与供水量与需水量比成正比的胁迫因子进行建模。为了说明在开花时间方面植物与植物的变异,应用了三个果穗的生长和发育。将这三个果穗出现的花丝的加权平均值用于确定最终可获得的收获指数。果穗生长的发生被设定在第十五营养阶段。使用以收获指数计的每日增量来模拟产量,该收获指数从潜在收获指数(马乔(muchow)等人,1990)更新为基于缺水对果穗生长和花丝出现的影响而在开花时期确定的收获指数。
近似贝叶斯计算(abc)
将五个性状鉴定为abc框架内研究的cgm的关键组分;总叶片数(tln)、最大叶片面积(am)、蒸气压差值(断点)(高于该蒸气压差值蒸腾降低到低于其潜在值)(vpdb)、花丝外露的最低果穗生物量(meb)、和从冠层发育完成(如通过旗叶扩展的完成测量的)的累积热单位、和花粉脱落的时间(tus)。性状tln和am一起影响冠层尺寸,其影响水分有限环境中的土壤水分平衡。性状vpdb影响冠层的蒸腾速率,并且也可以影响土壤水分平衡。当水分限制与开花期一致时,性状meb影响生殖弹性并最终影响核结实。tus允许开花时间的遗传变异来源,而不是与tln的变化相关。
将这五个性状作为cgm-wgp方法预测的潜在变量来处理。对于每个dh条目的每个性状的潜在值被模拟为性状特异的标记效应的线性函数:
ytlni=μtln+ziutln
yami=μam+zi-uam
yvpdbi=μvpds+ziuvpdb
ymebi=μmes+ziumeb
ytusi=μtus+ziutus
其中zi是dh条目i的观测的二等位基因的snp标记的向量,μtln、μam、μvpdb、μmeb和μtus是五个性状的截距,并且utln、uam、uvpdb、umeb和utus是标记效应的向量。符号θ用于表示联合参数向量[μtln,…,μtus,utln,…,utus]。
定义cgm性状的先验信息
独立的正态分布先验被用于θ的所有组分的五个性状。截距μ性状的先验是
表4:在cgm-wgp方法中使用的被鉴定为在两种干旱环境中影响谷物产量并作为作物生长模式内遗传变异的来源来处理的五个性状的先验参数值
使用不同的信息来源获得五个性状的先验值。对于tln、am和meb,在爱荷华州进行的实验中评估了106个dh系中38个dh系的子集的tln、am和meb。使用与用于获得谷物产量数据相同的测交种子来源获得这三个性状的数据。对于谷物产量,使用混合模型分析性状测量值,并且获得dh系的blup。对于性状tln、am和meb,然后将m性状计算为在爱荷华实验中包含的38个dh的子集上进行的测量的平均值。而且,对于这三个性状,将
对于性状vpdb和tus,不对dh条目进行直接测量。用于定义先验参数的所有信息都是基于玉米的已公开信息。对于vpdb性状,使用了由gholipoor等人(2013)报道的结果。对于tus,先验参数是基于指示3天tus间隔的公开信息(马乔(muchow)等人,1990)和指示叶片扩展和抗旱杂交种的脱落开始的同步终止的田间观测的组合来确定的。
如上所述实施abc。模拟模型运算符模型
cgm-wgp估计、预测和测试程序
拟合了cgm-wgp模型,并且使用来自fs或sfs环境的数据获得参数估计。使用50个dh条目的随机集作为训练集,在此将其称为估计集。然后使用剩余的56个dh条目来测试模型表现,并将其称为测试集。将数据取样到拟合cgm-wgp模型的环境将被称为估计环境。其他环境将被称为新环境。出于本文的目的,其他环境是新的,因为没有使用来自该其他环境的数据来选择cgm-wgp模型或估计参数。然后在估计环境和新环境两者中对选择的cgm-wgp模型进行了测试;例如基于在fs环境中的50个dh条目的样本选择该模型,在这种情况下,fs环境是估计环境,并且sfs环境是新环境,然后在fs估计环境和sfs新环境中对剩余的56个dh条目进行了测试。一旦选择了cgm-wgp模型,就使用参数估计来预测在fs和sfs两种环境中的估计集和测试集两者的dh条目的产量。对与估计环境相同的环境的预测将被称为观测环境预测(例如,使用用fs数据拟合的模型对fs进行预测)。对于没有数据用于拟合模型的环境的预测将被称为新环境预测(例如,使用用fs数据拟合的模型对sfs进行预测)。对于fs和sfs环境,将这个过程重复了20次。作为在特定环境中dh条目的预测谷物产量的点估计,我们使用了所讨论的dh条目的后验预测分布的平均值。使用该环境的天气、土壤、灌溉和管理数据,通过在接受的θ样本上估计cgmf(.)ik获得后验预测分布。
将cgm-wgp的预测准确度计算为在进行预测的环境中dh条目的预测和观测表现之间的皮尔森积差相关(pearsonproductmomentcorelation)。作为表现基准,也将基因组最佳线性无偏预测(gblup;meuwissen等人,2001)应用于所有数据集。
结果
应用于两个实验的灌溉时间表在实验过程中产生了类似的缺水时间模式(图18)。在两个实验中,最大缺水期与dh条目的开花时间一致。谷物产量数据的方差分析表明,dh条目之间存在显著的基因型变异(图19)。如通过从种植到花粉脱落(gdushd)的热单位测量的,也存在显著的开花时间的基因型变异。然而,在两种环境中在gdushd和谷物产量之间不存在线性或非线性关系(p>0.05)。因此,在两种干旱环境中,开花时间的变化不被认为对所选择的杂交的后代的谷物产量具有直接的影响。因此,尽管谷物产量变化的变异组分仍然可能与开花时间效应(以其他性状为条件)相关,但是dh条目的谷物产量变化的主要组分被认为与性状变异而不是开花时间相关。在两种环境之间的谷物产量的gei是显著的。gei变异组分比基因型变异组分小(图19)。在两个环境之间的谷物产量的遗传相关性虽然小于1.0,但估计为较高,为0.86。检测的谷物产量gei的一个组分与两种环境之间的基因型变异幅度的异质性有关。在两种环境之间比较谷物产量blup的散点图表明,在两种环境之间的dh条目的相对产量存在一些差异,但总体上普遍一致(图19)。鉴于这些谷物产量结果,数据集被认为适合于cgm-wgp方法的估计。值得注意的是,与在两种环境之间发生更大的等级变化水平的其他情况相比,在这两种环境之间的dh条目的相对较低的等级变化水平预期将提高gblup在这些环境之间成功预测的机会。对比在图19中显示的单个实例更宽范围的gei情景的探索在下面进一步讨论。在这里,我们专注于成功实施cgm-wgp方法对作为育种计划的一部分产生的经验数据集的要求。
用于运行cgm-wgp的环境输入的初始集导致在谷物产量预测和在sfs环境中观测的结果之间的一致性较差。预测的产量值一致地低于观测的产量。这导致了对这两种环境的环境输入的重新评价。最初的假设是相邻田间的土壤深度是相同的,并且不同的产量水平将由用于这两种环境的不同灌溉时间表来解释。对两个相邻田间的土壤特征的进一步研究揭示在相邻田间之间的土壤深度存在大约0.2m的显著差异(p<0.001)。一旦对cgm的输入进行了这个调整,sfs环境的预测产量与观测产量一致。这作为与在实践中应用cgm-wgp相关联的一些附加要求的实例而提供。虽然这可能被视为另外的成本,但也表明cgm-wgp对环境输入的响应,该环境输入是调整gei的影响的要求。
所有考虑的cgm-wgp情景的平均预测准确度为正(表5)。该结果表明,cgm和五个性状tln、am、meb、vpdb和tus提供了一个相关框架以通过应用abc算法获得的θ的参数的近似后验分布的形式定义捕获产量的遗传变异的模型。
表5.cgm-wgp和gblup方法获得的、在两种干旱环境(fs和sfs)中作为测交杂交种评估的dh条目的谷物产量的预测准确度,取20个重复的平均值。对于每个复制,106个dh条目属于估计集(50个dh条目)或测试集(56个dh条目)。对于每个实施,两种环境被定义为估计环境或预测环境。
对于估计环境和预测环境相同的情景实现了最高的预测准确度。dh条目的估计集和测试集都是这种情况。预期这个结果是因为环境中的预测不必适应不同环境之间发生的任何gei的效应。
与将cgm-wgp应用于dh条目的测试集相比,当将cgm-wgp应用于dh条目的估计集时,预测准确度始终更高(表5)。因此,当将选择的θ参数应用于从相同参考群体中取样的新dh条目时,出于预测目的而丧失模型适当性。不论估计环境和预测环境是相同(即fs至fs以及sfs至sfs)或者不同(即,fs至sfs以及sfs至fs),发生预测技能的这种损失。
对于所有考虑的情景,通过cgm-wgp实现的谷物产量的平均预测准确度与gblup实现的相似(表5)。虽然平均预测准确度相似,但是对于单个重复在cgm-wgp和gblup之间的预测准确度存在差异(图20)。这些差异表明,当将cgm-wgp和gblup方法应用于相同的估计数据集时,通过这些方法选择不同的产量遗传模型。通过两种预测方法选择不同遗传模型的结果是,不同dh条目的产量预测随着预测方法而改变,并且基于预测也可以改变个体dh条目的排名(图21)。为了进一步研究个体dh条目的产量预测的差异,可以对每个重复进行产量预测比较(图22)。因此,取决于育种者所应用的选择强度,即使两种预测方法的平均预测准确度相似,也可以选择不同的dh条目集。在cgm-wgp和gblup之间的另一个重要区别是在环境之间调整了谷物产量预测的规模。cgm-wgp方法当在两种环境之间移动时动态地调整了产量预测的规模,因此cgm-wgp产量预测的平均值接近于观测的产量值的平均值(图21a)。这些调整通过cgm中包含的性状和环境变量之间的关系来实现。gblup方法在选择的产量模型参数和在两种环境之间变化的环境变量之间没有任何关系。因此,当估计环境和测试环境不同并且gblup产量预测的平均值可能偏离观测的产量值的平均值时,没有调整通过gblup方法获得的产量预测的规模(图21b)。
实例8:基于生物模型的wgp应用于人类遗传学
由戈德斯坦(goldstein)和rypins(1992)描述的人类肾脏功能数学模型被用作生物模型来描述人类的尿钠排泄量(una,meq/l),该尿钠排泄量是作为生理性状平均动脉血压(map,mmhg)、醛固酮浓度(ald,ng/l)和血清钠(sna,meq/l)的函数。生理性状值是在以下范围内:map[50,120]、ald[40,125]和sna[139.75,140.25]。图23显示了观测的在una与这些生理性状之间的关系。
肾脏模型从此被表示为
其中,
群体、遗传和表型数据
合成的人类群体由1,550个个体组成。对于基因组,仅考虑遗传长度为0.596摩根(m)的一条染色体。这等于人类21号染色体的遗传长度(迪布(dib)等人,1996)。该染色体上填充有130个双等位基因基因座。基因型以标记之间(测量为r2)的连锁不平衡(ld)随着半衰期等于0.03m而呈指数衰减的方式随机地产生(图24)。这反映了在人类基因组中观测的快速ld衰减(戈达德(goddard)和海耶斯(hayes),2009)。次等位基因频率范围为从0.35至0.50,平均为0.42。杂合率接近50%。
假设30个基因座的随机样本对于上述三个生理性状(每个性状的基因座数为10个)是主效的。在所有随后的分析中,这些基因座被掩蔽,因此假设是未观测的。将剩余的100个基因座作为观测的单核苷酸多态性(snp)标记进行处理。
从标准的高斯分布抽取主效基因座的另外的替代效应。根据在1,550个个体中的每一个的基因座上观测的基因型通过总结这些效应来计算生理性状的原始遗传分数。随后,这些原始分数线性地重缩放到前面提到的范围。最后,通过使用生理性状值产生所有1,550个个体的观测una值作为进入f(·)i的输入。
统计模型和近似贝叶斯计算(abc)
生理性状map和ald被假设是未知的,并作为隐含变量来处理。它们被建模为性状特异的标记效应的线性函数
其中zi是个体i的观测的snp标记的基因型向量,μmap等表示截距,并且umap等表示标记效应。为简洁起见,θ将用于表示联合参数向量[μmap,μald,umap,uald]。
对于θ的所有组分使用独立的高斯分布先验。截距μ性状的先验为
将血清钠
如在先前的实例中所描述的,使用abc算法。模拟模型运算符模型(una|θ)包括肾模型f(·)i作为确定组分,并且高斯噪音变量与随机分量一样分布为
估计、预测和测试程序
将n=100个个体的随机子集用作估计集。将剩余的1,450个个体用于测试模型表现。作为预测una的点估计,我们使用了所讨论的个体的后验预测分布的平均值。通过在接受的θ样本上估计f(·)i来获得后验预测分布。预测准确度被计算为在预测和真实表现之间的皮尔森(pearson)相关性。作为表现基准,我们使用了基因组最佳线性无偏预测(gblup,meuwissen等人(2001))。对8个独立产生的数据集重复该程序。
结果
基于生物模型的肾脏-wgp方法在估计集中具有比基准方法gblup始终更高的平均预测准确度(表1)。这表明它确实导致更好的数据拟合。肾脏-wgp在测试集中也具有更高的平均预测准确度。尽管差异更小,但是它们是一致的,在8例中的6例中肾脏-wgp比gblup具有更高的准确度(对于平均预测准确度参见表1,并且对于在实例重复中的预测值与观测值参见图25)。
表1:对于基于生物模型的全基因组预测方法(肾脏-wgp)和基准方法(gblup),取8次重复的平均值,估计集和测试集中人类尿钠排泄量(una,meq/1)个体的预测准确度。
- 还没有人留言评论。精彩留言会获得点赞!