一种大黄酸酯衍生物纳米混悬剂及其制备方法与流程
本发明涉及医药领域,具体涉及一种大黄酸酯衍生物纳米混悬剂及其制备方法。
背景技术:
大黄酸是从大黄、何首乌、番泻叶、芦荟等中药中提取分离出来或经适当的化学反应制备而得,具有抗炎、抗肿瘤、抗菌、抗病毒、抗氧化、降糖调脂、保肝抗纤维化等多种药理活性,它具有类似四环素类的结构特征,具有碱金属粒子络合能力,使其具有亲骨性,并且在治疗骨关节炎、糖尿病肾病等疾病及协同抗肿瘤方面表现突出。大黄酸在水,乙醇等溶剂中溶解性差,无法将其制成注射剂;而口服大黄酸后体内消除较快,从而导致生物利用度低,限制了其在临床上的应用。近几年来关于大黄酸的结构修饰方面主要集中在7位酚羟基和三位羧基的,七位酚羟基主要是形成醚键或者与羧基发生酯化,从而提高大黄酸的抗炎活性,三位羧基多形成酰胺键和酯键。目前关于形成酰胺键的研究较多,主要用于提高大黄酸的抗肿瘤作用,而将大黄酸酯化,然后改变它的理化性质,再将其制成制剂的研究目前还没有出现过。
纳米混悬剂系采用少量表面活性剂稳定纯药物粒子所形成的一种亚微米胶体分散体系,纳米混悬剂能增加难溶性药物溶出度、提高药物的生物利用度、增加药物的稳定性及提高药效,同时纳米混悬剂中“纯的”药物纳米晶对胃肠道和黏膜组织具有较好的黏附性,可以延长药物在体内的滞留时间,进一步提高生物利用度。纳米混悬剂中的药物粒子通常以无定型和晶体两种形式存在,药物粒子的不同形态对于药物的溶出以及吸收有很大的影响。
技术实现要素:
本发明目的是提供一种大黄酸酯衍生物纳米混悬剂及其制备方法,采用大黄酸酯衍生物的形式提高大黄酸的脂溶性,再以大黄酸酯衍生物纳米混悬剂的形式提高其水溶性,最终提高大黄酸的生物利用度。
本发明采用的技术方案为:
一种大黄酸酯衍生物纳米混悬剂,由稳定剂和大黄酸酯衍生物组成,所述大黄酸酯衍生物为化合物A或化合物B,化合物A的结构式(Ⅱ)和化合物B的结构式(Ⅲ)如下所示:
所述的一种大黄酸酯衍生物纳米混悬剂,按重量百分比计算,包含1.8%-76%的大黄酸酯衍生物,5%-66%的稳定剂。
所述的一种大黄酸酯衍生物纳米混悬剂,所述稳定剂为泊洛沙姆、磷脂、十二烷基硫酸钠、聚乙烯吡咯烷酮PVP、维生素E聚乙二醇琥珀酸酯中的一种或两种以上组合。
所述的一种大黄酸酯衍生物纳米混悬剂,所述稳定剂为泊洛沙姆和磷脂复配,且二者的重量比为1:5~5:1。
所述的一种大黄酸酯衍生物纳米混悬剂,所述泊洛沙姆的型号为泊洛沙姆F68,泊洛沙姆F87,泊洛沙姆F108或泊洛沙姆F127。
所述的一种大黄酸酯衍生物纳米混悬剂,所述磷脂为大豆卵磷脂,蛋黄卵磷脂或氢化大豆磷脂。
所述的大黄酸酯衍生物纳米混悬剂的制备方法,步骤如下:
1)将大黄酸酯衍生物溶于有机溶剂中,得有机相;
2)将稳定剂溶于去离子水中,得到水相;
3)在冰水浴中,将有机相缓慢的滴加到水相中,搅拌10~30min,得到初混悬液;
4)在10000~20000转速下剪切1~5min进行预分散;
5)预分散后的混悬液经微射流在6900~14000psi压力下均质2~25次,最后得到大黄酸酯衍生物纳米混悬剂。
所述的制备方法,步骤1)的有机溶剂为乙醇,二氯甲烷,乙酸乙酯,二甲基甲酰胺DMF或二甲基亚砜DMSO。
本发明具有以下有益效果:
本发明可通过沉淀法和分散方法联用制备纳米混悬剂,分散方法为高速剪切,超声探头法,高压均质法中的任意一种或多种。本发明通过采用大黄酸3位羧基的酯化,引入酯键和不同长度的碳链,从而可以提高化合物的脂溶性,改变其油水分配系数;并以该大黄酸酯衍生物作为有效成分研制出了纳米混悬剂。大黄酸酯衍生物纳米混悬剂的制备解决了大黄酸生物利用低,临床应用开发困难的问题。本发明采用大黄酸酯衍生物,可以改善化合物的理化性质,对大黄酸的临床应用开发有重要意义。且本发明使用少量稳定剂和有机溶剂,安全性高,工艺简单。
本发明载药量高,适合临床上大剂量使用的药物。通过大鼠体内药动实验的研究,本发明能提高大黄酸的生物利用度。
本发明将大黄酸酯衍生物的合成同制剂结合,从化学性质因素和物理因素两方面进行研究,最后使得大黄酸的生物利用度得到较大范围的提高。本发明为难溶于水,难溶于有机溶剂的化合物临床应用提供了思路。
附图说明
图1为实施例10制备的大黄酸丁二醇酯纳米混悬剂(RH-BE-NS)透射电镜图。
图2为实施例10制备的大黄酸丁二醇酯纳米混悬剂粒径分布图。
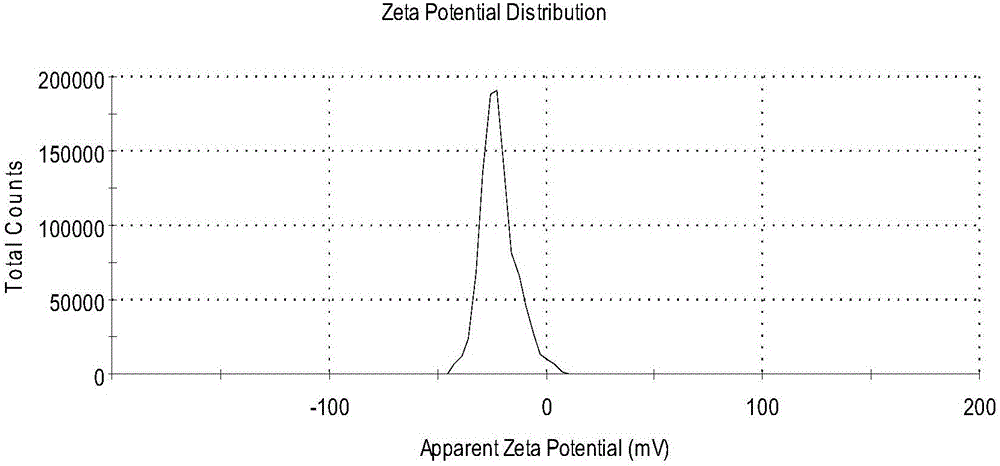
图3为实施例10制备的的大黄酸丁二醇酯纳米混悬剂的Zeta电位图。
图4表示本发明的大黄酸、大黄酸酯衍生物、大黄酸丁二醇酯纳米混悬剂药时曲线图。
图5表示本发明制备的大黄酸乙二醇酯(RH-EE)质谱图。
图6表示本发明制备的大黄酸丙二醇酯(RH-PE)质谱图。
图7表示本发明制备的大黄酸丁二醇酯(RH-BE)质谱图。
图8表示本发明制备的大黄酸一缩二乙二醇酯(RH-DE)质谱图。
图9表示本发明制备的大黄酸三缩四乙二醇酯(RH-TE)质谱图。
具体实施方式
大黄酸衍生物的制备
大黄酸与HO(CH2)nOH或H(OCH2CH2)nOH通过酯化反应形成羧酸酯衍生物。
具体步骤如下:将大黄酸和H(OCH2CH2)nOH或者HO(CH2)nOH加入到有机溶剂(甲苯或氯仿)中,然后缓慢滴加催化剂,80-90℃搅拌反应2~8h。待反应液降至室温,倒入冰水中,抽滤,得到滤液和滤饼。滤液中加入二氯甲烷萃取,直至二氯甲烷层不再有颜色,合并二氯甲烷层,干燥,减压浓缩得到产物为黄色固体。滤饼用饱和碳酸氢钠溶液多次打浆,洗去残留的大黄酸,过滤,滤饼干燥,得产物,产物均为黄色固体即大黄酸衍生物。由图5-9可知:上述方法成功合成了大黄酸酯衍生物-大黄酸乙二醇酯、大黄酸丙二醇酯、大黄酸丁二醇酯、大黄酸一缩二乙二醇酯和大黄酸三缩四乙二醇酯。
核磁结果:
大黄酸乙二醇酯(RH-EE)
1H NMR(600MHz,CDCl3)δ12.04(s,1H,OH),11.97(s,1H,OH),8.43(s,1H,Ar-H),7.97(s,1H,Ar-H),7.89(d,J=7.2Hz,1H,Ar-H),7.76-7.73(m,1H,Ar-H),7.35(d,J=8.4Hz,1H,Ar-H),4.54(t,2H,CH2CH2OH),4.03(t,2H,CH2CH2OH)。
大黄酸丙二醇酯(RH-PE)
1H NMR(600MHz,CDCl3)δ12.04(s,1H,OH),11.97(s,1H,OH),8.43(s,1H,Ar-H),7.97(s,1H,Ar-H),7.89(d,J=7.2Hz,1H,Ar-H),7.76-7.73(m,1H,Ar-H),7.35(d,J=8.4Hz,1H,Ar-H),4.54(t,2H,CH2CH2OH),4.03(t,2H,CH2CH2OH)。
大黄酸丁二醇酯(RH-BE)
1H NMR(600MHz,CDCl3)δ12.03(s,1H),11.98(s,1H),8.42(s,1H,Ar-H),7.94(s,1H,Ar-H)7.88(d,J=7.4Hz,1H,Ar-H),7.75-7.73(m,1H,Ar-H),7.34(d,J=8.4Hz,1H,Ar-H),7.27(s,1H,Ar-H),4.44(t,2H,COOCH2),3.76(t,2H,COOCH2CH2),1.95–1.91(m,2H,CH2OH),1.78–1.74(m,2H,CH2CH2OH).
大黄酸一缩二乙二醇酯(RH-DE)
1H NMR(600MHz,CDCl3)δ12.04(s,1H,OH),11.97(s,1H,OH),8.42(s,1H,Ar-H)7.94(s,1H,Ar-H),7.88(d,J=7.2Hz,1H,Ar-H),7.75-7.73(m,1H,Ar-H),7.35(d,J=8.4Hz,1H,Ar-H),4.58–4.55(m,COOCH2),3.90–3.88(m,2H,COOCH2CH2O),3.81–3.79(m,2H,CH2CH2OH),3.70–3.68(m,2H,CH2CH2OH),1.88(s,1H,CH2CH2OH).
大黄酸三缩四乙二醇酯(RH-TE)
1H NMR(600MHz,CDCl3)δ12.03(s,1H,OH),11.97(s,1H,OH),8.44(s,1H,Ar-H),7.97(s,1H,Ar-H),7.88(d,J=7.4Hz,1H,Ar-H),7.75-7.73(m,1H,Ar-H),7.34(d,J=8.4Hz,1H,Ar-H),4.56–4.54(m,2H,COOCH2CH2),3.89–3.87(m,2H,COOCH2CH2),3.74-3.72(m,12H).
本发明大黄酸衍生物纳米混悬剂制备实例(其中大黄酸衍生物以大黄酸丁二醇酯为例)
实施例1一种大黄酸酯衍生物纳米混悬剂
制备方法如下:
1)将10mg大黄酸丁二醇酯溶于1mL DMF中,超声至完全溶解后得到溶液A。
2)称取25mg的F68和25mg的大豆卵磷脂置于50mL去离子水中,超声至完全溶解得到溶液B。
3)将溶液B置于冰水浴中,在搅拌的条件下,将溶液A缓慢的加入溶液B中,搅拌30min,将得到的初混悬剂。
4)在20000转速下剪切2min。
5)然后马上在11850psi压力下进行微射流,循环15次,最后得到大黄酸酯衍生物纳米混悬剂,测得粒径为235nm,PDI为0.124,Zeta电位为-20.9mv。
实施例2一种大黄酸酯衍生物纳米混悬剂
方法同实施例1,仅将步骤1)中的有机溶剂由DMF改为乙醇,最后得到大黄酸酯衍生物纳米混悬剂,测得粒径为432nm,PDI为0.276,Zeta电位为-17.1mv。
实施例3一种大黄酸酯衍生物纳米混悬剂
方法同实施例1,仅将步骤1)中的有机溶剂由DMF改为二氯甲烷,最后得到大黄酸酯衍生物纳米混悬剂,测得粒径为500nm左右,PDI位0.875,Zeta点位为-21.5mv。
由实施例1-3可知,以DMF为有机溶剂时,制备出的纳米混悬剂粒径和PDI最小。当以二氯甲烷或乙醇为有机溶剂时,制备出的纳米混悬剂较DMF粒径分布较宽,PDI值较大;粒径都是400nm以上;但是以二氯甲烷和乙醇为有机溶剂时,有机溶剂较易旋出,有机溶剂残留较少;以乙醇为有机溶剂时制备出的纳米混悬剂较二氯甲烷为有机溶剂时制备出的纳米混悬剂稳定。
实施例4利用沉淀法制备大黄酸酯衍生物纳米混悬剂
将50mg大黄酸丁二醇酯溶于1mLDMF中得到溶液A。称取25mg泊洛沙姆F68,20mg卵磷脂溶于50mL水中得到溶液B。将溶液A缓慢滴加到溶液B中,搅拌30min,最后得到大黄酸酯衍生物混悬剂,测得其粒径为760nm,PDI为0.964,Zeta电位-21.8mv。
实施例5利用直接高压均质法制备大黄酸酯衍生物纳米混悬剂
此法不使用有机溶剂,处方量同实施例3,直接将50mg的大黄酸丁二醇酯加入到含有稳定剂的溶液B中,然后在20000转速下剪切4min,立即在11859psi压力下进行微射流,循环15次,最后制备出大黄酸酯衍生物纳米混悬剂。此法制备出的纳米混悬剂安全无毒,然而在制备的过程中会有损失,堵塞微射流,且制剂不稳定,容易聚集。测得其粒径为348m,PDI为0.453,Zeta电位-19.5mv。
实施例6利用超声探头法制备大黄酸酯衍生物纳米混悬剂
称取50mg大黄酸丁二醇酯置于1mL DMF中,超声至完全溶解得到溶液A。然后称取25mg F68,20mg卵磷脂置于40mL去离子水中,超声至完全溶解得到溶液B。将溶液A缓慢滴加到溶液B中,搅拌30min,立即用超声探头在315W的功率下超声4min,最后得到大黄酸酯衍生物纳米混悬剂。此法得到的纳米混悬剂虽然简单,药量损失少,但是得到制剂不稳定,易聚沉。测得其粒径为495nm,PDI为0.688,Zeta电位-23.5mv。
实施例7利用本发明沉淀法与高压均质结合法制备大黄酸酯衍生物纳米混悬剂
1)称取50mg大黄酸丁二醇酯置于1mL DMF中,超声至完全溶解得到溶液A。
2)称取25mg F68、20mg卵磷脂置于40mL去离子水中,超声至完全溶解得到溶液B。
3)将溶液B置于冰水浴中,在搅拌的条件下,将溶液A缓慢滴加到B中,搅拌30min,将得到的初混悬剂。
4)然后在20000转速下剪切4min。
5)立即在11859psi压力下进行微射流,循环15次,最后制备出大黄酸酯衍生物纳米混悬剂,沉淀法与高压均质法结合制备出的纳米混悬剂稳定,不易聚沉,且PDI较小,粒径均一,粒径为234nm,PDI为0.165,Zeta电位-21.5mv。
由实施例4-7,以直接高压均质法制备纳米混悬剂的方法简单易行,且不使用有机溶剂,但是对药物的损失较大,且容易改变药物的浓度,同时容易堵塞微射流,对于机器的损耗较大。以超声探头法制备纳米混悬剂是沉淀法与超声探头结合的方法,对于药物无损失,也不会改变药的浓度,但是制备出的制剂粒径不均匀。以沉淀法和高压均质法结合制备出纳米混悬剂的稳定性好,粒径均匀,对微射流损耗较小,但是需要使用有机溶剂,且药物损失较多。
实施例8一种大黄酸酯衍生物纳米混悬剂
方法同实施例7,仅将步骤1)中大黄酸丁二醇酯用量改为100mg最后得到纳米混悬剂,测得其粒径233.5nm,PDI为0.254,Zeta电位为-17.6mv。
实施例9一种大黄酸酯衍生物纳米混悬剂
方法同实施例7,仅将步骤1)中大黄酸丁二醇酯用量改为200mg最后得到纳米混悬剂,测得其粒径474.3nm,PDI为0.308,Zeta电位为-19.8mv。
由实施例7、8和9可知,大黄酸丁二醇酯的不同含量是影响纳米混悬剂粒径以及PDI的一个关键因素,随着大黄酸丁二醇酯含量的增加,纳米混悬剂的粒径以及PDI随之增大,Zeta电位随之减小。由于稳定剂是黏附在药物粒子的表面,以及分散在水溶液中,使得药物粒子不会发生聚集,保持一个稳定平衡状态。但是随着药物浓度增大,平衡被破坏,药物粒子虽然在高压均质下减小,但是会快会聚集,使得粒径增大,PDI值增大。
实施例10一种大黄酸酯衍生物纳米混悬剂
方法同实施例7,仅将步骤2)中的稳定剂25mg F68、20mg卵磷脂改为20mg F68、10mg卵磷脂,最后得到纳米混悬剂,测得粒径为256.3nm,PDI为0.154,Zeta电位为-22.6mv。
由图1可知,大黄酸丁二醇酯纳米混悬剂的药物粒子呈球形,粒子之间不黏连,粒径在230nm左右,说明大黄酸丁二醇酯纳米混悬剂稳定,未发生聚集。由图2可知,大黄酸丁二醇酯纳米混悬剂的粒径在230nm左右,与透射电镜测定结果接近。由图3可知,大黄酸丁二醇酯纳米混悬剂的Zeta电位绝对值大于20。
实施例11一种大黄酸酯衍生物纳米混悬剂
方法同实施例7,仅将步骤2)中的稳定剂25mg F68、20mg卵磷脂改为20mg F127、10mg卵磷脂,最后得到纳米混悬剂。最后得到纳米混悬剂,测得粒径为427nm,PDI为0.613,Zeta电位为-17.1mv。
实施例12一种大黄酸酯衍生物纳米混悬剂
方法同实施例7,仅将步骤2)中的稳定剂25mg F68、20mg卵磷脂改为20mg F68、10mg维生素E聚乙二醇琥珀酸酯,最后得到纳米混悬剂。测得粒径为496.5nm,PDI为0.482,Zeta电位为-20.8mv。
由实施例7、10-12可知,不同的稳定剂种类对于纳米混悬剂的粒径、Zeta电位,PDI的影响较大。F127和F68都属于亲水性泊洛沙姆,但是以F127和卵磷脂合用时,所制备出的纳米混悬剂粒径及PDI较F68大;以维生素E聚乙二醇琥珀酸酯和卵磷脂合用时,所制备出的纳米混悬剂粒径较大,但Zeta电位与F68结果接近,维生素E聚乙二醇琥珀酸酯虽然可以作为乳化剂,稳定剂,增溶剂,同时可以促进药物吸收,但是其不利于制备较小粒径的纳米混悬剂。
由上可知:本发明的优选配方如下:
表1为优选配方
大黄酸酯衍生物及大黄酸油水分配系数的测定
取200mL正辛醇,用等量的蒸馏水饱和(在振荡器中震荡24h),分别称取定量大黄酸以及大黄酸酯衍生物,将其分别加入到5mL蒸馏水饱和的正辛醇中,最后得到药物的正丁醇溶液,再加入等量的正辛醇饱和的水溶液,将其置于空气振荡器中,震荡24h,温度为常温,震荡速度,155rpm。最后用液相测定油相的浓度,然后根据油相计算出水相的药物浓度。
表2油水分配系数
结果分析,除了一缩二乙二醇酯衍生物外,其它产物的油水分配系数均大于大黄酸。大黄酸、大黄酸酯衍生物、大黄酸酯衍生物纳米混悬剂的体内药动学。
以大黄酸丁二醇酯纳米混悬剂为例研究
取42只SD大鼠,体重为250g±40g,随机分成7组,每组6只,给药前禁食12小时,但是自由饮水,给药前称重,7组大鼠分别口服灌胃为大黄酸(混悬于0.5%的CMCNa溶液),大黄酸酯衍生物(混悬于0.5%的CMCNa溶液),大黄酸丁二醇酯纳米混悬剂,然后于5min,10min,15min,20min,30min,1h,2h,3h,4h,6h,8h,10h,12h,24h眼眶取血1mL,置于肝素管中,在10000转速下离心10min,取上清,然后处理样品,用液相测定大黄酸的血药浓度。
表3药动数据参数
从表3中可以看出,大黄酸酯衍生物,随着碳链的增长,AUC值逐渐增大。
大黄酸三缩四乙二醇酯与其他酯衍生物相比,它对于大黄酸的AUC提高的最多,提高了2.17倍,但是它的Tmax最小,这是由于不仅引入了酯键和长碳链,还引入了醚键和羟基,它们与水形成氢键从而导致水溶性和脂溶性都有所提高导致的。
由上可知,大黄酸丁二醇酯纳米混悬剂大鼠口服灌胃后得到大黄酸的AUC为84.09,与大黄酸原料药(AUC0-∞=34.12)相比提高了2.46倍。
图4表示本发明的大黄酸、大黄酸酯衍生物、大黄酸丁二醇酯纳米混悬剂药时曲线图。由图4可知,将大黄酸酯衍生物制成纳米混悬剂后,大黄酸酯衍生物在体内可以迅速断开,形成大黄酸,从而使大黄酸的生物利用度得到了明显的提高。
- 还没有人留言评论。精彩留言会获得点赞!