异硫氰酸酯类前体化合物的结构、制备及其应用的制作方法
本发明涉及药物化合物领域,更具体地,涉及异硫氰酸酯类前体化合物的结构、制备及其应用。
背景技术:
在研究过程中发现有些药物化合物结构具有不稳定性,同时,在空气和常温环境下不易存放。而这些现有的抗癌药物在人体内并不易达到有效的药物浓度,且作用时间短,也就不能充分发挥抗癌效力。因此,研究抗肿瘤药物缓释系统成为了肿瘤药物治疗的重点,缓释药物能够增强对肿瘤细胞或组织的靶向性,同时延长药物释放时间,降低吸收速率,平稳血药浓度,减低毒副作用。本发明旨在寻找一种结构稳定、毒副作用小,靶向性强、治疗效果较好且能缓慢释放的前体药物。
CN101731258A公开了一种芳香基异硫氰酸酯和烯丙基异硫氰酸酯的组合物,其重量比为1∶9~9∶1,并且公开了上述组合物在制备防治土传病原菌、土壤线虫的农药熏蒸剂中的应用。
CN104173272A公开了一种异硫氰酸酯类化合物或其衍生物的缓释制剂,具体地,所述缓释制剂含有(a)治疗有效量的异硫氰酸酯类化合物或其衍生物;和(b)药学上可接受的载体,其提供的是一种组合物的形式。CN101822663B公开了异硫氰酸酯在制备防治耐药肿瘤药物的用途。CN101780066A公开了异硫氰酸酯在制备防治肿瘤侵袭转移药物的用途。CN102631340A公开了一种含异硫氰酸盐的抗肿瘤药物及其用途。CN104072395A公开了异硫氰酸酯衍生物对肿瘤细胞有杀伤效果。
但是到目前为止,尚不存在异硫氰酸酯类的前体药物,期望在这个领域有所改进。
技术实现要素:
本发明提供了一种结构稳定、毒副作用小、靶向性强、治疗效果较好且能缓慢释放的前体化合物,所述化合物的通式为:
其中R1为:C4-C6烷基、C1-C4烷氧基、C1-C6链烯基、C2-C10炔基、C1-C4烷酰基、C1-C4烷酰氧基、R2为: R3为:Cl、Br、烷基、C1-C6链烯基、C2-C10炔基、C1-C4烷酰基、C1-C4烷酰氧基;R4为:H、OH、CF3或甲氧基;X为:O、S或NH;n=0、1或2。
根据本发明的实施例,提供了化合物在用于预防或治疗癌症方面的应用。
在上述应用中,其中,所述癌症包括乳腺癌、白血病、增殖表皮癌、肝癌、胰腺癌、脑胶质瘤、肝癌、皮肤黑色素瘤、神经母细胞瘤、肺癌。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:在容器中加入2-二甲氨基硫代乙醇盐酸盐和乙醇溶液;随后加入5-[(4-异硫氰酸酯丁基)硫]-1-苯基-1H-四氮唑;用碱性溶液将反应液的pH值调至5-7,所述反应液在惰性气体保护下搅拌过夜;向所述反应液中滴加碱性溶液将pH值调至碱性;将所述反应液旋干,所得剩余物质溶于二氯甲烷,并进行洗涤,将所得有机相干燥并浓缩从而得到所述化合物的粗产品;以及对所述化合物的粗产品进行纯化。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:在容器中加入2-二甲氨基硫代乙醇盐酸盐和乙醇溶液;随后加入1-异硫氰酸-4-甲磺酰基丁烷;用碱性溶液将反应液的pH值调至5-7,在惰性气体保护下搅拌过夜;向所述反应液中滴加碱性溶液将pH值调至碱性;将所述反应液旋干,所得剩余物质溶于二氯甲烷,并进行洗涤,将所得有机相干燥并浓缩从而得到所述化合物的粗产品;以及对所述化合物的粗产品 进行纯化。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:将5-((4-异硫氰酸酯丁基)硫)-1-苯基-1H-四氮唑溶于二氯甲烷中;然后加入二甲氨基吡啶和2-(二甲氨基)乙基-3-巯基-2-甲基丙酯;体系在惰性气体保护下室温搅拌进行反应;旋除溶剂,得到所述化合物的粗产品;以及对所述化合物的粗产品进行纯化。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:将1-异硫氰酸-4-甲磺酰基丁烷溶于二氯甲烷;然后加入N,N-二甲氨基吡啶和2-(二甲氨基)乙基-3-巯基-2-甲基丙酯;体系在惰性气体保护下室温搅拌进行反应;旋除溶剂,得到所述化合物的粗产品;以及对所述化合物的粗产品进行纯化。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:将5-((4-异硫氰酸酯丁基)硫)-1-苯基-1H-四氮唑溶于二氯甲烷;然后加入三乙胺和2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫醇;体系在惰性气体保护下室温搅拌进行反应;旋除溶剂,得到所述化合物的粗产品;以及对所述化合物的粗产品进行纯化。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:将1-异硫氰酸-4-甲磺酰基丁烷溶于二氯甲烷中;然后加入三乙胺和2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫醇;体系在惰性气体保护下室温搅拌进行反应;旋除溶剂,得到所述化合物的粗产品;以及对所述化合物的粗产品进行纯化。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:将2-二甲氨基硫代乙醇盐酸盐溶于乙醇中;然后加入(4-溴苯基)(4-异硫氰酸酯丁基)硫醚;通过碱性溶液将反应液的pH值调至5-7,对所述反应液进行搅拌;旋除溶液,所得剩余物质溶于二氯甲烷,将有机相洗涤、干燥和浓缩,得到所述化合物的粗产品;以及对所述化合物的粗产品进行纯化。
根据本发明的另一实施例,提供了一种化合物的制备方法,所述方法包括:将2-二甲氨基硫代乙醇盐酸盐溶于乙醇中;然后加入2-((4-异硫氰酸 酯丁基)硫)苯并噻唑;通过碱性溶液将反应液的pH值调至5-7,对所述反应液进行搅拌;旋除溶液,所得剩余物质溶于二氯甲烷,将有机相洗涤、干燥和浓缩,得到所述化合物的粗产品;以及对所述化合物的粗产品进行纯化。
本发明提供的化合物结构稳定,能够缓慢释放出活性化合物,延长了药物释放时间,能有效抑制MCF-7、SUM-159、KG1a、HELA、HEPG2、PANC-1、SHG44、PLC/PRF5、A375、SH-SY5Y、NCI-H446等肿瘤细胞的存活和生长,并且耐药性低,毒副作用小。其可以用于预防或者治疗乳腺癌、白血病、增殖表皮癌、肝癌、胰腺癌、脑胶质瘤、肝癌、皮肤黑色素瘤、神经母细胞瘤、肺癌等癌症。
附图说明
图1和图2分别示出了化合物1c和化合物1d的1HNMR图。
图3和图4分别示出了化合物1e的1HNMR图和13CNMR图。
图5和图6分别示出了化合物1的1HNMR图和13CNMR图。
图7和图8分别示出了化合物2的1HNMR图和13CNMR图。
图9和图10分别示出了化合物3c的1HNMR图和13CNMR图。
图11和图12分别示出了化合物3d的1HNMR图和13CNMR图。
图13和图14分别示出了化合物3的1HNMR图和13CNMR图。
图15和图16分别示出了化合物4的1HNMR图和13CNMR图。
图17和图18分别示出了化合物5b的1HNMR图和13CNMR图。
图19和图20分别示出了化合物5d的1HNMR图和13CNMR图。
图21和图22分别示出了化合物5e的1HNMR图和13CNMR图。
图23和图24分别示出了化合物5的1HNMR图和13CNMR图。
图25和图26分别示出了化合物6的1HNMR图和13CNMR图。
图27和图28分别示出了化合物7的1HNMR图和13CNMR图。
图29和图30分别示出了化合物8的1HNMR图和13CNMR图。
图31示出了化合物1、化合物1e、卡马西平的标准曲线。
图32示出了化合物1在体外不同条件下的分解曲线。
图33示出了化合物3在体外不同条件下的分解曲线。
图34示出了化合物5在体外不同条件下的分解曲线。
图35示出了化合物6在体外不同条件下的分解曲线。
图36示出了化合物1e、1、3、5对MCF-7细胞的抑制。
图37示出了化合物2a、4、6对MCF-7细胞的抑制。
图38示出了化合物1e、1、3、5对sum159细胞的抑制。
图39示出了图39化合物2a、4、6对Sum159细胞的抑制。
图40示出了化合物1e、3、5对293T细胞的抑制。
图41示出了化合物2a、4、6对293T细胞的抑制。
图42示出了化合物2a、1e、1、3对KG1a细胞的抑制。
图43和图44示出了化合物1对Hela、HepG2、Panc-1、SHG44、PLC-PRF-5、A375、SH-SY5Y、NCI-H446细胞的抑制。
具体实施方式
下面的实施例可以使本领域技术人员更全面地理解本发明,但不以任何方式限制本发明。
药物的活性形式(I)及其前体药物(II)的通式如下所示:
其中R1为:C4-C6烷基、C1-C4烷氧基、C1-C6链烯基、C2-C10炔基、C1-C4烷酰基、C1-C4烷酰氧基、
R2为:
R3为:Cl、Br、烷基、C1-C6链烯基、C2-C10炔基、C1-C4烷酰基、C1-C4烷酰氧基;R4为:H,OH,CF3或甲氧基;
X为:O,S或NH;n=0,1,2。
下面描述各种化合物的合成步骤以及图谱。二甲氨基-(苯基-1H-四氮唑)异硫氰酸酯轭合物(以化合物1表示)的合成过程如下:
下面对各个步骤进行逐步描述。
2-[4-((1-苯基-1H-四氮唑-5-基)硫)丁基]异吲哚啉-1,3-二酮(化合物1c)的合成
在干燥的50mL两口圆底烧瓶中,将固体粉末状甲醇钠0.82g(15.03mmol)加入到20mL重蒸甲醇溶液中,待溶液搅拌澄清后,加入化合物1-苯基-1H-四氮唑-5-硫醇2.68g(15.03mmol),室温搅拌20分钟后加入化合物N-(4-溴丁基)邻苯二甲酰亚胺4.00g(14.18mmol)。通过TLC(石油醚:乙酸乙酯=3:1)检测反应,待反应结束后,向反应液加入10mL超纯水,继续搅拌10分钟后,旋除甲醇,析出大量白色固体粗产品,用水洗涤两次,所得固体放入真空干燥箱内45℃烘干,得到白色固体产物,重量为4.58g, 产率为85.0%。
1H-NMR(400MHz,CDCl3)δ7.86-7.82(m,2H),7.74-7.70(m,2H),7.59-7.52(m,5H),3.74(t,J=6.8Hz,2H),3.45(t,J=6.8Hz,2H),1.93-1.84(m,4H),化合物1c的1HNMR图在图1中示出。
4-[(1-苯基-1H-四氮唑-5-基)硫]丁基-1-胺(化合物1d)的合成
在500mL两口圆底烧瓶中,加入化合物2-[4-((1-苯基-1H-四氮唑-5-基)硫)丁基]异吲哚啉-1,3-二酮7.59g(20.0mmol)和300mL乙醇溶液,室温下加入5mL水合肼溶液(约100mmol),在50℃条件下搅拌4小时。通过TLC(二氯甲烷:甲醇=10:1,0.5%氨水)茚三酮显色检测反应,待反应完全结束后过滤除去白色固体,所得固体用乙醇洗涤两次,合并母液后经旋转蒸发仪旋干溶剂,得到油状液体,经硅胶柱(二氯甲烷:甲醇=40:1,0.5%氨水)除去大部分的水合肼后,得到淡黄色油状液体,重量为4.76g,产率为95.4%。
1H-NMR(400MHz,CDCl3)δ7.56-7.51(m,5H),3.40(t,J=7.6Hz,2H),2.79(t,J=6.8Hz,2H),2.67(br,2H),1.94-1.86(m,2H),1.68-1.61(m,2H),化合物1d的1HNMR图在图2中示出。
5-[(4-异硫氰酸酯丁基)硫]-1-苯基-1H-四氮唑(化合物1e)的合成
在250mL两口圆底烧瓶中,加入化合物4-[(1-苯基-1H-四氮唑-5-基)硫]丁基-1-胺1.15g(4.61mmol)、1M NaOH水溶液7.7mL(7.65mmol)和200mL二氯甲烷溶液,反应液在低温反应浴中降至0℃搅拌10分钟后,快速加入硫光气0.80g(6.92mmol),反应液搅拌10分钟后移至室温继续搅拌2小时。通过TLC(石油醚:乙酸乙酯=2:1)检测反应,待反应完全结束后,向溶液中加入饱和氯化钠水溶液,萃取分离有机相,经饱和氯化钠水溶液洗涤后,有机相用无水硫酸钠干燥,旋除溶剂,所得淡黄色固体经硅胶柱纯化(石油醚:乙酸乙酯=10:1),得到产物为淡黄色固体,重量为1.09g,产率为81%。
1H-NMR(400MHz,CDCl3)δ7.58-7.54(m,5H),3.59(t,J=6.4Hz,2H),3.43(t,J=6.8Hz,2H),2.03-1.96(m,2H),1.90-1.83(m,2H);13C-NMR(100 MHz,CDCl3)δ154.0,133.6,130.2,123.0,129.9,123.9,44.5,32.3,28.9,26.4,化合物1e的1HNMR图以及13CNMR图分别在图3和图4中示出。
二甲氨基-(苯基-1H-四氮唑)异硫氰酸酯轭合物(化合物1)的合成
在5mL两口圆底烧瓶中,加入化合物2-二甲氨基硫代乙醇盐酸盐100mg(0.706mmol)和1mL95%乙醇,随后加入化合物5-[(4-异硫氰酸酯丁基)硫]-1-苯基-1H-四氮唑171mg(0.588mmol),用1M的氢氧化钠水溶液将反应液调至pH6左右,反应液在氩气保护下升温至40℃搅拌过夜。通过TLC(二氯甲烷:甲醇=10:1,0.5%氨水)检测反应。待反应完全后,向反应液中滴加1M的氢氧化钠水溶液调pH至碱性,将反应液旋干,所得剩余物质溶于二氯甲烷,经饱和碳酸钠溶液和饱和食盐水各洗涤两次,所得有机相用无水硫酸钠干燥后浓缩,得到淡黄色油状粗产品,用硅胶柱纯化(二氯甲烷:甲醇=150:1),得到产品为油状粘稠液体,重量为27.3mg,产率为20.0%。
1H-NMR(400MHz,CDCl3)δ11.07(br,1H),7.55-7.51(m,5H),3.71-3.66(m,2H),3.39(t,J=7.2Hz,2H),3.01(t,J=5.2Hz,2H),2.72(t,J=5.6Hz,2H),2.30(s,6H),1.95-1.87(m,2H),1.82-1.75(m,2H);13C-NMR(100MHz,CDCl3)δ196.5,154.3,133.5,130.2,129.8,123.8,60.9,46.2,45.2,34.1,32.7,27.2,26.7,化合物1的1HNMR图以及13CNMR图分别在图5和图6中示出。
2-(二甲氨基)乙基[4-(甲基亚磺酰基)丁基]二硫代甲酸酯(化合物2)的合成过程如下:
在25mL两口圆底烧瓶中,加入化合物2-二甲氨基硫代乙醇盐酸盐 240mg(1.69mmol)和10mL 95%乙醇溶液,随后加入化合物1-异硫氰酸-4-甲磺酰基丁烷330mg(1.86mmol),用1M的氢氧化钠水溶液将反应液调至pH6左右,反应液在氩气保护下升温至40℃搅拌12小时。通过TLC(二氯甲烷:甲醇=10:1,0.5%氨水)检测反应,发现反应并不完全,随时间增长会出现较多副反应产品。向反应液中滴加1M的氢氧化钠水溶液调pH至碱性,将反应液旋干,所得剩余物质溶于二氯甲烷,经饱和碳酸钠溶液和饱和食盐水各洗涤两次,所得有机相用无水硫酸钠干燥后浓缩,得到淡黄色油状粗产品,用硅胶柱纯化(二氯甲烷:甲醇=150:1),得到产品为油状粘稠液体,重量为56.9mg,产率为11.9%。
1H-NMR(400MHz,CDCl3)δ11.06(br,1H),3.72-3.68(m,2H),3.05(t,J=5.2Hz,2H),2.76-2.72(m,4H),2.57(s,3H),2.34(s,6H),1.90-1.80(m,4H); 13C-NMR(100MHz,CDCl3)δ196.6,60.7,53.9,46.3,45.2,38.8,33.8,29.0,27.4,20.2,化合物2的1HNMR图以及13CNMR图分别在图7和图8中示出。
2-(二甲氨基)乙基-3-巯基-2-甲基丙基-(4-((苯并噻唑-2-基)硫)丁基)二硫代氨基甲酸酯(化合物3)的合成过程如下:
2-(二甲基氨基)乙基-3-(硫代乙酰基)-2-甲基丙酸酯(化合物3c)的合成
在一个25mL的两口瓶中,将乙酰巯基异丁酸200mg(1.23mmol)溶于15mL二氯甲烷,在0℃条件下,加入N,N-二甲氨基吡啶15mg(0.123mmol)和1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI)284mg(1.48mmol),保持0℃搅拌30分钟,然后加入二甲氨基乙醇121mg (1.36mmol),反应体系自然升到室温继续搅拌反应6h,通过TLC检测反应(二氯甲烷:甲醇=20:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋除溶剂,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=60:1,0.5%NH3·H2O)得到黄色粘稠液体,重量为250mg,产量为86.9%。
1H-NMR(400MHz,CDCl3)δ4.22(t,J=5.8Hz,2H),3.14–3.03(m,2H),2.72(td,J=7.3,6.2Hz,1H),2.62(t,J=5.8Hz,2H),2.32(s,9H),1.24(d,J=7.1Hz,3H);13C-NMR(100MHz,CDCl3)δ195.30,174.64,62.30,57.44,45.43,39.86,31.73,30.58,16.76,化合物3c的1HNMR图以及13CNMR图分别在图9和图10中示出。
2-(二甲基氨基)乙基-3-(巯基)-2-甲基丙酸酯(化合物3d)的合成
在一个25mL的两口瓶中,将2-(二甲氨基)乙基-3-乙酰巯基-2-甲基丙酯160mg(0.69mmol)溶于10mL甲醇中,加入碳酸氢钠69.1mg(0.82mmol),室温搅拌反应7h,通过TLC检测反应(二氯甲烷:甲醇=20:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋除溶剂,将所得油状物溶于10mL二氯甲烷中,加10mL H2O,萃取,有机相用无水硫酸钠干燥,过滤,用旋转蒸发仪旋除溶剂,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=20:1,0.5%NH3·H2O)得到黄色粘稠液体,重量为108mg,产率为82.3%。
1H-NMR(400MHz,CDCl3)δ4.29–4.18(m,2H),2.81–2.62(m,3H),2.59(d,J=5.7Hz,2H),2.30(s,6H),1.24(d,J=6.8Hz,3H);13C-NMR(100MHz,CDCl3)δ174.59,62.20,57.74,45.56,43.11,27.72,16.69,化合物3d的1HNMR图以及13CNMR图分别在图11和图12中示出。
2-(二甲氨基)乙基-3-巯基-2-甲基丙基-(4-((苯并噻唑-2-基)硫)丁基)二硫代氨基甲酸酯(化合物3)的合成
在一个10mL的两中瓶中,将5-((4-异硫氰酸酯丁基)硫)-1-苯基-1H-四氮唑100mg(0.34mmol)溶于3mL二氯甲烷中,加入二甲氨基吡啶13.5mg(0.11mmol)和2-(二甲氨基)乙基-3-巯基-2-甲基丙酯47mg (0.25mmol),体系在氩气保护下室温搅拌反应10h,通过TLC检测反应(二氯甲烷:甲醇=20:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋除溶剂,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到黄色粘稠固体,重量为105mg,产率为88.7%。
1H-NMR(400MHz,CDCl3)δ8.00(s,1H),7.57(s,5H),4.34–4.13(m,2H),3.82(dd,J=12.3,6.8Hz,2H),3.57–3.32(m,4H),2.96–2.84(m,1H),2.73–2.58(m,2H),2.32(d,J=12.8Hz,6H),1.98-1.91(m,2H),1.89–1.78(m,2H),1.35–1.17(m,2H);13C-NMR(100MHz,CDCl3)δ197.37,175.15,154.36,133.52,129.83,123.82,62.40,57.58,46.22,45.63,39.95,37.81,32.48,26.96,26.64,17.03,化合物3的1HNMR图以及13CNMR图分别在图13和图14中示出。
2-(二甲氨基)乙基-3-巯基-2-甲基丙基-(4-(甲基亚磺酰基)丁基)二硫代甲酸酯(化合物4)的合成过程如下:
在一个10mL的两中瓶中,将1-异硫氰酸-4-甲磺酰基丁烷60.3mg(0.34mmol)溶于5mL二氯甲烷,加入N,N-二甲氨基吡啶13.5mg(0.11mmol)和2-(二甲氨基)乙基-3-巯基-2-甲基丙酯47mg,(0.25mmol),体系在氩气保护下室温搅拌反应10h,通过TLC检测反应(二氯甲烷:甲醇=20:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋除溶剂,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到黄色粘稠固体,重量为105mg,产率为88.7%。
1H-NMR(400MHz,CDCl3)δ8.00(s,1H),7.57(s,5H),4.34–4.13(m,2H),3.82(dd,J=12.3,6.8Hz,2H),3.57–3.32(m,4H),2.96–2.84(m,1H),2.73–2.58(m,2H),2.32(s,6H),1.98-1.91(m,2H),1.89–1.78(m,2H),1.35–1.17(m,2H);13C-NMR(100MHz,CDCl3)δ197.37,175.15,154.36,133.52,129.83,123.82,62.40,57.58,46.22,45.63,39.95,37.81,32.48, 26.96,26.64,17.03,化合物4的1HNMR图以及13CNMR图分别在图15和图16中示出。
2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫基-(4-((苯并噻唑-2-基)硫)丁基)二硫代氨基甲酸酯(化合物5)的合成过程如下:
2-(2-(2-(二甲氨基)乙氧基)乙氧基)氯乙烷(化合物5b)的合成
在一个50mL的两口瓶中,将2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙醇410mg(2.31mmol)溶于20mL氯仿,反应体系置于0℃,缓慢滴加氯化亚砜825.6mg(6.94mmol),滴加完毕,将反应体系加热到70℃反应1h,通过TLC检测反应(二氯甲烷:甲醇=5:1,0.5%NH3·H2O),待反应完全后,加入20mL饱和碳酸钠溶液,萃取,有机相用无水硫酸钠干燥,过滤,滤液用旋转蒸发仪旋干,得到综红色液体,重量为430mg,产率为95%。
1H-NMR(400MHz,CDCl3)δ3.74(t,J=5.9Hz,2H),3.67(m,2H),3.64–3.57(m,6H),2.53(t,J=5.8Hz,2H),2.28(s,6H);13C-NMR(100MHz,CDCl3)δ71.44,70.77,70.47,69.51,58.92,45.95,42.77,化合物5b的 1HNMR图以及13CNMR图分别在图17和图18中示出。
S-(2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙基)乙酰硫酯(化合物5d)合成
在一个50mL的两口瓶中,将2-(2-(2-(二甲氨基)乙氧基)乙氧基)氯乙烷430mg(2.2mmol)溶于25mL N,N-二甲氨基甲酰胺,加入硫代乙酸钾301mg(2.64mmol),反应体系置于75℃反应12h,通过TLC检测反应(二氯甲烷:甲醇=5:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋干溶剂,所得红综色粘稠液体溶于20mL二氯甲烷,加入1M盐酸调节pH=1,加10mL水,萃取,收集水相,加入饱和碳酸钠溶液调pH=10,加入二氯甲烷(3×20mL)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液用旋转蒸发仪旋干,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=10:1)得到综红色液体,重量为345mg,产率为66.7%。
1H-NMR(400MHz,CDCl3)δ3.72(t,J=5.4Hz,2H),3.61(s,4H),3.58(t,J=6.5Hz,2H),3.07(t,J=6.4Hz,2H),2.77(t,J=5.3Hz,2H),2.48(s,6H),2.32(s,3H);13C-NMR(100MHz,CDCl3)δ195.58,70.44,70.25,69.82,68.06,58.17,45.14,30.68,28.90,化合物5d的1HNMR图以及13CNMR图分别在图19和图20中示出。
2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫醇(化合物5e)的合成
在一个25mL的两口瓶中,将S-(2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙基)乙酰硫酯250mg(1.06mmol)溶于10mL甲醇,加入水合肼271.3g(5.31mmol),反应体系在氩气保护下室温搅拌3h,通过TLC检测反应(二氯甲烷:甲醇=5:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋干溶剂,加入10mL饱和碳酸钠溶液和二氯甲烷,萃取,有机相用无水硫酸钠干燥,过滤,滤液用旋转蒸发仪旋干,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=10:1,0.5%NH3·H2O)得到综红色液体,重量为105mg,产率为51.1%。
1H-NMR(400MHz,CDCl3)δ3.60(m,8H),2.69(d,J=5.6Hz,2H),2.53(t,J=5.8Hz,2H),2.28(s,6H),1.60(br,1H);13C-NMR(100MHz,CDCl3)δ73.00,70.45,70.35,69.39,58.88,45.95,24.39,化合物5e的1HNMR图以及13CNMR图分别在图21和图22中示出。
2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫基-(4-((苯并噻唑-2-基)硫)丁基)二硫代氨基甲酸酯(化合物5)的合成
在一个10mL的两中瓶中,将5-((4-异硫氰酸酯丁基)硫)-1-苯基-1H-四氮唑100mg(0.34mmol)溶于3mL二氯甲烷,加入三乙胺11.6mg(0.11mmol)和2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫醇44mg(0.23mmol),体系在氩气保护下室温搅拌反应10h,通过TLC检测反应(二氯甲烷:甲醇=20:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋除溶剂,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到黄色粘稠固体,重量为78mg,产率为70.3%。
1H-NMR(400MHz,CDCl3)δ8.51(s,1H),7.57(s,5H),3.77(s,4H),3.69–3.64(m,2H),3.61(s,4H),3.41(d,J=7.1Hz,4H),2.59(t,J=5.7Hz,2H),2.33(s,6H),1.95(s,2H),1.81(s,2H);13C-NMR(100MHz,CDCl3)δ197.37,154.38,133.61,129.89,123.89,70.55,70.45,70.15,68.97,58.75,46.12,45.66,35.26,32.57,27.05,26.71,化合物5的1HNMR图以及 13CNMR图分别在图23和图24中示出。
2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫基-(4-(甲基亚磺酰基)丁基)二硫代甲酸酯(化合物6)的合成过程如下:
在一个25mL的两中瓶中,将1-异硫氰酸-4-甲磺酰基丁烷280mg(1.58mmol)溶于15mL二氯甲烷中,加入三乙胺26.6mg(0.26mmol)和2-(2-(2-(二甲氨基)乙氧基)乙氧基)乙硫醇254.4mg(1.32mmol),体系在氩气保护下室温搅拌反应10h,通过TLC检测反应(二氯甲烷:甲醇=20:1,0.5%NH3·H2O),待反应完全后,用旋转蒸发仪旋除溶剂,得到粗品,经硅胶柱层析纯化(二氯甲烷:甲醇=50:1)得到黄色粘稠固体,重量为250mg产率为64.5%。
1H-NMR(400MHz,CDCl3)δ8.66(s,1H),3.75(t,J=5.8Hz,4H),3.69–3.58(m,6H),3.37(t,J=5.7Hz,2H),2.79–2.71(m,2H),2.64(t,J=5.7Hz, 2H),2.58(s,3H),2.35(s,6H),1.87(br,4H);13C-NMR(100MHz,CDCl3)δ197.35,70.45,70.40,68.78,58.63,53.72,46.35,45.59,42.05,38.68,35.20,27.20,20.22,化合物6的1HNMR图以及13CNMR图分别在图25和图26中示出。
2-(二甲基氨基)乙基(4-((4-溴苯基)硫)丁基)二硫代氨基甲酸酯(化合物7)的合成过程如下:
在一5mL两口瓶中,将2-二甲氨基硫代乙醇盐酸盐121.0mg(0.854mmol)溶于2mL 95%乙醇中,加入(4-溴苯基)(4-异硫氰酸酯丁基)硫醚284.0mg(0.940mmol),加入1mol/L氢氧化钠溶液,将反应液调至PH6,反应液升温至40℃搅拌12小时。通过TLC检测反应(DCM:MeOH=10:1,0.5%NH3·H2O),旋除溶液,所得剩余物质溶于二氯甲烷,有机相经少量饱和碳酸钠溶液和饱和食盐水洗涤,所得有机相用无水硫酸钠干燥后浓缩,经硅胶柱纯化(DCM:MeOH=100:1),得到油状粘稠液体,重量为67.9mg,产率为19.5%。
1H-NMR(400MHz,CDCl3)δ11.04(bs,1H),7.37(d,J=8.4Hz,2H),7.16(d,J=8.4Hz,2H),3.66-3.61(m,2H),2.98(t,J=4.8Hz,2H),2.92(t,J=7.2Hz,2H),2.73(t,J=6.4Hz,2H),2.29(s,6H),1.77-1.65(m,4H);13C-NMR(100MHz,CDCl3)δ196.3,135.5,132.0,131.9,131.0,130.8,119.8,61.1,46.5,45.2,34.2,33.3,27.3,26.4,化合物7的1HNMR图以及13CNMR图分别在图27和图28中示出。
2-(二甲基氨基)乙基(4-((苯并噻唑-2-基)硫)丁基)二硫代氨基甲酸酯(化合物8)的合成过程如下:
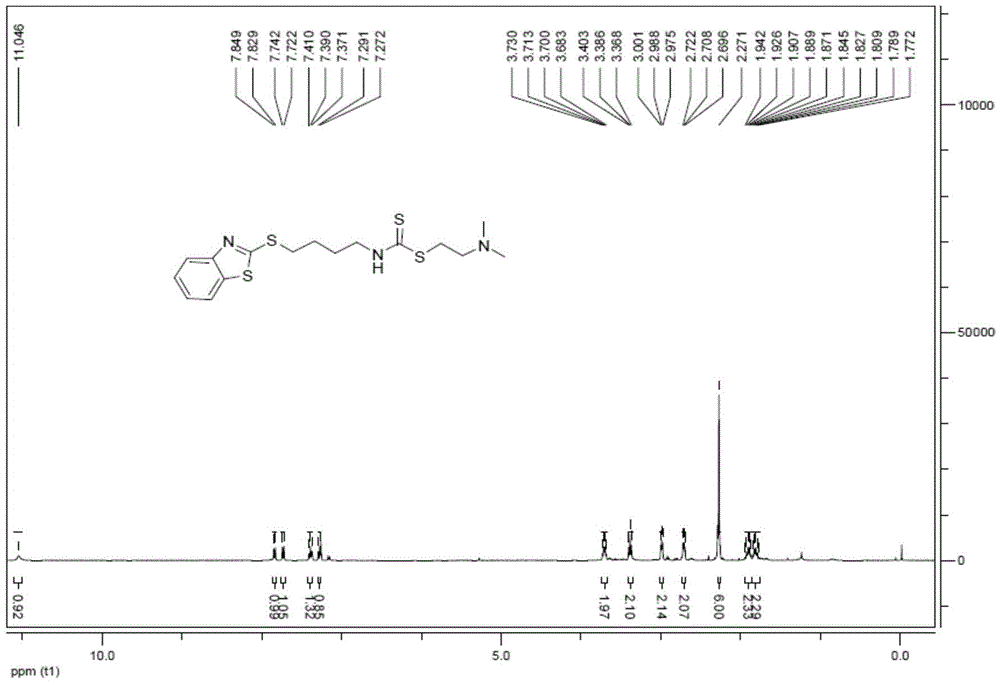
在一5mL两口瓶中,将2-二甲氨基硫代乙醇盐酸盐91.85mg(0.648mmol)溶于2mL 95%乙醇中,加入2-((4-异硫氰酸酯丁基)硫)苯并噻唑200.0mg(0.713mmol),加入1mol/L氢氧化钠溶液,将反应液调至PH6,反应液升温至40℃搅拌12小时。通过TLC检测反应(DCM:MeOH=10:1,0.5%NH3·H2O),旋除溶液,所得剩余物质溶于二氯甲烷,有机相经少量饱和碳酸钠溶液和饱和食盐水洗涤,所得有机相用无水硫酸钠干燥后浓缩,经硅胶柱纯化(DCM:MeOH=90:1),得到油状粘稠液体,重量为64.4mg,产率为25.7%。
1H-NMR(400MHz,CDCl3)δ11.05(bs,1H),7.84(d,J=8.0Hz,1H),7.73(d,J=8.0Hz,1H),7.39(t,J=8.0Hz,1H),7.28(d,J=7.6Hz,1H),3.73-3.68(m,2H),3.39(t,J=6.8Hz,2H),2.99(t,J=5.2Hz,2H),2.71(t,J=5.6Hz,2H),2.27(s,6H),1.94-1.87(m,4H),1.85-1.77(m,4H);13C-NMR(100MHz,CDCl3)δ196.3,166.7,153.2,135.2,126.1,124.3,121.5,121.0,61.0,46.6,45.2,34.1,32.9,27.2,27.0,化合物8的1HNMR图以及13CNMR图分别在图29和图30中示出。
缓释实验步骤及图谱
体外稳定性部分
本发明还提供了前体化合物在体外的稳定性实验方案,包括在pH7.4、pH5.4、pH4.4、pH3.4缓冲液及大鼠血浆中的缓释效应,实验采用内标法测定前体化合物在恒温37℃缓冲液及血浆中的浓度变化。首先,作出化合物1、化合物1e以及卡马西平的标准曲线(见图31),从而来确认化合物峰面积与浓度的线性关系。前体化合物的缓释实验方案具体以化合物5为例进行详述,其余类似或相同。具体步骤如下:
步骤1):用乙腈作溶剂分别精密配制50umol/L、100umol/L、200umol/L、 300umol/L、400umol/L、500umol/L、600umol/L的化合物1、化合物1e、卡马西平溶液,HPLC进样,测出不同浓度溶液的峰面积,重复3次,取平均值,作出化合物1、化合物1e和卡马西平的标准曲线(见图31)。
步骤2):用乙腈作溶剂分别精密配制600umol/L的化合物5溶液、600umol/L的化合物1e溶液、300umol/L的卡马西平溶液。
步骤3):分别取100uL新配制的化合物5溶液、化合物1e溶液和卡马西平溶液,加入300uL乙腈(C化合物5=100umol/L,C1e=100umol/L,C卡马西平=50umol/L),HPLC进样,测出峰面积分别为S化合物5、S1e、S卡马西平,通过峰面积计算校正因子f,(f化合物5=C化合物5*S卡马西平/(C卡马西平*S化合物5),f1e=C1e*S卡马西平/(C卡马西平*S1e))。
步骤4):取1mL新配制的化合物5溶液(600umol/L),加入9mL缓冲液(依次分别加pH7.4和pH5.4的PBS缓冲液;pH4.4和pH3.4乙酸/乙酸钠缓冲液),此时溶液浓度为C化合物5=60umol/L。
步骤5):将化合物5的缓冲液(60umol/L)放入37℃的恒温水浴锅中,保温处理,于设计时间点取500uL此缓冲液,加入100uL卡马西平溶液(300umol/L)(此时C化合物5=50umol/L,C卡马西平=50umol/L),HPLC进样,测出化合物5,化合物1e,卡马西平的峰面积S化合物5、S1e、S卡马西平,用内标法通过校正因子计算不同时刻化合物5的浓度(C化合物5=f化合物5*C卡马西平*S化合物5/S卡马西平,C1e=f1e*C卡马西平*S1e/S卡马西平),共24h,重复操作3次,取平均值,作出化合物5的缓释曲线图(见图34)
步骤6):取大鼠动脉血液,加抗凝剂,离心15min(4℃,12000rpm),取上层血浆,分装到小试管,-20℃冷冻保存备用。
步骤7):将冷冻保存的血浆从冰箱中取出,放在室温条件下自然解冻,精密配制600umol/L的化合物5的血浆样品,300umol/L的化合物1e乙腈溶液和300umol/L的卡马西平乙腈溶液。
步骤8):分别精密量取100uL新配制的化合物5血浆样品(600umol/L),200uL化合物1e乙腈溶液(300umol/L),200uL卡马西平乙腈溶液(300umol/L),加入700uL蛋白沉淀剂乙腈(此时C化合物5=50umol/L,C1e=50umol/L,C卡马西平=50umol/L),涡旋1min,离心10min(4℃,12000rpm), 取上层清液,微孔滤膜过滤,HPLC进样,测出峰面积分别为S化合物5、S1e、S卡马西平,通过峰面积计算校正因子f,(f化合物5=C化合物5*S卡马西平/(C卡马西平*S化合物5),f1e=C1e*S卡马西平/(C卡马西平*S1e))。
步骤9):将新配制的化合物5血浆样品(600umol/L)放入37℃恒温水浴锅中保温处理,于设计时间点取100uL此血浆样品,加入200uL卡马西平乙腈溶液(300umol/L),加入蛋白沉淀剂乙腈900uL,涡旋1min,离心10min(4℃,12000rpm),取上层清液,微孔滤膜过滤,HPLC进样,测出化合物5、化合物1e、卡马西平的峰面积S化合物5、S1e、S卡马西平,用内标法通过校正因子计算不同时刻化合物5和化合物1e的浓度(C化合物5=f化合物5*C卡马西平*S化合物5/S卡马西平,C1e=f1e*C卡马西平*S1e/S卡马西平),共24h,重复操作3次,取平均值,作出化合物5的分解曲线图(见图34)。
其中采用的色谱条件如下,流动相:水/乙腈=30/70(其中水中加入2‰三氟乙酸,pH1.35);流速:1mL/min;柱温:25℃;检测波长:240nm;进样量:20uL。
化合物5在pH为5.4的乙腈溶液里稳定存在,随着pH的升高不断分解为活性成分即化合物1e;当pH为7.4时在7h内化合物5和化合物1e达到平衡状态;化合物1e在乙腈溶解中稳定存在;化合物5在血浆中约10h完全分解为活性成分即化合物1e,化合物1e在10h内血浆内几乎完全代谢。
图32示出了化合物1在体外不同条件下的分解曲线。制作分解曲线的方法与化合物5类似,在此不再重复。
化合物1在酸性条件下能够稳定存在,随着pH的升高不断分解为活性成分即化合物1e;当pH为7.4时在10h内完全分解为活性成分即化合物1e;化合物1e在乙腈溶解中稳定存在;化合物1在血浆中约100min完全分解为活性成分即化合物1e,化合物1e在6h内血浆内几乎完全代谢。
图33示出了化合物3在体外不同条件下的分解曲线。制作分解曲线的方法与化合物5类似,在此不再重复。
化合物3在pH为3.4的乙腈溶液里稳定存在,随着pH的升高,不断分解为活性成分即化合物1e;当pH为7.4时分解为活性成分即化合物1e和中间体化合物1e*,并且7h内达到平衡;化合物1e在乙腈溶解中稳定存 在;在血浆中约25h完全分解为活性成分即化合物1e和中间体化合物1e*,中间体化合物1e*又不断转换为化合物1e,形成一个平衡。中间体化合物1e*的名称和结构如下式:
2-甲基-3-(4-((苯并噻唑-2-基)硫)丁基)二硫代氨基丙酸
图35示出了化合物6在体外不同条件下的分解曲线。制作分解曲线的方法与化合物1类似,在此不再重复。
化合物6在pH为4.4的乙腈溶液里稳定存在,随着pH的升高不断分解为活性成分即化合物2a;当pH为7.4时在25h内化合物6仍未完全分解为化合物2a;化合物2a在乙腈溶解中稳定存在;在血浆中约25h完全分解为活性成分即化合物2a,化合物2a在25h内血浆内几乎完全代谢。
通过实验发现,R2结构相同的化合物的缓释情况是类似的,其中,化合物2、化合物7和化合物8的缓释情况类似于化合物1,化合物4的缓释情况类似于化合物3,并且在此不再重复化合物2、化合物4、化化合物7和化合物8的缓释情况的描述。
活性测试及其图谱
药理活性部分
本发明采用MTT比色法测定细胞活性。
MTT比色法是一种检测细胞存活和生长的方法。其检测原理为活细胞线粒体中的琥珀酸脱氢酶能使外源性MTT还原为水不溶性的蓝紫色结晶甲瓒(Formazan)并沉积在细胞中,而死细胞无此功能。二甲基亚砜(DMSO)能溶解细胞中的甲瓒,用酶联免疫检测仪在490nm波长处测定其光吸收值,可间接反映活细胞数量。在一定细胞数范围内,甲瓒结晶形成的量与细胞数成正比。
MTT的化学名为3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐,商品名为噻唑蓝,是一种黄颜色的染料。
MTT粉末购买于Sigma公司,使用时用磷酸缓冲液(PBS)配制成浓度为5mg/ml的溶液,用0.22μm滤膜过滤以除去溶液里的细菌,然后于4℃下避光保存。具体以化合物1的sum159细胞为例,其余化合物进行类似或相同的处理。
MTT比色法测定细胞活性包括下面几个步骤:
步骤1):加药前一天下午用sum159细胞(购自北京金紫晶生物科技有限公司)铺96孔板。收集对数生长期的sum159细胞,活细胞计数后调整细胞浓度至2.5×104cells/mL。向96孔板中接种细胞,每孔加入100μL细胞悬液铺板,最终待测细胞为2500cells/孔。四周边缘孔不接种细胞,仅加入100μL细胞培养基(本实验中所使用的细胞培养基为改良型RPMI-1640(Hyclone)基础培养基,添加10%的胎牛血清(Hyclone))。5%CO2,37℃孵育过夜,以使细胞充分贴壁。
步骤2):次日上午加药。首先稀释药物,配制相应的药物浓度梯度。向前一天铺好的96孔板的细胞中加入100μL相应的浓度的药物,本实验中对于sum159细胞设置了9个浓度梯度,体系中药物终浓度梯度为:300μM、100μM、33.3333μM、11.1111μM、3.7037μM、1.2345μM、0.4115μM、0.1371μM、0.04572μM。每个浓度设置5个重复。同时设置不加药仅接种细胞的孔为对照组,对照组不加药,补加100μL的细胞培养基即可;设置未接种细胞仅加培养基的孔设为空白孔,也补加100μL细胞培养基。5%CO2,37℃培养箱孵育X(X=24,48,72,96)小时。
步骤3):X小时后每孔再加入20μL MTT溶液(5mg/ml,MTT),继续培养4小时。若药物与MTT能够反应,可先离心后弃去培养液,小心用PBS冲洗2-3遍后,再加入含MTT的培养液。
步骤4):4小时后终止培养,小心吸去孔内液体。每孔加入150μL二甲基亚砜,37℃培养箱孵育10分钟。采用酶联免疫检测仪MULTISKAN FC(Thermo scientific)测定490nm处各孔的吸光值,测量时以空白孔作为调零孔。
步骤5):处理数据。首先采用下列公式计算抑制率:
抑制率=1-加药组OD值/对照组OD值
然后以Log C(药物浓度对数)为横坐标,抑制率为纵坐标,用数据处理软件SPSS软件(IBM公司)进行几率单位加权回归法(Bliss法)进行数据处理,作图,得到IC50值。
根据上述测试方法,测得各化合物在48小时对MCF-7、SUM-159、KG1a和297T细胞的抑制IC50及IC90以及化合物1对HELA、HEPG2、PANC-1等其它多种细胞的抑制作用,见表1、表2和图36至图44,其中分别以化合物2a和1e为缓释出的活性物质,为阳性标样:
表1----MCF-7、SUM-159、KG1a和293T活性测试结果
表2----化合物1对各种细胞的活性测试结果
根据以上描述,可以理解,本发明化合物能够缓慢释放出活性化合物, 从而能有效抑制MCF-7、SUM-159、KG1a、HELA、HEPG2、PANC-1、SHG44、PLC/PRF5、A375、SH-SY5Y、NCI-H446等肿瘤细胞的存活和生长,并且耐药性低,毒副作用小。其可以用于预防或者治疗乳腺癌、白血病、增殖表皮癌、肝癌、胰腺癌、脑胶质瘤、肝癌、皮肤黑色素瘤、神经母细胞瘤、肺癌等癌症。
本领域技术人员应理解,以上实施例仅是示例性实施例,在不背离本发明的精神和范围的情况下,可以进行多种变化、替换以及改变。
- 还没有人留言评论。精彩留言会获得点赞!