释放一氧化氮的药物组合物的制作方法与工艺
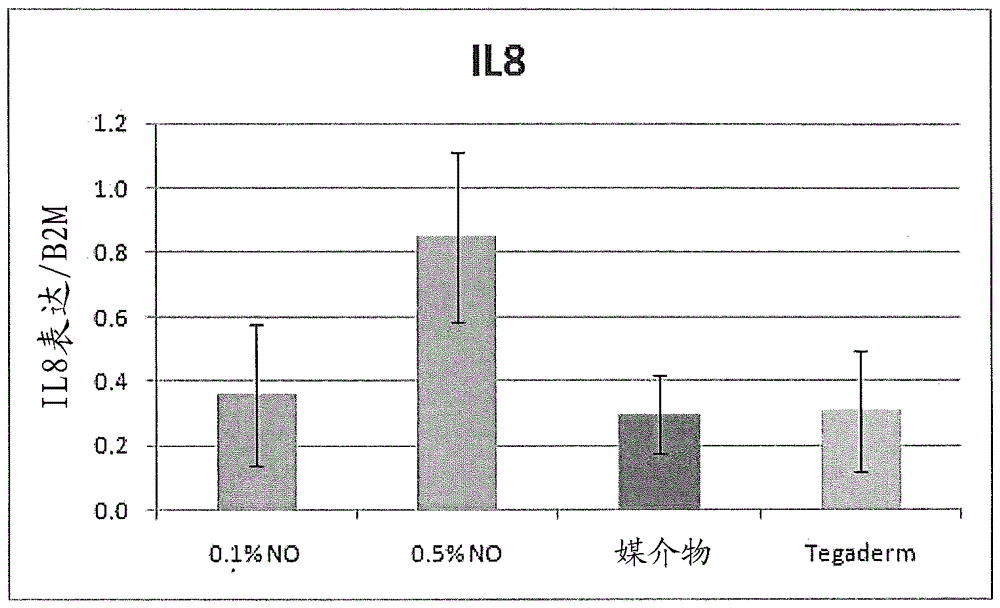
释放一氧化氮的药物组合物相关申请数据本申请要求2012年3月14日提交的美国临时专利申请序列号61/610,563的权益,所述申请的公开内容通过引用以其整体并入本文。发明领域本发明总体涉及释放一氧化氮的药物组合物和使用其的方法。发明背景许多皮肤疾病或病症由具有来自各种炎症和驻留细胞的介质的相关释放的炎症所导致。中性粒细胞、肥大细胞和淋巴细胞协调炎症应答,所述炎症应答导致炎性介质的显著释放和许多自由基的生成。其中炎症是重要组分的皮肤疾病包括,但不限于,痤疮和酒渣鼻,异位性皮炎,接触性皮炎,药疹,牛皮癣,脂溢性皮炎,结缔组织疾病(诸如狼疮和硬皮病),其它自身免疫疾病,诸如发疱性疾病大疱性类天疱疮或天疱疮,色素性疾病(诸如炎症后色素沉着,黑斑病和白癜风),荨麻疹(urticaria)或荨麻疹(hives),与皮肤感染相关的炎症,诸如体癣或手指或脚趾甲的真菌感染,等等。炎症对于大多数这些疾病是一个重要步骤。用于治疗炎性皮肤状况的新组合物和制备和/或使用此类组合物的方法可以是期望的。类似地,许多伤口(慢性或急性的)可具有炎症方面。在炎症状况和伤口中,应当提供治疗剂的递送,而不显著刺激或恶化炎症状况。此外,如果活性成分水分活化的,则含水媒介物可能是不适当的。单独的矿物油软膏可以在应用之前保护水分活化剂免受水分影响,但它们也可以降低活性剂在应用时的有效性。因此,用于递送水分活化的活性剂的新组合物,在一些情况下,适合用于治疗具有对于疾病的炎性方面的适应症。本发明通过提供释放一氧化氮的药物组合物和使用其的方法而解决了以前本领域中的不足之处。发明概述本发明的第一个方面包含用于局部递送水分活化的活性药物成分的药物组合物,所述组合物包含:疏水性基料和两亲性化合物。本发明的第二个方面包含用于局部递送水分活化的活性药物成分的药物组合物,所述组合物包含:以约0.1%至约35%的浓度存在于所述组合物中的水分活化的活性药物成分;以约30%至约60%的浓度存在于所述组合物中的疏水性聚合物;以约1%至约30%的浓度存在于所述组合物中的矿物油;以约1%至约20%的浓度存在于所述组合物中的两亲性化合物;以约1%至约25%的浓度存在于所述组合物中的共溶剂;和以约1%至约25%的浓度存在于所述组合物中的湿润剂。本发明的进一步方面包含治疗个体的皮肤的方法,所述方法包括以有效治疗个体的皮肤的量局部施用本发明的药物组合物。现在将针对本文所述的其它实施方案,更详细地描述本发明的前述方面和其它方面。应当理解的是,本发明可以不同形式体现且不应解释为限于本文所述的实施方案。相反,提供这些实施方案使得本公开内容将透彻且完整,并且将本发明的范围充分传达给本领域技术人员。附图简述下面附图被提供以说明本发明概念的各个方面,并不意在限制本发明的范围,除非本文中指明。图1显示用于制备根据本发明的一些实施方案的药物组合物的操作流程图。图2显示根据本发明概念的一些实施方案的具有2%Nitricil™的软膏和具有2%Nitricil™的局部凝胶对铜绿假单胞菌(Pseudomonasaeruginosa)的时间杀灭的图。图3显示根据本发明概念的一些实施方案的有和没有2%Nitricil™的软膏对铜绿假单胞菌的时间杀灭的图。图4显示根据本发明概念的一些实施方案的有和没有2%Nitricil™的软膏对铜绿假单胞菌的时间杀灭的图。图5显示在生理条件下根据本发明的一些实施方案的两种类型的Nitricil™(NVN1和NVN4)软膏的释放概况的图。图6显示用于根据本发明的一些实施方案的软膏的5.5-kg规模制备的工艺流程图。图7显示释放一氧化氮的Nitricil™NVN1软膏对伤口再上皮形成(re-epithelialization)的影响的图。图8显示用0.1%和0.5%Nitricil™NVN1、媒介物和Tegaderm处理的伤口中通过qPCT测量的伤口组织中IL-8的表达水平的图。图9显示通过上皮下混合的白细胞浸润的存在和量评估的白细胞浸润的图。发明详述本发明现在将在下文中更全面地描述。然而,本发明可以以不同的形式体现并且不应被解释为限于本文所述的实施方案。相反,提供这些实施方案使得本公开内容将透彻且完整,并且将本发明的范围充分传达给本领域技术人员。本文发明的说明书中使用的术语仅以描述具体实施方案为目的,而并不意图限制本发明。除非上下文另有明确指示,否则如本发明的说明书和随附权利要求中所使用的单数形式“一(a)”、“一(an)”和“所述(the)”还旨在包括复数形式。除非另有定义,否则本文中所使用的所有术语(包括技术术语和科学术语)具有与本发明所属领域的普通技术人员通常理解的相同的含义。将进一步理解,术语,诸如常用字典中定义的那些,应当被理解为具有与它们在本申请和相关领域的背景下的含义一致的含义,并且不应被解释为理想化的或过于正式的意义,除非本文中明确如此定义。本发明的说明书中使用的术语仅用于描述具体实施方案的目的,且不旨在限制本发明。本文中提到的所有出版物、专利申请、专利和其它参考文献都通过引用以其整体并入。在冲突术语的情况下,以本说明书为准。此外,如本文中所使用,“和/或”是指且涵盖,一个或多个相关所列项的任何和所有可能组合,以及当以替代方式(“或”)解释时缺乏组合。除非上下文另有说明,否则特别意指本文描述的本发明的各个特征可以以任何组合使用。此外,本发明还包括,在本发明的一些实施方案中,可以排除或省略本文所述的任何特征或特征的组合。为了说明,如果说明书陈述复合物包含组分A、B和C,则特别意指可以省略和排除保护任何A、B或C或其组合。如本文中所使用,过渡短语“基本上由......组成”(和语法变体)应当被解释为涵盖所述材料或步骤,“和对要求保护的发明的一种或多种基本和新颖特性没有实质性影响的那些”。参见,关于Herz,537F.2d549,551-52,190U.S.P.Q.461,463(CCPA1976)(原文强调);还参见MPEP§2111.03。因此,如本文中所使用的术语“基本上由......组成”不应被理解为等同于“包含”。当是指可测量的值诸如化合物的量或浓度、剂量、时间、温度等时,如本文中所使用的术语“约”意指涵盖指定量的±20%、±10%、±5%、±1%、±0.5%、或者甚至±0.1%的变化。本文中对于可测量的值提供的范围可以包括其中任何其它范围和/或个别值。本发明提供了可以局部施用的药物组合物。本发明的药物组合物可以包含疏水性基料和两亲性化合物,基本上由其组成,或由其组成。在本发明的具体实施方案中,药物组合物进一步包含水分活化的活性药物成分。本发明的药物组合物可以包含软膏、油膏、乳膏,等等(and/orthelike)。如本文中所使用的“疏水性基料”是指天然和/或合成的脂肪、蜡、油,等等。本发明的药物组合物中可以使用任何合适的疏水性基料。在本发明的某些实施方案中,药物组合物包含两种或更多种疏水性基料,诸如,但不限于,2、3、4、5、或更多种疏水性基料。示例性疏水性基料包括,但不限于,支链和无支链的烃、支链和无支链的烃蜡、凡士林、烃凝胶、液体石蜡、白矿脂、矿脂、微晶蜡、小烛树蜡(andelillawax)、巴西棕榈蜡、羊毛脂(羊毛蜡)、羊毛蜡醇、细茎针草蜡、软木蜡、guaruma蜡、米糠蜡、甘蔗蜡、浆果蜡、小冠巴西棕榈蜡(ouricurywax)、大豆蜡、霍霍巴油、尾羽油脂、纯地蜡、石蜡、微蜡、植物油、动物油、巴西棕榈蜡、蜂蜡、可可脂、硬脂、矿物油、植物油、鳄梨油、琉璃苣油、芥花油、蓖麻油、春黄菊油、椰子油、玉米油、棉籽油、菜籽油、月见草油、红花油、向日葵油、大豆油、甜杏仁油、棕榈油、棕榈仁油、牛蒡种子油、芝麻油、琉璃苣籽油(borgoofficialisseedoil)、油菜油茶油(brassicacampestrisoleiferaoil)、油鲱油(brevoortiaoil)、牛脚油(bubulumoil)、岩蔷薇油(cistusladaniferusoil)、油棕油(elaeisguineensisoil)、杏仁油、松油、橄榄油、花生油、小麦胚芽油、葡萄籽油、蓟油、猪油、牛脂、棕榈油精、雾冰草脂(illipebutter)、牛油树脂、可可脂、烛果脂、婆罗双树脂(salbutter)、卵磷脂、日本蜡羊毛脂、部分氢化的植物油、疏水性聚合物、及其任何组合。在本发明的一些实施方案中,疏水性基料可以包含疏水性聚合物。本发明的药物组合物中可以使用任何合适的疏水性聚合物。示例性的疏水性聚合物包括但不限于烃聚合物和/或共聚物、芳香族聚氨酯、硅橡胶、聚硅氧烷、聚己内酯、聚碳酸酯、聚氯乙烯、聚乙烯、聚-L-丙交酯、聚-DL-乙交酯、聚醚醚酮(PEEK)、聚酰胺、聚酰亚胺和聚乙酸乙烯酯。在本发明的具体实施方案中,本发明的药物组合物包含一种或多种烃聚合物和/或共聚物。在某些实施方案中,本发明的药物组合物包含一种或多种烃聚合物和/或共聚物,诸如,但不限于,以商标Versagel®从Indianapolis,IN的CalumetSpecialtyProductsPartners商购的那些和/或以商品名CrodabaseSQ从EastYorkshire,UnitedKingdom的CrodaInternationalPlc商购的那些。在本发明的一些实施方案中,疏水性聚合物可以在药物组合物中充当增稠剂和/或胶凝剂。具体而言,疏水性聚合物可以充当粘弹性物质,并且可以在应用部位保留所述组合物,连同其中分散的任何化合物(例如,活性药物成分等)。疏水性聚合物可以以约30重量%至约60重量%或其中的任何范围,诸如,但不限于,约35重量%至约55重量%或约40重量%至约50重量%的浓度存在于本发明的药物组合物中。在本发明的具体实施方案中,疏水性基料包含一种或多种植物和/或矿物油。本发明的药物组合物中可以使用任何合适的油。示例性的矿物油包括,但不限于,轻质矿物油、白色矿物油、石蜡油、环烷油(naphtenicoils)、芳香油、及其任何组合。油(例如,植物和/或矿物油)可以以约1重量%至约30重量%或其中的任何范围,诸如,但不限于,约5重量%至约20重量%或约5重量%至约15重量%的浓度存在于本发明的药物组合物中。在本发明的一些实施方案中,疏水性基料,诸如,但不限于,油(例如,植物和/或矿物油)可以用来调整所述药物组合物的粘度和/或覆盖性(spreadability)。例如,低粘度疏水性基料,诸如轻质矿物,可用于使药物组合物,诸如,包含高粘度疏水性基料的药物组合物变薄(即,降低粘度)。这可以使得能够在宽区域应用本发明的药物组合物,并可以用于在应用部位保持其中分散的任何化合物(例如,活性药物成分等)。在本发明的某些实施方案中,疏水性基料包含矿物油和疏水性聚合物。疏水性基料可以以约35重量%至约90重量%或其中的任何范围,诸如,但不限于,约40重量%至约80重量%或约50重量%至约70重量%的浓度存在于本发明的药物组合物中。在本发明的某些实施方案中,疏水性基料以约45重量%至约55重量%的浓度存在于本发明的药物组合物中。如本文中所使用的“两亲性化合物”是指包含亲水性和疏水性特性的化合物。两亲性化合物可以包含两种或更多种化合物,其中每种可以提供亲水性特性和/或疏水性特性。在一些实施方案中,两亲性化合物包含一种具有亲水性和疏水性特性的化合物。在本发明的具体实施方案中,两亲性化合物可以吸收水分,而基本上不吸收气态水分。水分的吸收可以允许在与水分接触之后,但不在与气态水分接触之后,本发明的药物组合物中水分活化的活性药物成分的活化。如本文中所使用的“基本上吸收”(及其语法变体)是指吸收的气态水分的量多于两亲性化合物的2重量%。因此,本发明的两亲性化合物吸收两亲性化合物的小于约2重量%、1.5重量%、1重量%、0.5重量%、0.25重量%或其中的任何范围的气态水分。在本发明的一些实施方案中,两亲性化合物可以防止和/或尽可能减少本发明的药物组合物显著吸收气态水分,由此水分可以以小于约2%存在于本发明的药物组合物中。如本文中所使用的“水分”是指液体,诸如,但不限于,体液,诸如,但不限于,血液、汗液、粘液、唾液、皮脂、泪液、渗出物、和/或阴道分泌物;水;除氧水;盐水溶液;酸性或碱性缓冲溶液;和/或其任何组合。如本文中所使用的“气态水分”是指气相中的水分。例如,气态水分,包括,但不限于,水蒸汽。因此,在本发明的一些实施方案中,两亲性化合物可以防止和/或尽可能减少水蒸汽的吸收,由此,当活性药物成分(API)包含水分活化的药物成分时,本发明的药物组合物中的API不被气态水分(例如水蒸气)所活化。相反,当本发明的药物组合物与水分接触时,两亲性化合物可以吸收和/或允许水分(例如,水,体液等)被吸收,从而当API包含水分活化的活性药物成分时活化所述API。在本发明的具体实施方案中,两亲性化合物吸收小于约2重量%或约1重量%的水蒸汽。这可以尽可能减少和/或防止本发明的药物组合物吸收水蒸汽,因此水可以以小于约2重量%或约1重量%的水存在于本发明的药物组合物中。在本发明的某些实施方案中,两亲性化合物吸收小于约0.5重量%的水蒸汽,因此本发明的药物组合物可以包含小于约0.5重量%的水。两亲性化合物可以具有12至20或其中的任何范围的亲水亲油平衡(HLB)值,诸如,但不限于,15至20或18至20。在本发明的某些实施方案中,两亲性化合物包含19的HLB值。示例性的两亲性化合物包括,但不限于,脂肪酸酯。本发明的药物组合物中可以存在一种或多种脂肪酸酯,诸如2、3、4或更多种脂肪酸酯。示例性的脂肪酸酯包括,但不限于,C6-C22烷基和/或烯基脂肪酸酯类,诸如月桂酸甲酯,月桂酸乙酯,肉豆蔻酸乙酯,棕榈酸乙酯,亚油酸乙酯,异丁酸丙酯,月桂酸异丙酯,肉豆蔻酸异丙酯,棕榈酸异丙酯,肉豆蔻酸油醇酯(oleylmyristate),硬脂酸油醇酯和油酸油醇酯;醚-酯类,诸如乙氧基化脂肪醇的脂肪酸酯;多元醇酯类,诸如乙二醇单-和二-脂肪酸酯,二甘醇单和二脂肪酸酯;聚乙二醇(6-2000)脂肪酸单-和/或二酯类,诸如PEG-6月桂酸酯,PEG-6硬脂酸酯,PEG-8二月桂酸酯,PEG-8二硬脂酸酯等;聚乙二醇甘油脂肪酸酯类,诸如PEG-20-月桂酸甘油酯,PEG-20-硬脂酸甘油酯和PEG-20-油酸甘油酯;丙二醇单-和二脂肪酸酯;聚丙二醇2000单油酸酯;聚丙二醇2000单硬脂酸酯;乙氧基化的丙二醇单硬脂酸酯;甘油基单-和二脂肪酸酯类;聚甘油脂肪酸酯类,诸如聚甘油基-10月桂酸酯等;乙氧基化的单硬脂酸甘油酯;1,3-丁二醇单硬脂酸酯;1,3-丁二醇二硬脂酸酯;聚氧乙烯多元醇脂肪酸酯;脱水山梨糖醇脂肪酸酯类,包括脱水山梨糖醇三油酸酯和脱水山梨糖醇单月桂酸酯;聚乙二醇脱水山梨糖醇脂肪酸酯类,诸如PEG-6脱水山梨糖醇单油酸酯;聚氧乙烯脱水山梨醇脂肪酸酯,包括聚氧乙烯(20)脱水山梨糖醇单月桂酸酯;蔗糖脂肪酸酯,诸如蔗糖单棕榈酸酯和蔗糖单硬脂酸酯;蜡酯类,诸如蜂蜡,鲸蜡,肉豆蔻酸肉豆蔻酯,硬脂酸硬脂基酯和山萮酸二十烷醇酯;聚乙二醇烷基醚类,诸如PEG-10油基醚或PEG-9十六烷基醚;聚乙二醇烷基酚类,诸如PEG-10-100壬基酚;聚氧乙烯-聚氧丙烯嵌段共聚物,诸如泊洛沙姆188;甾醇酯类,诸如胆固醇脂肪酸酯,及其任何组合。脂肪酸酯可以包含聚乙二醇(PEG)甘油酯。PEG甘油酯的聚乙二醇部分可以提供两亲性化合物的亲水性特性,并且可以包括,但不限于,PEG5-1000或其中的任何范围,以及其任何组合。PEG甘油酯的甘油酯部分可以提供两亲性化合物的疏水性特性,并且可以包括,但不限于,天然的和/或氢化的油,诸如但不限于蓖麻油,氢化蓖麻油,维生素A,维生素D,维生素E,维生素K,植物油(例如,玉米油,橄榄油,花生油,棕榈仁油,杏仁油,扁桃仁油,等),以及其任何组合。示例性的聚乙二醇(PEG)甘油酯包括,但不限于,PEG-20蓖麻油,PEG-20氢化蓖麻油,PEG-20玉米甘油酯,PEG-20扁桃仁甘油酯;PEG-23三油酸酯,PEG-40棕榈仁油,PEG-8辛酸/癸酸甘油酯,PEG-6辛酸/癸酸甘油酯,月桂酰聚乙二醇-32甘油酯,硬脂酰聚乙二醇甘油酯,生育酚PEG-1000琥珀酸脂,以及其任何组合。在本发明的一些实施方案中,脂肪酸酯包含PEG5-30(即,PEG5、6、7、8、9、10,等等)和辛酸/癸酸甘油酯。在本发明的具体实施方案中,药物组合物包含PEG-5-辛酸/癸酸甘油酯,PEG-6-辛酸/癸酸甘油酯,PEG-7-辛酸/癸酸甘油酯,和/或PEG-8-辛酸/癸酸甘油酯。在本发明的某些实施方案中,药物组合物包含一种或多种脂肪酸酯,诸如,但不限于以商标SOFTIGEN®从Hamburg,Germany的Sasol商购的那些。两亲性化合物可以以约1重量%至约30重量%或其中的任何范围,诸如,但不限于,约2重量%至约20重量%或约5重量%至约15重量%的浓度存在于本发明的药物组合物中。在本发明的某些实施方案中,两亲性化合物以约10重量%的浓度存在于本发明的药物组合物中。本发明的药物组合物可以进一步包含一种或多种赋形剂。用于药物组合物中使用的赋形剂是本领域中众所周知的,且实例可见HandbookofPharmaceuticalExcipients(Rowe,R.C.等人,APhAPublications;第5版,2005)。赋形剂的类别可以包括,但不限于,软化剂、湿润剂、共溶剂、pH调节剂、防水剂、消泡剂、表面活性剂、增溶剂、润湿剂、渗透增强剂、抗氧化剂、和/或溶剂。赋形剂可以以任何合适的浓度存在于本发明的药物组合物中。在本发明的具体实施方案中,药物组合物可以进一步包含共溶剂。共溶剂可以以约1重量%至约30重量%或其中的任何范围,诸如,但不限于,约2重量%至约20重量%或约5重量%至约15重量%的浓度存在于本发明的药物组合物中。在本发明的某些实施方案中,共溶剂以约10重量%至约15重量%的浓度存在于本发明的药物组合物中。示例性的共溶剂包括,但不限于,脂肪酸酯、丙二醇、甘油、聚乙二醇。在本发明的一些实施方案中,共溶剂可以包含中性油。在本发明的某些实施方案中,共溶剂包含辛酸和/或癸酸甘油三酯,诸如,但不限于以商标MIGLYOL®从Hamburg,Germany的Sasol商购的那些。本发明的药物组合物可以包含湿润剂。可以使用任何合适的湿润剂或湿润剂的组合。湿润剂可以以约1重量%至约25重量%或其中的任何范围,诸如,但不限于,约2重量%至约20重量%或约5重量%至约15重量%的浓度存在于本发明的药物组合物中。在本发明的某些实施方案中,湿润剂以约10重量%至约15重量%的浓度存在于本发明的药物组合物中。示例性的湿润剂包括,但不限于,二元醇类,诸如多元醇,二甘醇单乙醚和甲氧基聚乙二醇;甘油类,诸如丙二醇,甘油,异丙醇,乙醇,乙二醇,聚乙二醇,乙氧基二甘醇或其混合物;糖多元醇类,诸如山梨醇,木糖醇和麦芽糖醇;多元醇类,诸如聚葡萄糖;二甲基异山梨醇(dimethylisosorbide);皂树提取物(quillaia);尿素;以及其任何组合。在本发明的具体实施方案中,湿润剂包括亚烷基二醇,诸如己二醇,丁二醇,戊二醇,以及其任何组合。本发明的药物组合物可以包含活性药物成分(API)。API可以以任何合适的浓度存在于本发明的药物组合物中。在本发明的具体实施方案中,API包含水分活化的活性药物成分,诸如,但不限于,释放一氧化氮的化合物和/或水溶性API。在本发明的一些实施方案中,本发明的药物组合物可以通过控制药物组合物吸收的水分的量提供API的受控和/或持续释放。在本发明的具体实施方案中,水分活化的API以约0.1重量%至约70重量%或其中的任何范围,诸如,但不限于,约1重量%至约50重量%或约2重量%至约30重量%的浓度存在于组合物中。为了避免本发明的药物组合物中的砂砾感,API的最大粒度可以小于约100µm,并且在一些实施方案中,小于约20µm,并且在进一步实施方案中,小于约10µm。根据本发明的实施方案的组合物中可以包括任何合适的活性药物成分(API)或API的组合。API的实例包括,但不限于,抗微生物剂、抗痤疮剂、抗炎剂、止痛剂、麻醉剂、抗组胺剂、防腐剂、免疫抑制剂、抗出血剂、血管扩张剂、创伤愈合剂、抗生物膜剂及其混合物。抗微生物剂的实例包括,但不限于,青霉素和相关药物、碳青霉烯类、头孢菌素类和相关药物、红霉素、氨基糖苷、杆菌肽、短杆菌肽、莫匹罗星、氯霉素、甲砜霉素、夫西地酸钠、林可霉素、克林霉素、大环内酯类、新生霉素、多粘菌素类、利福霉素、壮观霉素、四环素、万古霉素(vanomycin)、替考拉宁、链阳性菌素、抗叶酸剂,包括磺酰胺、甲氧苄氨嘧啶及其组合和乙胺嘧啶、合成抗菌剂,包括硝基呋喃、扁桃酸乌洛托品和马尿酸乌洛托品、硝基咪唑、喹诺酮、氟喹诺酮、异烟肼、乙胺丁醇、吡嗪酰胺、对氨基水杨酸(PAS)、环丝氨酸、卷曲霉素、乙硫异烟胺、丙硫异烟胺、氨硫脲、紫霉素、扁枝衣霉素(eveminomycin)、糖肽、甘氨酰四环素(glyclyclycline)、酮内酯、噁唑烷酮;亚胺培南(imipenen)、丁胺卡那霉素、奈替米星、磷霉素、庆大霉素、头孢曲松、Ziracin、利奈唑胺、Synercid、氨曲南和甲硝唑、依匹普林、山费培南钠、比阿培南、达内霉素(Dynemicin)、头孢瑞南、头孢噻利、山费培南西酯(Sanfetrinemcelexetil)、头孢匹罗、Mersacidin、利福拉齐、Kosan、来那培南、Veneprim、硫培南、利替培南酯(ritipenamacoxyl)、Cyclothialidine、美加球菌素A、卡芦莫南、头孢唑兰和头孢他美。局部抗痤疮剂的实例包括,但不限于,阿达帕林、壬二酸、过氧化苯甲酰、克林霉素和克林霉素磷酸酯、强力霉素、红霉素、角质层分离剂,诸如水杨酸和视黄酸(Retin-A'')、诺孕酯、有机过氧化物、类视黄醇,诸如异维甲酸和维甲酸、磺胺醋酰钠(sulfacetamidesodium)和他扎罗汀。特别的抗痤疮剂包括阿达帕林、壬二酸、过氧化苯甲酰、克林霉素(例如克林霉素磷酸酯)、强力霉素(例如一水合强力霉素)、红霉素、异维甲酸、诺孕酯、磺胺醋酰钠、他扎罗汀、依曲替酯和阿曲汀(acetretin)。抗组胺剂的实例包括,但不限于,盐酸苯海拉明、水杨酸苯海拉明、苯海拉明、盐酸氯苯吡胺、马来酸氯苯吡胺、盐酸异西喷地、盐酸曲吡那敏、盐酸异丙嗪、盐酸甲吡咯嗪等。局部麻醉剂的实例包括盐酸地布卡因、地布卡因、盐酸利多卡因、利多卡因、苯佐卡因、对丁基氨基苯甲酸2-(二乙基氨基)乙酯盐酸化物(p-buthylaminobenzoicacid2-(die-ethylamino)ethylesterhydrochloride)、盐酸普鲁卡因、丁卡因、盐酸丁卡因、盐酸氯普鲁卡因、盐酸羟基普鲁卡因、甲哌卡因、盐酸可卡因、盐酸哌罗卡因、达克罗宁和盐酸达克罗宁。防腐剂的实例包括,但不限于,醇、季铵化合物、硼酸、氯己定和氯己定衍生物、碘、酚、萜烯、杀菌剂、消毒剂,包括硫柳汞、酚、百里酚、苯扎氯铵、苄索氯铵、氯己定、聚维酮碘、西吡氯铵、丁香油酚和三甲基溴化铵。抗炎剂的实例包括,但不限于,非类固醇抗炎剂(NSAIDs);丙酸衍生物,诸如布洛芬和萘普生;乙酸衍生物,诸如吲哚美辛;烯醇酸衍生物,诸如美洛昔康、对乙酰氨基酚;水杨酸甲酯;单乙二醇水杨酸酯;阿司匹林;甲芬那酸;氟灭酸;吲哚美辛;双氯芬酸;阿氯芬酸;双氯芬酸钠;布洛芬;酮洛芬;萘普生;普拉洛芬;非诺洛芬;舒林酸;芬氯酸;环氯茚酸;氟比洛芬;芬替酸;丁苯羟酸;吡罗昔康;苯基丁氮酮;羟基保泰松;氯非宗;喷他佐辛;嘧吡唑;盐酸羟哌苯噻酮;类固醇,诸如丙酸氯倍他索、二丙酸倍他米松酯(bethamethasonedipropionate)、丙酸卤贝他索酯(halbetasolproprionate)、双醋二氟拉松、醋酸氟轻松、哈西奈德、安西缩松、去羟米松、曲安奈德、糠酸莫米松、丙酸氟替卡松、二丙酸倍他米松、曲安奈德、丙酸氟替卡松、地奈德、氟轻松、戊酸氢化可的松、泼尼卡酯、丙炎松、氟轻松(fluocinoloneacetonide)、氢化可的松和本领域中已知的其它、氢化泼尼松、地塞米松、氟轻松、醋酸氢化可的松、醋酸氢化泼尼松、甲泼尼龙、醋酸地塞米松、倍他米松、戊酸倍他米松、氟米松(flumetasone)、氟米龙(fluorometholone)、二丙酸倍氯米松、氟轻松醋酸酯、局部皮质类固醇,并可以是较低效力的皮质类固醇之一,诸如氢化可的松、氢化可的松-21-单酯(例如氢化可的松-21-乙酸酯、氢化可的松-21-丁酸酯、氢化可的松-21-丙酸酯、氢化可的松-21-戊酸酯等)、氢化可的松-17,21-二酯(例如氢化可的松-17,21-二乙酸酯、氢化可的松-17-乙酸酯-21-丁酸酯、氢化可的松-17,21-二丁酸酯等)、阿氯米松、地塞米松、氟米松、氢化泼尼松或甲泼尼龙,或可以是较高效力的皮质类固醇,诸如丙酸氯倍他索、苯甲酸倍他米松、二丙酸倍他米松、双醋二氟拉松、氟轻松醋酸酯、糠酸莫米松、曲安奈德。止痛剂的实例包括,但不限于,阿芬太尼、苯佐卡因、丁丙诺啡、布托啡诺、氨苯丁酯、辣椒素、可乐定、可待因、地布卡因、脑啡肽、芬太尼、氢可酮、氢吗啡酮、吲哚美辛、利多卡因、羟甲左吗喃、度冷丁、美沙酮、吗啡、尼可吗啡、鸦片、奥布卡因、羟考酮、羟吗啡酮、喷他佐辛、普莫卡因、丙美卡因、丙氧芬、丙氧间卡因、舒芬太尼、丁卡因和曲马多。麻醉剂的实例包括,但不限于,醇诸如酚;苯甲酸苄酯;炉甘石;氯二甲苯酚;达克罗宁;氯胺酮;薄荷醇;普莫卡因;间苯二酚;三氯生;普鲁卡因药物,诸如苯佐卡因、布比卡因、氯普鲁卡因;辛可卡因;可卡因;地昔卡因;二胺卡因;地不卡因;依替卡因;海克卡因;左布比卡因;利多卡因;甲哌卡因;羟乙卡因;丙胺卡因;普鲁卡因;丙美卡因;丙氧卡因;吡咯卡因;利索卡因;罗多卡因;罗哌卡因;丁卡因;及衍生物,诸如药学上可接受的盐和酯,包括盐酸布比卡因、盐酸氯普鲁卡因、环己氨磺酸二胺卡因、盐酸地布卡因、盐酸达克罗宁、盐酸依替卡因、盐酸左布比卡因、盐酸利多卡因、盐酸甲哌卡因、盐酸普莫卡因、盐酸丙胺卡因、盐酸普鲁卡因、盐酸丙美卡因、盐酸丙氧卡因、盐酸罗哌卡因和盐酸丁卡因。抗出血剂的实例包括,但不限于,凝血酶、维生素K1、硫酸鱼精蛋白、氨基己酸、氨甲环酸、肾上腺色腙、肾上腺色腙磺酸钠(carbaxochromesodiumsulfanate)、芸香苷和橙皮苷。在本发明的一些实施方案中,活性药物成分(API)包含释放一氧化氮(NO)的化合物,基本上由其组成,或由其组成。本发明的药物组合物中可以使用任何合适的释放NO的化合物。在本发明的一些实施方案中,释放NO的化合物包含包括NO供体基团(donorgroup)的小分子化合物。如本文中所使用的“小分子化合物”是指具有小于500道尔顿的分子量的化合物,并包括有机和/或无机小分子。在本发明的一些实施方案中,释放NO的化合物包含包括NO供体基团的大分子。如本文中所使用的“大分子”是指具有500道尔顿或更大的分子量的化合物。可使用任何合适的大分子,包括交联或非交联的聚合物、树枝状聚物(dendrimer)、金属化合物、有机金属化合物、基于无机的化合物及其它大分子支架。在一些实施方案中,所述大分子具有范围为约0.1nm-约100μm的公称直径,并且可包括两个或更多个大分子的聚集物,其中所述大分子结构用NO供体基团进一步修饰。在本发明的某些实施方案中,释放NO的化合物的NO供体在暴露于外部条件(诸如光、热、水、酸、碱,等等)之后释放一氧化氮。例如,在本发明的一些实施方案中,释放NO的化合物包含作为NO供体的二醇二氮烯鎓(diazeniumdiolate)官能团。二醇二氮烯鎓官能团可在特定条件下(诸如在暴露于水之后)产生一氧化氮。作为另一个实例,在本发明的一些实施方案中,释放NO的化合物可以包含,但不限于,作为NO供体的亚硝基硫醇(nitrosothiol)官能团。所述NO供体可在特定条件下(诸如在暴露于光之后)产生一氧化氮。其它NO供体基团的实例包括,但不限于,亚硝胺、羟基亚硝胺、羟胺和羟基脲。NO供体和/或释放NO的化合物的任何合适的组合可用于本发明的药物组合物中。此外,可通过共价和/或非共价相互作用将所述NO供体掺入小分子和/或大分子中和/或掺至小分子和/或大分子上。在本发明的一些实施方案中,释放NO的化合物可呈释放NO的颗粒的形式,诸如在美国公开号2009/0214618中所述的那些,所述申请的公开内容通过引用以其整体并入本文。此类颗粒可通过其中所述方法制备。释放NO的化合物可以通过任何合适的机制,包括经由与水反应和/或热降解,释放一氧化氮。释放NO的化合物中可以包括的释放NO的官能团的实例包括,但不限于,二醇二氮烯鎓、亚硝胺、羟基亚硝胺、亚硝基硫醇、羟胺、羟基脲、和金属亚硝酰基络合物。也可以利用能够以治疗的方式释放一氧化氮的其它释放NO的官能团,诸如酸化的亚硝酸盐。释放NO的化合物可以是小分子化合物、寡聚物和/或聚合物,并且可以是任何合适的物理形式,诸如,但不限于,颗粒、涂层、薄膜、液体、溶液等。在一些实施方案中,释放一氧化氮的化合物包含如上所述的二醇二氮烯鎓官能化的聚硅氧烷大分子。释放NO的化合物的其它非限制性实例包括如美国专利公开号2006/0269620或2010/0331968中所述的释放NO的沸石;如在美国专利申请公开号2010/0239512或2011/0052650中所述的释放NO的金属有机框架(MOFs);如题为"TunableNitricOxide-ReleasingMacromoleculesHavingMultipleNitricOxideDonorStructures"的美国临时专利申请序列号61/526,918中所述的释放NO的多供体化合物;如美国公开号2009/0214618中所述的释放NO的树枝状聚物或金属结构;如美国公开号2011/0086234中所述的释放一氧化氮的涂层;和如美国公开号2010/0098733中所述的化合物。本段落中每篇参考文献的公开内容都通过引用以其整体并入本文。此外,可以如2012年1月20日提交的题为"TemperatureControlledSol-GelCo-Condensation"的国际申请号PCT/US2012/022048中所述制备释放NO的大分子,所述申请的公开内容通过引用以其整体并入本文。作为实例,在本发明的一些实施方案中,释放NO的颗粒包括装载NO的沉淀二氧化硅。所述装载NO的沉淀二氧化硅可由一氧化氮供体修饰的硅烷单体形成为共缩合的硅氧烷网络。在本发明的一个实施方案中,所述一氧化氮供体为N-二醇二氮烯鎓。在一些实施方案中,可通过预装载(pre-charging)法由氨基烷氧基硅烷形成一氧化氮供体,且共缩合的硅氧烷网络可由包括烷氧基硅烷和氨基烷氧基硅烷的硅烷混合物的缩合来合成,以形成一氧化氮供体修饰的共缩合的硅氧烷网络。如本文中所使用的“预装载法”意指在与烷氧基硅烷共缩合之前将氨基烷氧基硅烷用一氧化氮“预处理”或“预装载”。在一些实施方案中,预装载性一氧化氮可通过化学法实现。在另一个实施方案中,“预装载”法可用于产生经NO供体更密集官能化的共缩合的硅氧烷网络和材料。所述共缩合的硅氧烷网络可以是大小一致的二氧化硅颗粒、多种大小的二氧化硅颗粒的集合、无定形二氧化硅、热解法二氧化硅、纳米晶体二氧化硅、陶瓷二氧化硅、胶态二氧化硅、二氧化硅涂层、二氧化硅薄膜、有机修饰的二氧化硅、介孔二氧化硅、硅胶、生物活性玻璃或者任何合适形式或状态的二氧化硅。在一些实施方案中,烷氧基硅烷为具有式Si(OR)4的四烷氧基硅烷,其中R为烷基。R基团可以是相同的或不同的。在一些实施方案中,将四烷氧基硅烷选为原硅酸四甲酯(TMOS)或原硅酸四乙酯(TEOS)。在一些实施方案中,氨基烷氧基硅烷具有式:R"-(NH-R')n-Si(OR)3,其中R为烷基,R'为亚烷基、支链亚烷基或亚芳烷基(aralkylene),n为1或2,且R"选自烷基、环烷基、芳基和烷基胺。在一些实施方案中,氨基烷氧基硅烷可选自N-(6-氨基己基)氨基丙基三甲氧基硅烷(AHAP3);N-(2-氨基乙基)-3-氨基丙基三甲氧基硅烷(AEAP3);(3-三甲氧基甲硅烷基丙基)二-亚乙基三胺(DET3);(氨基乙基氨基甲基)苯乙基三甲氧基硅烷(AEMP3);[3-(甲基氨基)丙基]三甲氧基硅烷(MAP3);N-丁基氨基-丙基三甲氧基硅烷(n-BAP3);叔丁基氨基-丙基三甲氧基硅烷(t-BAP3);N-乙基氨基异丁基三甲氧基硅烷(EAiB3);N-苯基氨基-丙基三甲氧基硅烷(PAP3);和N-环己基氨基丙基三甲氧基硅烷(cHAP3)。在一些实施方案中,氨基烷氧基硅烷具有式NH[R'-Si(OR)3]2,其中R为烷基,且R'为亚烷基。在一些实施方案中,氨基烷氧基硅烷可选自双(3-三乙氧基甲硅烷基丙基)胺、双-[3-(三甲氧基甲硅烷基)丙基]胺和双-(3-三甲氧基甲硅烷基)丙基]乙二胺。在一些实施方案中,如上文所述,使氨基烷氧基硅烷针对NO释放预装载,并且用二醇二氮烯鎓取代氨基。因此,在一些实施方案中,氨基烷氧基硅烷具有式:R''-N(NONO-X+)-R'-Si(OR)3,其中R为烷基,R'为亚烷基或亚芳烷基,R''为烷基或烷基胺,X+为选自Na+、K+和Li+的阳离子。硅氧烷网络的组成(例如,氨基烷氧基硅烷的量或化学组成)和一氧化氮装载条件(例如,溶剂和碱)可改变,以使一氧化氮释放的量和持续时间最优化。因此,在一些实施方案中,可改变二氧化硅颗粒的组成来调节自二氧化硅颗粒释放NO的半衰期。在另一个实施方案中,氨基烷氧基硅烷的氨基被二醇二氮烯鎓取代,并且所述氨基烷氧基硅烷具有式R''-N(NONO-X+)-R'-Si(OR)3,其中:R为烷基,R'为亚烷基或亚芳烷基,R''为烷基或烷基胺,X+为选自Na+和K+的阳离子。在本发明的一些实施方案中,释放NO的颗粒的粒度在20nm-10μm的范围。可以调整粒度以尽可能减少或防止毒性和穿透通过表皮(或缺乏抵抗力的真皮)并进入血管。在具体的实施方案中,粒度分布在小于约10µm的平均粒度左右,以允许颗粒进入毛囊(follicle)。在进一步实施方案中,粒度分布在小于约8µm的平均粒度左右。在其它实施方案中,粒度分布在大于约10µm的平均粒度左右,以防止颗粒进入毛囊。在又进一步实施方案中,可以提供平均粒度分布在两种或更多种平均粒度左右的颗粒的混合物。例如,可以将具有分布在小于约10µm的平均粒度左右以允许颗粒进入毛囊的粒度的颗粒的混合物与具有分布在大于约10µm的平均粒度左右以防止颗粒进入毛囊的粒度的颗粒混合。所述颗粒可以具有相同的一氧化氮释放概况或不同的一氧化氮释放概况。例如,较小颗粒可以具有经调整以增强其调节皮脂产生和/或异常角质化的能力的释放概况,且较大颗粒可以具有经调整以增强其杀灭细菌、促进伤口愈合、减少疤痕或由一氧化氮提供的其它所需治疗效果的能力的释放概况。还可以提供其它组合和多种组合。可以通过任何合适的方法制备本发明的药物组合物。然而,在一些实施方案中,本发明的组合物可以通过分别在2011年7月5日和2012年3月13日提交的均题为"MethodsofManufacturingTopicalCompositionsandApparatusForSame,"的美国临时专利申请序列号61/504,626和61/610,179(律师案卷号9729-26PR2)中描述的方法进行制备,所述申请通过引用以其整体并入本文。在本发明的某些实施方案中,制备本发明的药物组合物的方法包括将包含疏水性基料、两亲性化合物和任选的共溶剂的第一赋形剂组合物均质化。机械置顶搅拌装置可以用来混合第一赋形剂组合物,直到实现所需均匀性和/或一致性。均质化速度和/或速率可以是恒定的、变化的、增加的和/或降低的,以实现期望的均匀性和/或一致性。在本发明的具体实施方案中,混合本发明的第一赋形剂组合物,直到所述组合物在视觉上是均匀的。在本发明的一些实施方案中,所述方法进一步包括单独将包含疏水性基料、活性药物成分和任选湿润剂的第二赋形剂组合物均质化。然后可以将第一赋形剂组合物和第二赋形剂组合物组合以形成本发明的药物组合物。图1是本发明的示例性实施方案的操作流程图。如图1中可见,通过分配用于在过程中使用的原材料而开始操作(方框100)。然后设定混合容器的温度(方框110)。将疏水性基料和两亲性化合物添加至容器(方框120),任选地添加共溶剂(方框125),以形成第一赋形剂组合物。然后设定均质化参数(例如,均质化速率,时间等),并且开始第一赋形剂组合物的均质化(方框130)。在本发明的具体实施方案中,在添加第一赋形剂组合物的组分之后,相比于初始速度增加均质化速度并维持,直到获得视觉上均匀的组合物。在分开的容器中,将疏水性基料和活性药物成分(API),诸如,但不限于水分活化的API,组合(方框140),任选地与湿润剂(方框145)组合,以形成第二赋形剂组合物。然后设定均质化参数(例如,均质化速率,时间等),并且开始第二赋形剂组合物的均质化(方框150)。接下来,将第一和第二赋形剂组合物组合并均质化,直到获得期望的均匀性(方框160)。可以使用任何合适的均质化机械装置。均质化装置的实例包括机械置顶搅拌,诸如螺旋桨、锚、针距叶片(pitchblade)、转子-定子、旋转叶片、超声装置、在线和高压均化器。可以使用任何这些方法,并且多种方法可以在一些实施方案中组合使用。预混料组合物的均质化可以提供具有所需API稳定性和混合均匀性的最终局部组合物。在本发明的一些实施方案中,可以使用在线均化器。在本发明的具体实施方案中,可以使用使活性药物成分(例如,水分活化的活性药物成分)维持低于其中活性药物成分可能降解的温度的均质化方法和/或装置。如果在特定温度维持特定的持续时间,则活性药物成分可能在该温度降解。因此,在本发明的一些实施方案中,将活性药物成分在特定温度维持的持续时间低于其中所述活性成分在该温度可能降解的时间期间。在本发明的某些实施方案中,对于整个均质化过程,保持活性药物成分的温度不超过其中活性药物成分可能降解的温度。在本发明的一些实施方案中,在约-15℃至约30℃的范围内或其中的任何范围的温度进行均质化。在本发明的具体实施方案中,在室温进行均质化。在本发明的一些实施方案中,在干燥的惰性气氛中进行均质化,从而使得均质化容器中基本上不存在水和氧气。本发明的药物组合物可用于通过局部施用药物组合物来治疗个体的皮肤。因此,本发明的另一个方面包含治疗个体的皮肤的方法,所述方法包括将本发明的药物组合物局部施用于个体的皮肤。在本发明的一些实施方案中,当本发明的药物组合物中存在水分活化的活性药物成分时,所述方法可以进一步包括在局部施用组合物的步骤之前、之后和/或期间使水分(例如,水)接触组合物和/或应用部位。在本发明的一些实施方案中,在施用本发明的药物组合物之前,水分,诸如,但不限于水和/或体液,已经存在于应用部位。可以处理个体的皮肤的任何部分,包括,但不限于,个体的粘膜(包括体腔)、指甲和/或头皮。然而,在本发明的一些实施方案中,通过本文所述的方法治疗一个或多个个体的附肢。此外,在本发明的一些实施方案中,通过本文所述方法治疗个体的躯干。本发明可用于兽医和医疗应用两者中。适于用本发明的方法实施方案治疗的个体包括但不限于鸟类和哺乳动物个体。本发明的哺乳动物包括,但不限于,犬、猫科动物、牛科动物、山羊、马、绵羊、猪、啮齿类动物(例如大鼠和小鼠)、兔类动物、灵长类动物(例如猿猴和人),非人灵长类动物(例如,猴、狒狒、黑猩猩、大猩猩)等,以及子宫内的哺乳动物。需要根据本发明治疗的任何哺乳动物个体都是合适的。两种性别以及处于任何发育阶段(即新生儿、婴幼儿、少年、青少年、成人)的人个体均可根据本发明治疗。在本发明的一些实施方案中,个体是哺乳动物,且在某些实施方案中,个体是人。人个体包括所有年龄的男性和女性,包括胎儿、新生儿、婴幼儿、少年、青少年、成人和老年个体以及怀孕个体。在本发明的具体实施方案中,个体是人青少年和/或成人。根据本发明的说明性鸟类包括鸡、鸭、火鸡、鹅、鹌鹑、雉、平胸类鸟(例如鸵鸟)和驯养的鸟(例如鹦鹉和金丝雀),以及蛋中的鸟。本发明的方法也可在动物个体特别是哺乳动物个体(诸如小鼠、大鼠、狗、猫、家畜和马)上实施,以用于兽医目的和/或用于药物筛选和药物开发目的。在本发明的具体实施方案中,个体“需要”本发明的方法,例如,个体已经被诊断患有以下疾病或病症,处于以下疾病或病症的风险,和/或据信具有以下疾病或病症,所述疾病或病症可以使用本发明的方法进行治疗。在本发明的一些实施方案中,个体具有皮肤病症,诸如,但不限于,痤疮,特应性皮炎和/或牛皮癣。在本发明的其它实施方案中,个体具有伤口,诸如,但不限于,褥疮、烧伤、和/或糖尿病足溃疡。在本发明的一些实施方案中,个体具有炎性皮肤状况或病症。如本文中所使用的“治疗(Treat)”、“治疗(treating)”或“…的治疗(treatmentof)”(及其语法变体)是指为个体赋予益处的任何类型的治疗,并且可意指个体的状况的严重度得到降低,至少部分改善或缓减和/或至少一种临床症状中的一些减轻、缓解或减少得到实现和/或存在所述疾病或病症的进程的延迟。在本发明的具体实施方案中,与不存在本发明的方法的皮肤病症的严重度相比,个体中的皮肤病症的严重度得到降低。在本发明的其它实施方案中,本发明的方法改善伤口愈合和/或防止免于感染。下面非限制性实施例中更详细地解释本发明。实施例实施例1表1和2描述了根据本发明的实施方案制备的各种药物组合物。表1:包含制剂1的药物组合物。表2:包含制剂2的药物组合物。实施例2制备如美国公开号2009/0214618和2012年1月20日提交的题为“TemperatureControlledSol-GelCo-Condensation”的国际申请号PCT/US2012/022048中所述的具有2%Nitricil™且包含MAP3的软膏制剂。表3-6显示了产生的软膏和凝胶的制剂。如本文所述制备表4和5中的软膏制剂。还制备没有Nitricil™活性成分的表4中的软膏版本,其中用矿物油和轻质矿物油代替活性成分。如美国专利申请序列号12/860,457中所述制备表3中所述的软膏。如美国临时专利申请序列号61/504,628中所述产生表6中的局部凝胶。表3:包含制剂11-15-12的软膏表4:包含制剂TO-005的软膏表5:包含制剂TO-006的软膏表6:醇凝胶。将表4和5中软膏制剂的功效与表6中显示醇凝胶制剂(已知针对铜绿假单胞菌(Pseudomonasaeruginosa)是杀细菌的)和表3中显示的较早软膏制剂进行比较。所有制剂都含有如上所述的2%Nitricil™,且以用于测试的PBS中50mg/ml的稀释度(相当于1mg/ml的Nitricil™浓度)进行测试。图2显示测试的结果。软膏制剂T0-005在一小时之内针对铜绿假单胞菌是杀细菌的。图3比较了有和没有Nitricil™的T0-005制剂的时间杀灭。T0-005媒介物制剂没有表现出任何抗细菌活性(图3,底部)。实施例3Nitricil™如实施例2中所述产生。表7中所述的制剂如本文所述制备。表7:包含制剂T0-2的软膏通过用轻质矿物油代替Nitricil™而制备T0-2制剂的媒介物版本。所有制剂都含有如上所述的2%Nitricil™,且以用于测试的PBS中50mg/ml的稀释度(相当于1mg/ml的Nitricil™浓度)测试针对铜绿假单胞菌的时间杀灭。图4显示测试的结果。软膏制剂T0-2在一小时之内针对铜绿假单胞菌是杀细菌的,而媒介物则不是。实施例4在猪动物模型中评价根据本发明构思的局部软膏在减少伤口中的铜绿假单胞菌的功效。如所述制备软膏,其中关于2%和4%制剂如表1中所述。不具有Nitricil™的软膏被用作媒介物对照。在三只动物的脊柱旁和胸部区域制造测量为10mmx7mmx0.5mm深的矩形伤口。伤口彼此间隔15mm的未受伤的皮肤。将25μl含有106cfu/ml铜绿假单胞菌的悬浮液接种到每个伤口中。然后将所有伤口在接种的30分钟内用聚氨酯薄膜敷料(Tegaderm;3M,St.Paul,MN)来覆盖,并使其原地停留48小时。48小时后,将聚氨酯薄膜敷料去除,并回收三个伤口用于基线细菌计数。将剩余伤口分成四组,每组8个伤口,用200mg处理以覆盖受伤区域和周围未受伤皮肤。该处理通过用无菌特氟龙刮刀轻轻铺开,并用薄膜敷料重新包敷。处理应用后每天更换敷料。在受伤后第4天开始,如下面“回收部分”中所述在每天敷料更换前,从每组回收四个伤口。将三个伤口在接种后培养48小时,用于细菌的基线计数。对于每个治疗组,在治疗后第4和7天回收四个伤口。为了从伤口回收细菌,在伤口区域周围放置无菌手术钢筒(22mm内径)。将一(1)ml所有目的中和剂溶液移取到筒中,并用无菌特氟龙刮刀擦洗该部位30秒。从所有培养样品进行连续稀释,并使用SpiralPlater系统(SpiralBiotech,Norwood,MA)评估微生物污染的程度。使用具有CN补充的假单胞菌琼脂基料从伤口分离铜绿假单胞菌。将所有板在37℃有氧孵育过夜(24小时),其后计数活菌落数目。表8显示2%和4%软膏制剂、媒介物对照和未处理对照在第4天和第7天的计数结果。如表8中可见,2%和4%软膏两者到第7天都实现了显著的病原体减少。表8:软膏针对铜绿假单胞菌的功效。实施例5还利用2只动物在相同的猪动物模型中测试实施例4的制剂针对金黄色葡萄球菌MRSA的功效。实验程序如实施例4中所述,除了攻击病原体的改变。表9显示2%软膏、4%软膏、媒介物对照和未处理对照在第4天和第7天的结果。如表9中可见,2%和4%软膏两者到第7天都防止病原体生长并降低计数。表9:软膏针对金黄色葡萄球菌(Staphylococcusaureus)MRSA的功效。实施例6如美国专利申请公开号2009/0214618和2012年1月20日提交的题为“TemperatureControlledSol-GelCo-Condensation,”的PCT专利申请号PCT/US12/22048(其公开内容如同以其整体描述地通过引用并入本文)中所述制造包含MAP3的释放一氧化氮的大分子化合物(Nitricil™NVN1)。将所得大分子颗粒球磨,以提供8至10µm的平均粒度,以提供活性药物成分(API)。图5是Nitricil™NVN1和NVN4在pH7.4和37℃在释放的前200分钟的释放概况的图。Nitricil™NVN4是包含1:1比率的AEP3/TEOS的释放一氧化氮的大分子化合物,并且如美国专利申请公开号2009/0214618和2012年1月20日提交的题为“TemperatureControlledSol-GelCo-Condensation”的PCT专利申请号PCT/US12/22048中所述制造以提供API。下表10中提供了Nitricil™NVN1的整体释放动力学。表10:Nitricil™NVN1在pH7.4和37℃的半衰期和效力将Nitricil™NVN1配制为如表11中所述的两种成品剂型的软膏。表11:Nitricil™NVN1软膏制剂通过增加矿物油的量代替API的重量来配制安慰剂软膏。实施例7称重为22±2g的BALB/c来源的雄性小鼠由BioLascoTaiwan(在CharlesRiverLaboratories技术许可下)提供。在整个实验过程中将动物圈养在清洁区域下的独立通风笼架(IVCRacks,36MiniIsolator系统)中。将每5只小鼠保持在用高压釜灭菌的动物笼(以cm计,26.7长x20.7宽x14.0高)中,并以12小时光/暗周期保持在受控的温度(20-24℃)和湿度(50%-80%)下。所述动物可随意自由接触灭菌的标准实验室食物[MF-18(OrientalYeastCo.,Ltd.Japan)]和无菌自来水。通常根据实验室动物护理和使用指南(NationalAcademyPress,Washington,D.C.,2010)进行这项工作的所有方面,即动物的圈养、实验和处置。使用称重为22±2g的5只BALB/c雄性小鼠的组。通过将噁唑酮(100µL,丙酮中1.5%)应用于其预先剃净的腹部表面而使动物致敏。七天后,在噁唑酮(1%,20µL/耳)攻击之前30min和之后15min,将测试物质(20mg/耳)和媒介物(20μL/耳)局部施用于右耳的前表面和后表面。在噁唑酮攻击后24小时用Dyer模型微米测量计(micrometergauge)测量耳肿胀作为炎症指标。通过从右耳(处理的耳朵)减去左耳(正常对照)的厚度而计算耳水肿。根据下式计算抑制百分比:(Ic–It)/Icx100,其中Ic和It是指对照和处理小鼠中分别耳厚度(mm)的增加。使用单因素ANOVA和Dunnett氏检验来确定媒介物对照和处理组之间的统计显著性。显著性被设定在P<0.05。在BALB/c小鼠中的噁唑酮诱导的耳肿胀测定法(过敏性接触性皮炎模型)中评估实施例6中描述的测试物品(0.2%和2%Nitricil™NVN1软膏)的可能抗炎活性。在用第二次应用噁唑酮攻击之前30分钟和之后15分钟时各自局部(TOP)施用测试物质和媒介物。24小时后测量测试物质对耳肿胀的影响,结果总结在下表12中。表12:释放NO的组合物的体内抗炎功效注:负值表明没有抑制或刺激。使用单因素ANOVA和Dunnett氏检验来确定媒介物对照(或对应的安慰剂对照)和处理组之间的差异。*P<0.05,相对于媒介物A或对应的安慰剂对照。相对于媒介物对照A(丙酮/乙醇:1/1)和安慰剂软膏,0.2%和2%Nitricil™NVN1软膏的局部施用没有与显著抑制相关。安慰剂软膏处理没有表现出对噁唑酮诱导耳肿胀的显著效果。阳性对照地塞米松(0.1mg/耳×2)与噁唑酮诱导的耳肿胀的显著抑制相关。实施例8使用冷加工法,如表13中所述制备软膏制剂。选择这些制剂用于规模放大。表13:Nitricil™NVN1软膏制剂(TO-007和TO-008)。使用具有8-L混合容器的罗斯双轴混合机(RossDualShaftMixer)(型号:CDA-2)将表13中提供的制剂的开发过程中使用的实验室规模过程规模放大至5.5-kg规模。搅拌和均质化系统含有两个独立驱动的顶部进入搅拌器,如下所述:1.以约23-225rpm的速度范围驱动的三翼锚式搅拌器。锚被设计为具有三角形横截面,并且包括用于擦拭混合罐的侧壁和底部的固定的特氟龙刮板。2.以约1,000-10,000rpm的速度范围驱动的高速分散机,2”直径叶片。制造四批软膏,以确定材料添加的顺序,以及合适的混合速度(锚式搅拌器和高速分散机),和用于小规模过程的混合时间。表14中提供了开发批次制剂的总结,图6中提供了制造的过程流程图。表14:用于局部软膏的批次配方表15和16中提供了所述批次的分析结果。表15:用于安慰剂软膏的分析结果。测试建议的说明方法参考结果外观无色至灰白色,半透明至不透明软膏METH-014白色,不透明的凝胶通过HPLC不存在NVN1从样品色谱来看不存在NVN1METH-058符合含水量报道值,%w/wMETH-0030.2表观pH报道值METH-0066.4表16:对于2%、6%、12%和20%Nitricil™NVN1软膏的分析结果。实施例9在BALB/c小鼠中评价Nitricil™软膏,以确定Nitricil™软膏在体内的潜在抗炎特性。称重为22±2g的BALB/c衍生的雄性小鼠由BioLascoTaiwan(在CharlesRiverLaboratories技术许可下)提供。在整个实验过程中将动物容纳在清洁区域下的独立通风笼架(IVCRacks,36MiniIsolator系统)中。将每5只小鼠保持在用高压釜灭菌的动物笼(以cm计,26.7长x20.7宽x14.0高)中,并以12小时光/暗周期保持在受控的温度(20-24℃)和湿度(50%-80%)下。所述动物可随意自由接触灭菌的标准实验室食物[MF-18(OrientalYeastCo.,Ltd.Japan)]和无菌自来水。通常根据实验室动物护理和使用指南(NationalAcademyPress,Washington,D.C.,2011)进行这项工作的所有方面,即动物的圈养、实验和处置。在本研究中测试Nitricil™局部软膏(1%和4%)和安慰剂软膏。表17中提供了Nitricil™局部软膏制剂和安慰剂软膏的组合物。地塞米松(0.1mg/耳)被用作阳性对照。地塞米松是一种用于治疗各种炎症和自身免疫性疾病的有效糖皮质激素类固醇。表17:本研究中使用的软膏制剂的组合物。所用的测试系统是7天噁唑酮诱导的耳肿胀测定法。噁唑酮诱导的耳肿胀可用作炎症模型。噁唑酮是诱导迟发型超敏反应的过敏原,并且因此最常用作适应性免疫应答驱动的炎症模型(例如,过敏性接触性皮炎,牛皮癣等)。在该测定法中,通过将噁唑酮一次局部应用于其预先剃净的腹部表面而使小鼠(每组5只)对噁唑酮(100µL,丙酮中1.5%)致敏。七天后,通过将噁唑酮第二次应用于耳朵而攻击动物。在第二次噁唑酮(1%,20µL/耳)攻击之前30min和之后15min(引发阶段),将测试物品(20mg/小鼠)和媒介物(20μL/耳)局部(TOP)施用于右耳的前表面和后表面。在噁唑酮攻击后24小时用Dyer模型微米测量计测量耳肿胀作为炎症指标。通过从右耳(处理的耳朵)减去左耳(正常对照)的厚度而计算耳水肿(表18)。额外组用地塞米松(已知的抗炎剂)(阳性对照)处理,以验证测定有效性。根据下式计算抑制百分比:(Ic–It)/Icx100,其中Ic和It是指对照和处理小鼠中分别耳厚度(mm)的增加。使用单因素ANOVA和Dunnett氏检验来确定媒介物对照和处理组之间的统计显著性。显著性被设定在P<0.05。表18:处理信息和肿胀结果。注:使用单因素ANOVA和Dunnett氏检验来确定安慰剂/媒介物对照和处理组之间的差异。*P<0.05,相对于媒介物(丙酮/乙醇/1:1)。†P<0.05,相对于安慰剂软膏。相对于媒介物对照(丙酮/乙醇:1/1)和安慰剂软膏两者,1%Nitricil™NVN1局部软膏和4%Nitricil™NVN1局部软膏的局部施用都与噁唑酮诱导的耳肿胀的显著(P<0.05)抑制相关。1%Nitricil™NVN1局部软膏相对于丙酮/乙醇媒介物使耳肿胀抑制57%,而相对于安慰剂软膏抑制58%。4%Nitricil™NVN1局部软膏相对于丙酮/乙醇媒介物使耳肿胀抑制59%,而相对于安慰剂软膏抑制60%。相对于丙酮/乙醇媒介物,安慰剂软膏对耳肿胀没有影响。相对于丙酮/乙醇媒介物,地塞米松(阳性对照)使耳肿胀抑制86%。与安慰剂软膏对照或媒介物(丙酮/乙醇:1/1)相比,以1%和4%的Nitricil™局部软膏的局部施用引起小鼠中噁唑酮诱导的耳肿胀的显著(P<0.05)抑制。因此,1%和4%的Nitricil™局部软膏两者显著抑制过敏性接触性皮炎的体内模型中的炎症。在该测试的条件下,4%Nitricil™NVN1局部软膏没有比1%Nitricil™NVN1局部软膏更显著有效。相对于媒介物对照(丙酮/乙醇:1/1),安慰剂软膏组不具有任何影响。阳性对照地塞米松(0.1mg/小鼠×2)与小鼠中噁唑酮诱导的耳肿胀的显著抑制相关。表19显示来自本研究、实施例7中所述的研究、和如实施例8中所述的用Nitricil™软膏制剂的随后研究的噁唑酮诱导的耳肿胀结果的百分比抑制与乙醇/丙酮媒介物制剂或安慰剂制剂的比较。对于Nitricil™NVN4软膏制剂,所述制剂与实施例8中对于Nitricil™NVN1软膏制剂提供的那些类似,所述Nitricil™NVN1软膏制剂具有对于轻质矿物油进行的微小调整,以负责制剂中Nitricil™的量的差异。表19:来自各个研究的噁唑酮诱导的耳肿胀结果的百分比抑制的比较。*相对于乙醇/丙酮媒介物或安慰剂制剂的显著(P<0.05)抑制。实施例10在猪部分-厚度伤口模型中的伤口愈合研究使用Nitricil™NVN1软膏,诸如实施例8中所述的TO-007软膏制剂,在猪模型中处理部分厚度伤口。用以下制剂处理部分厚度伤口:含有0.1%、0.5%、1%、或4%Nitricil™NVN1的软膏制剂,媒介物软膏,用于标准闭合(occlusion)的Tegaderm作为阳性对照,或置于空气暴露作为阴性对照。来自八-动物伤口愈合研究的结果显示于图7中。较低剂量,0.1%和0.5%Nitricil™NVN1软膏,表明再上皮形成的快得多的速率。最低剂量(0.1%)的所有20个伤口到第6天完全愈合,比对应的软膏媒介物或Tegaderm闭合的护理标准快2整天。尽管没有在热损伤模型中收集该数据,但这清楚地表明一氧化氮刺激更快愈合的能力。在伤口形成后第2天、第4天和第7天从每个处理组中的所有动物取两个活检样品。通过伤口的中心(包括两侧的正常邻近皮肤)获得用于组织学的楔形活检样品。从伤口的另一半取钻取活检样品,用于RNA分离和随后的RT-PCR分析。对于任何处理组没有观察到上皮厚度的任何差异,显示出调节的愈合过程和上皮中细胞没有过度增殖。与其它处理组相比,用0.5%Nitricil™NVN1软膏处理的伤口在第2天表达升高水平的IL-8mRNA(图8)。用0.5%Nitricil™NVN1软膏处理2天后,IL-8(中性粒细胞化学引诱物)的表达在伤口中被显著诱导(p≤0.05)。一氧化氮可以激活IL-8启动子,且IL-8进而可以抑制中性粒细胞中iNOS的表达。这种信号传导作用足以促进愈合,但没有促进中性粒细胞的过度招募,并引起持续的炎症应答(图9)。对于任何治疗,经由组织学测量的白细胞浸润不是显著性差异的。以上是说明本发明的,而不应被解释为限制本发明。本发明由以下权利要求以及其中包括的权利要求的等同方案所定义。本文引用的所有出版物、专利申请、专利、专利公开和其它参考文献对于其中提供参考的句子和/或段落相关的教导通过引用以其整体并入本文。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1