一种在碳纤维表面接枝羟基封端超支化聚合物的方法与流程
本发明涉及一种碳纤维表面的改性方法。
背景技术:
近年来,碳纤维(cf)由于其高强度、高模量而被广泛用作先进复合材料的理想增强体,碳纤维增强聚合物(cfrp)复合材料的性能很大程度上依赖于界面性能,高质量界面是载荷从树脂均匀转移到碳纤维的保障。但由于碳纤维表面光滑呈现化学惰性、表面能低,与树脂基体的浸润性和粘结性差,最终影响了复合材料的界面性能及其它性能的发挥,所以碳纤维表面处理对其增强材料的发展至关重要,目前未改性的碳纤维与树脂基体界面结合弱,未改性的碳纤维复合材料的界面剪切强度约为49mpa,因此需要对碳纤维的表面进行改性,目前对碳纤维的表面改性技术主要包括表面氧化技术、化学气相沉积技术以及表面涂层技术等,这些方法基本能改善碳纤维与树脂的浸润性,但是存在生产工艺复杂,成本高和反应时间较长等问题。因此,目前我们致力于找到一种条件温和、绿色环保、高效的碳纤维表面改性方法,旨在避免单丝纤维本体强度下降的同时,在其表面形成更多的活性位点,来提高碳纤维与树脂的界面结合强度。
接枝聚合物技术作为一个新兴的研究方向,是将大分子接枝在碳纤维表面上,以改善碳纤维表面活性和粗糙度,进而改善其界面结合性能。羟基封端超支化聚合物(hthbp)由于具有大量的末端极性羟基、孔洞结构和高树脂相容性而受到广泛的关注,它不仅提供了能与碳纤维和树脂基体反应的大量活性位点,而且会促进固化剂和环氧树脂的反应,因此可成为功能化碳纤维的理想材料,但存在羟基封端超支化聚合物合成工艺较为复杂和合成原料有毒等问题。
技术实现要素:
本发明的目的是要解决碳纤维和树脂基体界面结合弱和羟基封端超支化聚合物的合成工艺复杂,合成原料有毒,制约其改性碳纤维的问题,而提供一种在碳纤维表面接枝羟基封端超支化聚合物的方法。
一种在碳纤维表面接枝羟基封端超支化聚合物的方法,是按以下步骤完成的:
一、制备羟基封端超支化聚合物:
①、将异佛尔酮二异氰酸酯和n,n-二甲基乙酰胺加入到玻璃容器中,得到异佛尔酮二异氰酸酯溶液;再将装有异佛尔酮二异氰酸酯溶液的玻璃容器置于0℃~5℃的冰水浴中;
步骤一①中所述的异佛尔酮二异氰酸酯的质量与n,n-二甲基乙酰胺的体积比为(6g~12g):50ml;
②、将三羟甲基氨基甲烷加入到n,n-二甲基乙酰胺中,再超声混合10min~20min,得到三羟甲基氨基甲烷溶液;
步骤一②中所述的三羟甲基氨基甲烷的质量与n,n-二甲基乙酰胺的体积比为(3g~6g):50ml;
③、将步骤一①中置于0℃~5℃的冰水浴的玻璃容器中的异佛尔酮二异氰酸酯溶液进行搅拌,搅拌速度为300r/min~400r/min,再将三羟甲基氨基甲烷溶液以10滴/min~15滴/min的滴加速度滴加到温度为0℃~5℃、搅拌速度为300r/min~400r/min的异佛尔酮二异氰酸酯溶液中,得到反应液ⅰ;
步骤一③中所述的异佛尔酮二异氰酸酯溶液与三羟甲基氨基甲烷溶液的体积比为1:1;
④、将反应液ⅰ的温度升温至5℃~10℃,再在温度为5℃~10℃下搅拌反应5h~6h,再将反应液ⅰ的温度升温至35℃~40℃,再向35℃~40℃反应液ⅰ中加入二月桂酸二丁基锡,最后在温度为35℃~40℃和搅拌速度为300r/min~400r/min下搅拌反应10h~15h,得到反应液ⅱ;
步骤一④中所述的二月桂酸二丁基锡的质量与反应液ⅰ的体积比为(0.08g~0.12g):100ml;
⑤、将反应液ⅱ加入到去离子水中析出沉淀,再静置25min~30min,再在离心速度为6000r/min~8000r/min下进行离心,得到固体反应产物;
⑥、使用去离子水对固体反应产物清洗3次~5次,再将去离子水清洗后的固体反应产物放入冷冻干燥机中,再在温度为-10℃~-5℃下冷冻干燥36h~48h,得到干燥的白色固体物质,即为羟基封端超支化聚合物;
二、碳纤维的抽提处理:
①、将碳纤维放入装有丙酮的索氏提取器中,再将丙酮加热至75℃~85℃,丙酮不断蒸出并在索氏提取器中冷凝,使碳纤维表面的杂质在蒸馏的丙酮中不断得到清洗,清洗时间为48h~72h,再将碳纤维取出,得到去除表面杂质的碳纤维;将去除表面杂质的碳纤维取出,再置于温度为70℃~80℃的烘箱中干燥2h~4h,得到抽提处理后的碳纤维;
三、氧化:
①、将抽提处理后的碳纤维浸入到过硫酸钾/硝酸银混合水溶液中,加热至60℃~80℃,再在温度为60℃~80℃的条件下恒温1h~2h,得到氧化后的碳纤维;所述的过硫酸钾/硝酸银混合水溶液中过硫酸钾的浓度为0.1mol/l~0.2mol/l;所述的过硫酸钾/硝酸银混合水溶液中硝酸银的浓度为0.0001mol/l~0.05mol/l;
步骤三①中所述的抽提处理后的碳纤维的质量与过硫酸钾/硝酸银混合水溶液的体积比为(0.3g~1.5g):(300ml~500ml);
②、室温条件下将步骤三①得到的氧化后的碳纤维在蒸馏水中浸泡5min~10min,将经蒸馏水中浸泡后的碳纤维取出,弃除蒸馏水;
步骤三②中所述的氧化后的碳纤维的质量与蒸馏水的体积比为(0.3g~1.5g):(300ml~500ml);
③、重复步骤三②3次~5次,得到蒸馏水清洗后的氧化碳纤维;
④、将步骤三③得到的蒸馏水清洗后的氧化碳纤维在温度为70℃~80℃的条件下干燥2h~4h,得到干燥后的氧化碳纤维;
⑤、将步骤三④得到的干燥后的氧化碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃~100℃的条件下使用无水乙醇清洗氧化碳纤维,清洗时间为2h~4h,得到无水乙醇清洗的氧化的碳纤维;
⑥、将步骤三⑤得到的无水乙醇清洗的氧化的碳纤维在温度为70℃~80℃的条件下干燥2h~4h,得到干燥的氧化碳纤维;
四、接枝:
①、将羟基封端超支化聚合物加入到n,n-二甲基乙酰胺中,再超声分散15min~30min,然后加入干燥的氧化碳纤维、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和4-二甲氨基吡啶,最后在温度为100℃~150℃和搅拌速度为300r/min~400r/min的条件下搅拌回流12h~24h,得到处理后的碳纤维;
步骤四①中所述的羟基封端超支化聚合物的质量与n,n-二甲基乙酰胺的体积比为(1g~1.5g):(30ml~60ml);
步骤四①中所述的干燥的氧化碳纤维的质量与n,n-二甲基乙酰胺的体积比为(1.0~2.0g):(30ml~60ml);
步骤四①中所述的1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐的质量与n,n-二甲基乙酰胺的体积比为(0.5g~1.0g):(30ml~60ml);
步骤四①中所述的4-二甲氨基吡啶的质量与n,n-二甲基乙酰胺的体积比为(0.05~0.1g):(30ml~60ml);
②、将处理后的碳纤维取出后浸入到蒸馏水中10min~15min;
③、循环步骤四③3次~5次,得到蒸馏水清洗的处理后的碳纤维;
④、将蒸馏水清洗的处理后的碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃~100℃的条件下使用无水乙醇清洗蒸馏水清洗的处理后的碳纤维,清洗时间为2h~4h,得到无水乙醇清洗的处理后的碳纤维;
⑤、将无水乙醇清洗的处理后的碳纤维置于温度为75℃~80℃的烘箱中干燥3h~5h,得到表面接枝羟基封端超支化聚合物的碳纤维。
本发明的原理及优点:
一、本发明使用了无毒和高活性的三羟甲基氨基甲烷(toam)作为合成原料,大量缩短了合成时间,减少了环境污染;
二、本发明采用共价接枝法,将羟基封端超支化聚合物接枝到碳纤维表面,制备了一种不仅表面含有大量极性末端羟基并且易于与基体形成共价键的碳纤维;
三、由于羟基封端的超支化聚合物具有大量的极性羟基和孔洞结构,当被接枝到碳纤维表面后,碳纤维与树脂之间的浸润性和粘结性有显著提高,这将大大改善复合材料的界面性能,进而提高复合材料的力学性能和热稳定性;
四、本发明制备的表面接枝羟基封端超支化聚合物的碳纤维的界面剪切强度(ifss)由原丝(未接枝的碳纤维)的48.8mpa提高到60.9mpa~87.8mp,提高了25%~79.9%;层间剪切强度(ilss)由原丝(未接枝的碳纤维)的58.6mpa提高到69.7mpa~81.8mpa,提高了19%~39.6%。
本发明可获得一种在碳纤维表面接枝羟基封端超支化聚合物的方法。
附图说明
图1为实施例一步骤二③得到的抽提处理后的碳纤维的xps谱图;
图2为图1的分峰谱图,图中1为c1s(1),2为c1s(2),3为c1s(3);
图3为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的xps谱图;
图4为图3的分峰谱图,图中4为c-n,5为c-o,6为c=o,7为ncoo;
图5为实施例一步骤二③得到的抽提处理后的碳纤维的sem图;
图6为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的sem图;
图7为界面剪切强度柱状图,图中1为实施例一步骤二③得到的抽提处理后的碳纤维的界面剪切强度,2为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的界面剪切强度;
图8为层间剪切强度柱状图,图中1为实施例一步骤二③得到的抽提处理后的碳纤维的层间剪切强度,2为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的层间剪切强度。
具体实施方式
具体实施方式一:本实施方式是一种在碳纤维表面接枝羟基封端超支化聚合物的方法,是按以下步骤完成的:
一、制备羟基封端超支化聚合物:
①、将异佛尔酮二异氰酸酯和n,n-二甲基乙酰胺加入到玻璃容器中,得到异佛尔酮二异氰酸酯溶液;再将装有异佛尔酮二异氰酸酯溶液的玻璃容器置于0℃~5℃的冰水浴中;
步骤一①中所述的异佛尔酮二异氰酸酯的质量与n,n-二甲基乙酰胺的体积比为(6g~12g):50ml;
②、将三羟甲基氨基甲烷加入到n,n-二甲基乙酰胺中,再超声混合10min~20min,得到三羟甲基氨基甲烷溶液;
步骤一②中所述的三羟甲基氨基甲烷的质量与n,n-二甲基乙酰胺的体积比为(3g~6g):50ml;
③、将步骤一①中置于0℃~5℃的冰水浴的玻璃容器中的异佛尔酮二异氰酸酯溶液进行搅拌,搅拌速度为300r/min~400r/min,再将三羟甲基氨基甲烷溶液以10滴/min~15滴/min的滴加速度滴加到温度为0℃~5℃、搅拌速度为300r/min~400r/min的异佛尔酮二异氰酸酯溶液中,得到反应液ⅰ;
步骤一③中所述的异佛尔酮二异氰酸酯溶液与三羟甲基氨基甲烷溶液的体积比为1:1;
④、将反应液ⅰ的温度升温至5℃~10℃,再在温度为5℃~10℃下搅拌反应5h~6h,再将反应液ⅰ的温度升温至35℃~40℃,再向35℃~40℃反应液ⅰ中加入二月桂酸二丁基锡,最后在温度为35℃~40℃和搅拌速度为300r/min~400r/min下搅拌反应10h~15h,得到反应液ⅱ;
步骤一④中所述的二月桂酸二丁基锡的质量与反应液ⅰ的体积比为(0.08g~0.12g):100ml;
⑤、将反应液ⅱ加入到去离子水中析出沉淀,再静置25min~30min,再在离心速度为6000r/min~8000r/min下进行离心,得到固体反应产物;
⑥、使用去离子水对固体反应产物清洗3次~5次,再将去离子水清洗后的固体反应产物放入冷冻干燥机中,再在温度为-10℃~-5℃下冷冻干燥36h~48h,得到干燥的白色固体物质,即为羟基封端超支化聚合物;
二、碳纤维的抽提处理:
①、将碳纤维放入装有丙酮的索氏提取器中,再将丙酮加热至75℃~85℃,丙酮不断蒸出并在索氏提取器中冷凝,使碳纤维表面的杂质在蒸馏的丙酮中不断得到清洗,清洗时间为48h~72h,再将碳纤维取出,得到去除表面杂质的碳纤维;将去除表面杂质的碳纤维取出,再置于温度为70℃~80℃的烘箱中干燥2h~4h,得到抽提处理后的碳纤维;
三、氧化:
①、将抽提处理后的碳纤维浸入到过硫酸钾/硝酸银混合水溶液中,加热至60℃~80℃,再在温度为60℃~80℃的条件下恒温1h~2h,得到氧化后的碳纤维;所述的过硫酸钾/硝酸银混合水溶液中过硫酸钾的浓度为0.1mol/l~0.2mol/l;所述的过硫酸钾/硝酸银混合水溶液中硝酸银的浓度为0.0001mol/l~0.05mol/l;
步骤三①中所述的抽提处理后的碳纤维的质量与过硫酸钾/硝酸银混合水溶液的体积比为(0.3g~1.5g):(300ml~500ml);
②、室温条件下将步骤三①得到的氧化后的碳纤维在蒸馏水中浸泡5min~10min,将经蒸馏水中浸泡后的碳纤维取出,弃除蒸馏水;
步骤三②中所述的氧化后的碳纤维的质量与蒸馏水的体积比为(0.3g~1.5g):(300ml~500ml);
③、重复步骤三②3次~5次,得到蒸馏水清洗后的氧化碳纤维;
④、将步骤三③得到的蒸馏水清洗后的氧化碳纤维在温度为70℃~80℃的条件下干燥2h~4h,得到干燥后的氧化碳纤维;
⑤、将步骤三④得到的干燥后的氧化碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃~100℃的条件下使用无水乙醇清洗氧化碳纤维,清洗时间为2h~4h,得到无水乙醇清洗的氧化的碳纤维;
⑥、将步骤三⑤得到的无水乙醇清洗的氧化的碳纤维在温度为70℃~80℃的条件下干燥2h~4h,得到干燥的氧化碳纤维;
四、接枝:
①、将羟基封端超支化聚合物加入到n,n-二甲基乙酰胺中,再超声分散15min~30min,然后加入干燥的氧化碳纤维、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和4-二甲氨基吡啶,最后在温度为100℃~150℃和搅拌速度为300r/min~400r/min的条件下搅拌回流12h~24h,得到处理后的碳纤维;
步骤四①中所述的羟基封端超支化聚合物的质量与n,n-二甲基乙酰胺的体积比为(1g~1.5g):(30ml~60ml);
步骤四①中所述的干燥的氧化碳纤维的质量与n,n-二甲基乙酰胺的体积比为(1.0~2.0g):(30ml~60ml);
步骤四①中所述的1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐的质量与n,n-二甲基乙酰胺的体积比为(0.5g~1.0g):(30ml~60ml);
步骤四①中所述的4-二甲氨基吡啶的质量与n,n-二甲基乙酰胺的体积比为(0.05~0.1g):(30ml~60ml);
②、将处理后的碳纤维取出后浸入到蒸馏水中10min~15min;
③、循环步骤四③3次~5次,得到蒸馏水清洗的处理后的碳纤维;
④、将蒸馏水清洗的处理后的碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃~100℃的条件下使用无水乙醇清洗蒸馏水清洗的处理后的碳纤维,清洗时间为2h~4h,得到无水乙醇清洗的处理后的碳纤维;
⑤、将无水乙醇清洗的处理后的碳纤维置于温度为75℃~80℃的烘箱中干燥3h~5h,得到表面接枝羟基封端超支化聚合物的碳纤维。
本实施方式的原理及优点:
一、本实施方式使用了无毒和高活性的三羟甲基氨基甲烷(toam)作为合成原料,大量缩短了合成时间,减少了环境污染;
二、本实施方式采用共价接枝法,将羟基封端超支化聚合物接枝到碳纤维表面,制备了一种不仅表面含有大量极性末端羟基并且易于与基体形成共价键的碳纤维;
三、由于羟基封端的超支化聚合物具有大量的极性羟基和孔洞结构,当被接枝到碳纤维表面后,碳纤维与树脂之间的浸润性和粘结性有显著提高,这将大大改善复合材料的界面性能,进而提高复合材料的力学性能和热稳定性;
四、本实施方式制备的表面接枝羟基封端超支化聚合物的碳纤维的界面剪切强度(ifss)由原丝(未接枝的碳纤维)的48.8mpa提高到60.9mpa~87.8mp,提高了25%~79.9%;层间剪切强度(ilss)由原丝(未接枝的碳纤维)的58.6mpa提高到69.7mpa~81.8mpa,提高了19%~39.6%。
本实施方式可获得一种在碳纤维表面接枝羟基封端超支化聚合物的方法。
具体实施方式二:本实施方式与具体实施方式一不同点是:步骤一①中所述的异佛尔酮二异氰酸酯的质量与n,n-二甲基乙酰胺的体积比为(10g~12g):50ml。其它步骤与具体实施方式一相同。
具体实施方式三:本实施方式与具体实施方式一或二之一不同点是:步骤一②中所述的三羟甲基氨基甲烷的质量与n,n-二甲基乙酰胺的体积比为(5g~6g):50ml。其它步骤与具体实施方式一或二相同。
具体实施方式四:本实施方式与具体实施方式一至三之一不同点是:步骤一④中所述的二月桂酸二丁基锡的质量与反应液ⅰ的体积比为(0.1g~0.12g):100ml。其它步骤与具体实施方式一至三相同。
具体实施方式五:本实施方式与具体实施方式一至四之一不同点是:步骤四①中所述的羟基封端超支化聚合物的质量与n,n-二甲基乙酰胺的体积比为(1.2g~1.5g):(45ml~60ml)。其它步骤与具体实施方式一至四相同。
具体实施方式六:本实施方式与具体实施方式一至五之一不同点是:步骤一②中所述的超声功率为343w~346.5w。其它步骤与具体实施方式一至五相同。
具体实施方式七:本实施方式与具体实施方式一至六之一不同点是:步骤四①中所述的干燥的氧化碳纤维的质量与n,n-二甲基乙酰胺的体积比为(1.5~2.0g):(30ml~60ml)。其它步骤与具体实施方式一至六相同。
具体实施方式八:本实施方式与具体实施方式一至七之一不同点是:步骤四①中所述的1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐的质量与n,n-二甲基乙酰胺的体积比为(0.5g~0.67g):(50ml~60ml)。其它步骤与具体实施方式一至七相同。
具体实施方式九:本实施方式与具体实施方式一至八之一不同点是:步骤四①中所述的超声功率为343w~346.5w。其它步骤与具体实施方式一至八相同。
具体实施方式十:本实施方式与具体实施方式一至九之一不同点是:步骤四①中所述的4-二甲氨基吡啶的质量与n,n-二甲基乙酰胺的体积比为(0.05~0.067g):(50ml~60ml)。其它步骤与具体实施方式一至九相同。
采用以下实施例验证本发明的有益效果:
实施例一:一种在碳纤维表面接枝羟基封端超支化聚合物的方法是按以下步骤完成的:
一、制备羟基封端超支化聚合物:
①、将12g异佛尔酮二异氰酸酯和50mln,n-二甲基乙酰胺加入到三口烧瓶中,得到异佛尔酮二异氰酸酯溶液;再将装有异佛尔酮二异氰酸酯溶液的玻璃容器置于0℃的冰水浴中;
②、将6g三羟甲基氨基甲烷加入到50mln,n-二甲基乙酰胺中,再在超声功率为346.5w下超声混合20min,得到三羟甲基氨基甲烷溶液;
③、将步骤一①中置于0℃的冰水浴的玻璃容器中的异佛尔酮二异氰酸酯溶液进行搅拌,搅拌速度为400r/min,再将步骤一②中得到的三羟甲基氨基甲烷溶液以15滴/min的滴加速度滴加到温度为0℃、搅拌速度为400r/min的异佛尔酮二异氰酸酯溶液中,得到反应液ⅰ;
④、将反应液ⅰ的温度升温至5℃,再在温度为5℃下搅拌反应6h,再将反应液ⅰ的温度升温至40℃,再向40℃反应液ⅰ中加入0.1g二月桂酸二丁基锡,最后在温度为40℃和搅拌速度为400r/min下搅拌反应15h,得到反应液ⅱ;
⑤、将步骤一④中得到的反应液ⅱ加入到500ml去离子水中析出沉淀,再静置30min,再在离心速度为8000r/min下进行离心,得到固体反应产物;
⑥、使用去离子水对固体反应产物清洗5次,再将去离子水清洗后的固体反应产物放入冷冻干燥机中,再在温度为-10℃下冷冻干燥48h,得到干燥的白色固体物质,即为羟基封端超支化聚合物;
二、碳纤维的抽提处理:
①、将碳纤维放入装有丙酮的索氏提取器中,再将丙酮加热至82℃,丙酮不断蒸出并在索氏提取器中冷凝,使碳纤维表面的杂质在蒸馏的丙酮中不断得到清洗,清洗时间为72h,再将碳纤维取出,得到去除表面杂质的碳纤维;将去除表面杂质的碳纤维取出,再置于温度为70℃的烘箱中干燥4h,得到抽提处理后的碳纤维;
三、氧化:
①、将抽提处理后的碳纤维浸入到过硫酸钾/硝酸银混合水溶液中,加热至70℃,再在温度为70℃的条件下恒温1h,得到氧化后的碳纤维;所述的过硫酸钾/硝酸银混合水溶液中过硫酸钾的浓度为0.1mol/l;所述的过硫酸钾/硝酸银混合水溶液中硝酸银的浓度为0.01mol/l;
步骤三①中所述的抽提处理后的碳纤维的质量与过硫酸钾/硝酸银混合水溶液的体积比为1.5g:500ml;
②、室温条件下将步骤三①得到的氧化后的碳纤维在蒸馏水中浸泡10min,将经蒸馏水中浸泡后的碳纤维取出,弃除蒸馏水;
步骤三②中所述的氧化后的碳纤维的质量与蒸馏水的体积比为1.5g:500ml;
③、重复步骤三②5次,得到蒸馏水清洗后的氧化碳纤维;
④、将步骤三③得到的蒸馏水清洗后的氧化碳纤维在温度为70℃的条件下干燥4h,得到干燥后的氧化碳纤维;
⑤、将步骤三④得到的干燥后的氧化碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃的条件下使用无水乙醇清洗氧化碳纤维,清洗时间为2h,得到无水乙醇清洗的氧化的碳纤维;
⑥、将步骤三⑤得到的无水乙醇清洗的氧化的碳纤维在温度为70℃的条件下干燥4h,得到干燥的氧化碳纤维;
四、接枝:
①、将1.5g羟基封端超支化聚合物加入到60mln,n-二甲基乙酰胺中,再在超声功率为346.5w下超声30min,然后加入1.5g干燥的氧化碳纤维、0.67g1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和0.067g4-二甲氨基吡啶,最后在温度为100℃和搅拌速度为400r/min的条件下搅拌回流24h,得到处理后的碳纤维;
②、将处理后的碳纤维取出后浸入到蒸馏水中15min;
③、循环步骤四③5次,得到蒸馏水清洗的处理后的碳纤维;
④、将蒸馏水清洗的处理后的碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃的条件下使用无水乙醇清洗蒸馏水清洗的处理后的碳纤维,清洗时间为2h,得到无水乙醇清洗的处理后的碳纤维;
⑤、将无水乙醇清洗的处理后的碳纤维置于温度为80℃的烘箱中干燥4h,得到表面接枝羟基封端超支化聚合物的碳纤维,表示为cf-hthbp。
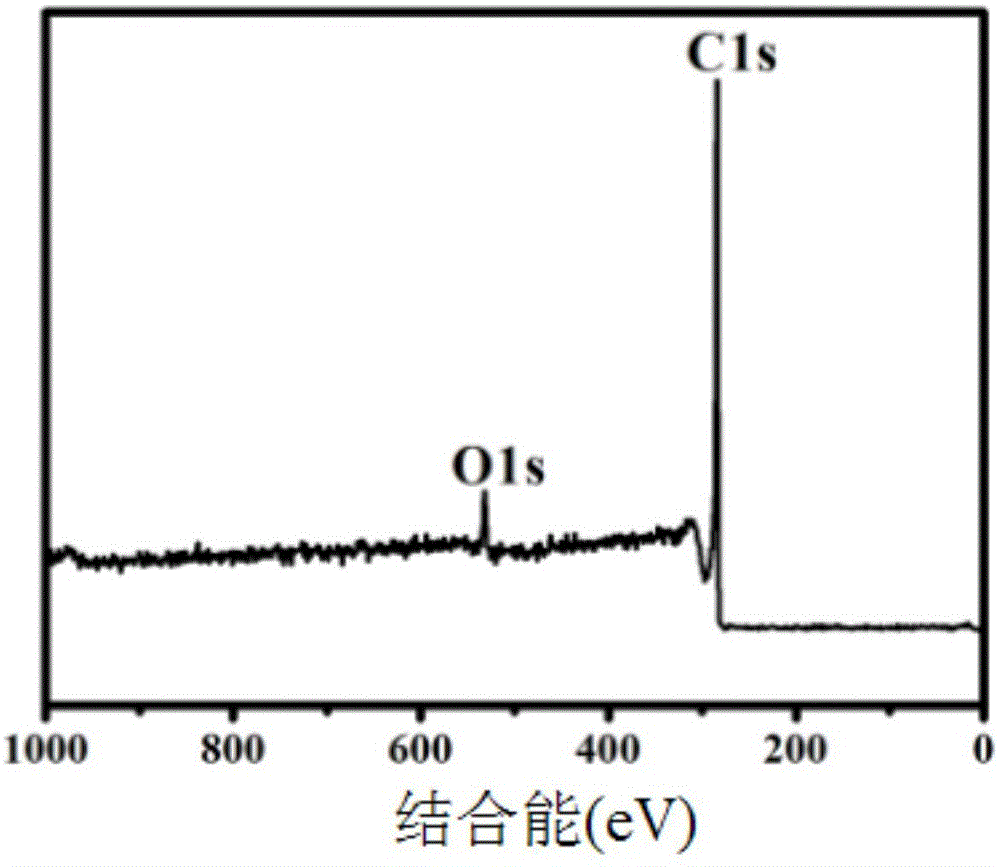
图1为实施例一步骤二③得到的抽提处理后的碳纤维的xps谱图;
图2为图1的分峰谱图,图中1为c1s(1),2为c1s(2),3为c1s(3);
图3为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的xps谱图;
图4为图3的分峰谱图,图中4为c-n,5为c-o,6为c=o,7为ncoo;
表1为碳纤维改性前后元素含量变化。碳纤维表面接枝羟基封端超支化聚合物(hthbp)后,o含量显著增加,由未处理的3.32%提高到20.19%。从cf-hthbp的分峰图中可以看出,在结合能为289ev处出现了ncoo新峰,这些结果表明羟基封端超支化聚合物(hthbp)已共价接枝到碳纤维表面,即hthbp中的羟基和氧化碳纤维中的羧基发生反应生成了酯键,该键的相对含量为4.67%。
表1
注:cf为实施例一步骤二③得到的抽提处理后的碳纤维,cf-hthbp为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维。
图5为实施例一步骤二③得到的抽提处理后的碳纤维的sem图;
图6为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的sem图;
从图5~图6可以看出,未接枝的碳纤维(实施例一步骤二③得到的抽提处理后的碳纤维)表面较为光滑。而实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维(cf-hthbp)表面形成了一层覆盖物,这是由于羟基封端超支化聚合物(hthbp)均匀的共价接枝在碳纤维表面,这将提高纤维表面粗糙度和比表面积,有利于增大纤维与树脂之间的机械互锁作用和物理缠结密度,从而形成粘结强度更大的界面结合。
表2为实施例一步骤二③得到的抽提处理后的碳纤维(cf)和实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维(cf-hthbp)的接触角和表面能的变化,由表2可以得出,碳纤维接枝hthbp后,在水和二碘甲烷中的接触角都明显的降低,其极性分量、色散分量和表面能都有相应的提高,表面能由29.97mn/m升高到61.19mn/m提高了104.2%。这说明hthbp接枝到碳纤维表面使纤维表面极性基团和粗糙度有较大幅度的提高,从而碳纤维表面与树脂的润湿性有明显的改善。
表2
界面剪切强度测试(一):
(一)本实验采用fa620型复合材料界面评价装置(日本东荣株式会社)。首先将碳纤维单丝用双面胶牢固地粘贴在金属支架上,再称取质量比为100:32的环氧树脂e-51和固化剂h-256并混合均匀,并用钢针蘸取一滴点在碳纤维单丝表面,环氧树脂会因表面张力作用形成树脂微滴,随后在90℃、120℃和150℃下分别恒温保持2h、2h和3h进行固化,从而制得碳纤维/环氧树脂微滴复合材料。在测试过程中,选取直径80μm左右的树脂微滴作为测试对象,树脂球直径太大容易将纤维拉断而树脂球并未与纤维脱离,若树脂球太小,设备刀口夹不住,测试过程没有测试到力的作用刀口就从树脂球滑过,载荷加载速度为0.5μm·s-1,每组样品测出50个有效数据并计算其平均值,所述的碳纤维为实施例一步骤二③得到的抽提处理后的碳纤维(cf)。界面剪切强度(ifss)可根据公式(1)得到:
式中fmax——纤维拔脱时的载荷峰值(n);
d——纤维单丝直径(m);
l——环氧树脂微滴包埋长度(m)。
按照上述方法对实施例一步骤二③得到的抽提处理后的碳纤维(cf)的界面剪切强度进行测试;
界面剪切强度测试(二):本测试与界面剪切强度测试(一)的不同点是:所述的碳纤维为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维(cf-hthbp)。其他步骤及测试方法均与界面剪切强度测试(一)相同。
界面剪切强度测试(一)和界面剪切强度测试(二)测试结果如图7所示;
图7为界面剪切强度柱状图,图中1为实施例一步骤二③得到的抽提处理后的碳纤维的界面剪切强度,2为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的界面剪切强度;
从图7可以看出,碳纤维接枝hthbp后的界面剪切强度(ifss)由原丝的48.8mpa提高到87.8mpa,提高了79.9%。
层间剪切强度测试(一):
(一)本实验在电子万能试验机gt-7000-a2x(中国台湾)上采用三点弯曲法测试碳纤维复合材料的ilss。首先在20cm长的框架上缠绕20圈的碳纤维,再称取质量比为100:32的环氧树脂e-51和固化剂h-256并混合均匀,随后使基体树脂充分浸润碳纤维,接着将其置于模具中,把模具放到真空干燥箱中进行真空除泡,然后将模具放到热压机上按如下工艺固化,在90℃、120℃和150℃下分别恒温保持2h、2h和3h进行固化,从而制得碳纤维/环氧树脂复合材料。在测试过程中,试样尺寸为20mm×6mm×2mm,试样跨度与厚度比是5,载荷加载速度为2mm·min-1,测试在室温下进行,每组样品测出50个有效数据并计算其平均值,所述的碳纤维为实施例一步骤二③得到的抽提处理后的碳纤维(cf)。层间剪切强度(ilss)可根据公式(2)得到:
式中f——断层时最大载荷(n);
b——试样横截面宽度(mm);
h——试样横截面厚度(mm)。
按照上述方法对实施例一步骤二③得到的抽提处理后的碳纤维(cf)的层间剪切强度进行测试;
层间剪切强度测试(二):本测试与层间剪切强度测试(一)的不同点是:所述的碳纤维为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维(cf-hthbp)。其他步骤及测试方法均与层间剪切强度测试(一)相同。
层间剪切强度测试(一)和层间剪切强度测试(二)测试结果如图8所示;
图8为层间剪切强度柱状图,图中1为实施例一步骤二③得到的抽提处理后的碳纤维的层间剪切强度,2为实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的层间剪切强度。
从图8可以看出,碳纤维接枝hthbp以后的层间剪切强度(ilss)由原丝的58.6mpa提高到81.8mpa,提高了39.6%。
实施例一步骤四⑤得到的表面接枝羟基封端超支化聚合物的碳纤维的界面剪切强度和层间剪切强度均提高很多,这是因为碳纤维表面经接枝hthbp后含有大量极性羟基,可以与树脂基团参加化学反应。与此同时,碳纤维表面粗糙度的增加可以增强纤维与树脂机械互锁作用和物理缠结密度,从而使碳纤维增强复合材料有更好的界面结合,这将有助于最终复合材料的界面性能的提高。
实施例二:一种在碳纤维表面接枝羟基封端超支化聚合物的方法是按以下步骤完成的:
一、制备羟基封端超支化聚合物:
①、将6g异佛尔酮二异氰酸酯和50mln,n-二甲基乙酰胺加入到三口烧瓶中,得到异佛尔酮二异氰酸酯溶液;再将装有异佛尔酮二异氰酸酯溶液的玻璃容器置于5℃的冰水浴中;
②、将3g三羟甲基氨基甲烷加入到50mln,n-二甲基乙酰胺中,再在超声功率为343w下超声混合10min,得到三羟甲基氨基甲烷溶液;
③、将步骤一①中置于5℃的冰水浴的玻璃容器中的异佛尔酮二异氰酸酯溶液进行搅拌,搅拌速度为300r/min,再将步骤一②中得到的三羟甲基氨基甲烷溶液以15滴/min的滴加速度滴加到温度为5℃、搅拌速度为300r/min的异佛尔酮二异氰酸酯溶液中,得到反应液ⅰ;
④、将反应液ⅰ的温度升温至10℃,再在温度为10℃下搅拌反应5h,再将反应液ⅰ的温度升温至35℃,再向35℃反应液ⅰ中加入0.1g二月桂酸二丁基锡,最后在温度为35℃和搅拌速度为300r/min下搅拌反应10h,得到反应液ⅱ;
⑤、将步骤一④中得到的反应液ⅱ加入到500ml去离子水中析出沉淀,再静置25min,再在离心速度为7000r/min下进行离心,得到固体反应产物;
⑥、使用去离子水对固体反应产物清洗5次,再将去离子水清洗后的固体反应产物放入冷冻干燥机中,再在温度为-10℃下冷冻干燥36h,得到干燥的白色固体物质,即为羟基封端超支化聚合物;
二、碳纤维的抽提处理:
①、将碳纤维放入装有丙酮的索氏提取器中,再将丙酮加热至82℃,丙酮不断蒸出并在索氏提取器中冷凝,使碳纤维表面的杂质在蒸馏的丙酮中不断得到清洗,清洗时间为48h,再将碳纤维取出,得到去除表面杂质的碳纤维;将去除表面杂质的碳纤维取出,再置于温度为80℃的烘箱中干燥2h,得到抽提处理后的碳纤维;
三、氧化:
①、将抽提处理后的碳纤维浸入到过硫酸钾/硝酸银混合水溶液中,加热至60℃,再在温度为60℃的条件下恒温2h,得到氧化后的碳纤维;所述的过硫酸钾/硝酸银混合水溶液中过硫酸钾的浓度为0.2mol/l;所述的过硫酸钾/硝酸银混合水溶液中硝酸银的浓度为0.005mol/l;
步骤三①中所述的抽提处理后的碳纤维的质量与过硫酸钾/硝酸银混合水溶液的体积比为1.5g:300ml;
②、室温条件下将步骤三①得到的氧化后的碳纤维在蒸馏水中浸泡5min,将经蒸馏水中浸泡后的碳纤维取出,弃除蒸馏水;
步骤三②中所述的氧化后的碳纤维的质量与蒸馏水的体积比为1.5g:500ml;
③、重复步骤三②3次,得到蒸馏水清洗后的氧化碳纤维;
④、将步骤三③得到的蒸馏水清洗后的氧化碳纤维在温度为80℃的条件下干燥2h,得到干燥后的氧化碳纤维;
⑤、将步骤三④得到的干燥后的氧化碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃的条件下使用无水乙醇清洗氧化碳纤维,清洗时间为2h,得到无水乙醇清洗的氧化的碳纤维;
⑥、将步骤三⑤得到的无水乙醇清洗的氧化的碳纤维在温度为80℃的条件下干燥2h,得到干燥的氧化碳纤维;
四、接枝:
①、将1g羟基封端超支化聚合物加入到30mln,n-二甲基乙酰胺中,再在超声功率为343w下超声30min,然后加入1.5g干燥的氧化碳纤维、0.5g1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和0.05g4-二甲氨基吡啶,最后在温度为150℃和搅拌速度为400r/min的条件下搅拌回流12h,得到处理后的碳纤维;
②、将处理后的碳纤维取出后浸入到蒸馏水中10min;
③、循环步骤四③4次,得到蒸馏水清洗的处理后的碳纤维;
④、将蒸馏水清洗的处理后的碳纤维置于装有无水乙醇的索氏提取器中,在温度为90℃的条件下使用无水乙醇清洗蒸馏水清洗的处理后的碳纤维,清洗时间为2h,得到无水乙醇清洗的处理后的碳纤维;
⑤、将无水乙醇清洗的处理后的碳纤维置于温度为80℃的烘箱中干燥4h,得到表面接枝羟基封端超支化聚合物的碳纤维,表示为cf-hthbp。
实施例二的实验结果如下:
碳纤维改性前后元素变化:cf-hthbp和cf相比,其表面的含氧元素升高到了14.36%,由原来的氧元素/碳元素之比0.034变为了0.18。
碳纤维改性前后微观形貌变化:纤维氧化后表面缺陷较多,对纤维本体强度造成损坏。
碳纤维改性前后的接触角及表面能:在水和二碘甲烷中的接触角较大,从而导致碳纤维与树脂的润湿性较差。
碳纤维界面剪切强度和层间剪切强度分析:界面剪切强度(ifss)由原丝的48.8mpa提高到60.9mpa,提高了25%。层间剪切强度(ilss)由原丝的58.6mpa提高到69.7mpa,提高了19%。
通过分析可知,采用实施例一的效果更好。
- 还没有人留言评论。精彩留言会获得点赞!