曲霉明素A及其衍生物、合成方法与应用与流程
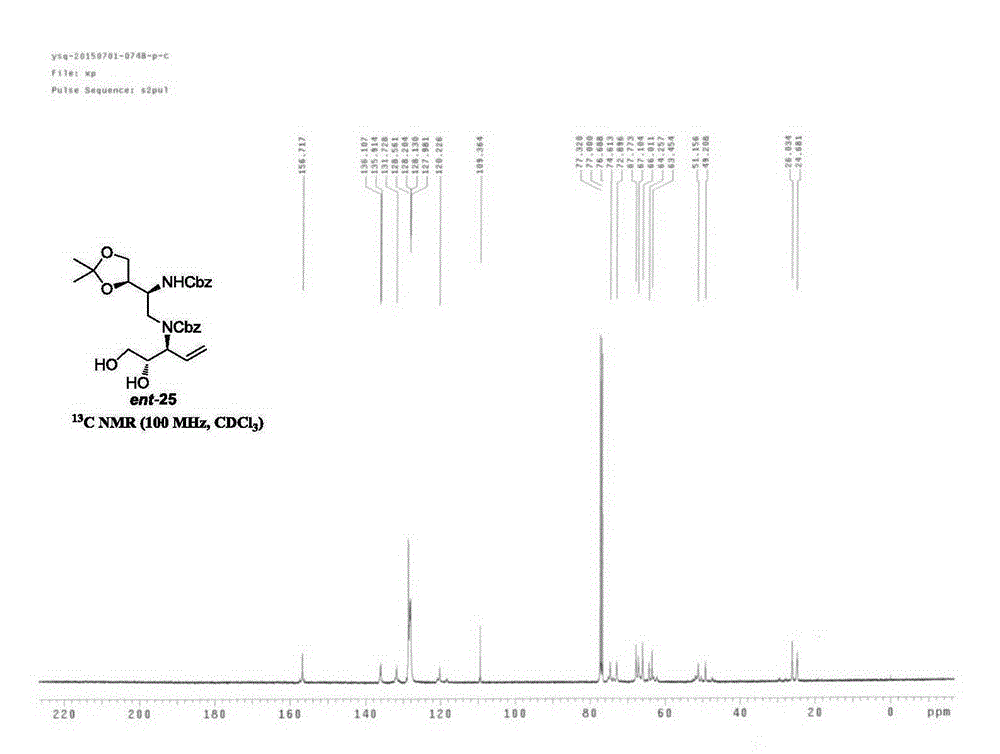
本发明涉及药物化学领域,具体涉及一种具有特定立体构型的曲霉明素A及其衍生物,还涉及所述曲霉明素A及其衍生物的化学合成方法及其在制备药物中的应用。
背景技术:
:内酰胺酶是革兰阴性杆菌对内酰胺类抗生素耐药的主要机制,细菌产生的β-内酰胺酶大部分系活性部位带丝氨酸残基的酶类,也有一小部分是活性部位为金属离子的酶类,称为金属β-内酰胺酶(metallo-β-lactamase,MBL)。金属β-内酰胺酶的酶活性中心需金属锌离子的参与而发挥催化活性,故称为金属β-内酰胺酶。其底物为包括碳青霉烯类在内的一大类β-内酰胺抗生素,其活性不被常见的β-内酰胺酶酶抑制剂如克拉维酸等所抑制。NDM-1(NewDelhimetallo-β-lactamase-1)是金属β-内酰胺酶中的一种常见类型。这类酶可在铜绿假单胞菌、嗜麦芽窄食单胞菌、粘质沙雷菌、肠杆菌属菌、肺炎克雷伯菌、嗜水气单胞菌和不动杆菌、脆弱类杆菌属、等细菌中检出。由于金属β-内酰胺酶可分解β-内酰胺环结构,而含有β-内酰胺环结构的抗生素仍是人类对抗致病细菌最常用的广谱抗生素,包括青霉素、碳青霉烯、头孢菌素等,以及新发展的头霉素类、硫霉素类、单环β-内酰胺类等其他非典型β-内酰胺类抗生素,都含有β-内酰胺环结构,因此,携带β-内酰胺酶(如NDM-1)的细菌可以使绝大部分的抗生素失效,这些临床最常用的抗生素一旦失效会对人类生命安全造成严重威胁。因此,如何有效抑制NDM-1酶的活性已成为全球性亟待解决的健康问题。技术实现要素:本发明的第一目的是提供曲霉明素A及其衍生物,所述曲霉明素A及其衍生物具有特定立体构型,对于NDM-1酶具有快速有效的抑制作用。具体的,本发明提供曲霉明素A及其衍生物,其结构如通式I所示:本发明所述各化学式中,(S)或(R)代表相应位置手性碳的立体构型为S或R。所述通式I中,R1代表C(O)R5或CH(NH2)C(O)R5;R2、R3、R4、R5彼此独立地代表OH、(NH)OH、OR6或NR7R8;R6代表C1~C10的烷基或C1~C6的烯基;R7、R8彼此独立地代表H、C1~C10的烷基或C1~C6的烯基;m=1或2。优选地,所述通式I中,R1代表C(O)R5或CH(NH2)C(O)R5;R2、R3、R4、R5彼此独立地代表OH、(NH)OH或OMe;m=1或2。本发明提供的曲霉明素A及其衍生物可选自如下结构的化合物:其中,I-1表示的化合物即为本发明所述的曲霉明素A,其中三个手性碳的立体构型分别为S、S、S,可采用(S,S,S)-AMA表示。本发明提供的曲霉明素A及其衍生物对NDM-1酶具有快速有效的抑制作用。本发明的第二目的是提供所述曲霉明素A及其衍生物的合成方法。本发明所述通式I中,当R1代表CH(NH2)C(O)R5时,所述曲霉明素A及其衍生物的合成方法为:所述合成方法包括以下具体步骤:(1)化合物ent-22与化合物ent-23进行还原氨化反应,所得产物再与苄氧甲酰氯进行氨基保护反应,得化合物ent-25;(2)所述化合物ent-25与2,2-二甲氧基丙烷进行双羟基保护反应,得化合物ent-26;(3)所述化合物ent-26与臭氧进行氧化双键切断反应,再与L-天冬氨酸双叔丁基酯盐或L-谷氨酸双叔丁基酯盐进行还原氨化反应,所得产物与苄氧甲酰氯进行氨基保护反应,得化合物11;(4)所述化合物11水解脱去丙叉基保护基,得化合物12;(5)所述化合物12进行双羟基氧化切断反应后,再与琼斯试剂进行醛基氧化反应,得化合物13;(6)所述化合物13在酸性条件下水解完全脱去保护基,或先进行羧酸衍生化反应后再完全脱去保护基,即得。上述步骤(1)中,化合物ent-22可由以下方法合成:以化合物ent-21为原料,进行氧化反应,得化合物ent-22。该步骤可优选为:将化合物ent-21溶于二氯 甲烷,加入戴斯马丁氧化剂DMP,在常温下反应,得到化合物ent-22。上述步骤(6)还可以包括:以所述完全脱去保护基的产物为原料,进行羧酸衍生化反应。所述羧酸衍生化反应是指在羧酸基团上进行常规的烃化、酰化、酯化、缩合等反应,即可获得一系列衍生物。按照上述方法,以含有相应基团的具体化合物为原料,可以合成本发明所述化合物I-1、I-2、I-3和I-5。具体而言,本发明所述化合物I-1,即曲霉明素A可由以下方法合成:包括以下具体步骤:(1)将化合物ent-22和ent-23溶于甲醇,加入氰基硼氢化钠和乙酸,室温下进行还原氨化反应;将所得产物溶于乙酸乙酯和饱和碳酸氢钠溶液,加入苄氧甲酰氯,室温下进行氨基保护反应,纯化后,得化合物ent-25;(2)将所述化合物ent-25溶于二氯甲烷,加入樟脑磺酸和2,2-二甲氧基丙烷,室温下进行双羟基保护反应,纯化后,得化合物ent-26;(3)将所述化合物ent-26溶于甲醇,通入臭氧进行氧化双键切断反应,再在无臭氧剩余的环境下加入L-天冬氨酸双叔丁基酯盐酸盐和氰基硼氢化钠,室温下进行还原氨化反应;将所得产物溶于甲苯和水,加入苄氧甲酰氯和碳酸钾,室温下进行氨基保护反应,纯化后,得化合物32;(4)将所述化合物32溶于四氢呋喃和水,加入乙酸或稀盐酸,在加热条件下水解脱去丙叉基保护基,纯化后,得化合物33;(5)将所述化合物33溶于丙酮和水,加入高碘酸钠,在室温下进行双羟基氧化切断反应;所得产物在室温下与琼斯试剂进行醛基氧化反应,纯化后,得化合物34;(6)将所述化合物34溶于二氯甲烷,加入苯甲醚和三氟甲磺酸,水解完全脱去保护基,纯化后,得化合物I-1,即(S,S,S)-AMA。本发明所述衍生物I-2可由以下方法合成:按照合成I-1所述的步骤(1)~(5)合成得到化合物34;(6)将化合物34溶于四氢呋喃,与氯甲酸乙酯和N-甲基吗啉在零摄氏度下反应,纯化后,得到化合物35;将所述化合物35溶于二氯甲烷,加入苯甲醚和三氟甲磺酸,水解完全脱去保护基,纯化后,得到 衍生物I-2的钠盐,经转化,即得衍生物I-2。本发明所述衍生物I-3可以化合物I-1为原料进行羧酸衍生化得到;具体为:将化合物I-1与CH2N2进行甲酯化反应,得衍生物I-3。本发明所述衍生物I-5可由以下方法合成:包括以下具体步骤:按照合成I-1所述的步骤(1)和(2)合成得到化合物ent-26;(3)将所述化合物ent-26溶于甲醇,通入臭氧进行氧化双键切断反应,再在无臭氧剩余的环境下加入L-谷氨酸双叔丁基酯盐酸盐和氰基硼氢化钠,室温下进行还原氨化反应;将所得产物溶于甲苯和水,加入苄氧甲酰氯和碳酸钾,室温下进行氨基保护反应,纯化后,得化合物45;(4)将所述化合物45溶于四氢呋喃和水,加入乙酸或稀盐酸,在加热条件下水解脱去丙叉基保护基,纯化后,得化合物46;(5)将所述化合物46溶于丙酮和水,加入高碘酸钠,在室温下进行双羟基氧化切断反应;所得产物在室温下与琼斯试剂进行醛基氧化反应,纯化后,得化合物47;(6)将所述化合物47溶于二氯甲烷,加入苯甲醚和三氟甲磺酸,水解完全脱去保护基,纯化后,得化合物I-5。本发明所述通式I中,当R1代表C(O)R5时,所述曲霉明素A及其衍生物的合成方法为:所述方法包括以下具体步骤:(1)以化合物37为原料,与高碘酸钠进行氧化双键切断反应,再与L-天冬氨酸双叔丁基酯盐或L-谷氨 酸双叔丁基酯盐进行还原氨化反应,所得产物与苄氧甲酰氯进行氨基保护反应,得化合物14;(2)所述化合物14与臭氧进行氧化双键切断反应,再与琼斯试剂进行醛基氧化反应,得化合物15;(3)所述化合物15在酸性条件下水解完全脱去保护基,或先进行羧酸衍生化反应后再完全脱去保护基,即得。上述步骤(1)中,化合物37可由以下方法合成得到:以化合物36为原料,与苄氧甲酰氯进行氨基保护反应,得化合物37。该步骤可优选为:将化合物36溶于乙酸乙酯和饱和碳酸氢钠溶液,加入苄氧甲酰氯,室温下进行氨基保护反应,纯化后,得化合物37。上述步骤(3)还可以包括:以所述完全脱去保护基的产物为原料,进行羧酸衍生化反应。所述羧酸衍生化反应是指在羧酸基团上进行常规的烃化、酰化、酯化、缩合等反应,即可获得一系列衍生物。按照上述方法,以含有相应基团的具体化合物为原料,可以合成本发明所述化合物I-4。具体而言,本发明所述衍生物I-4可由以下方法合成得到:包括以下具体步骤:(1)将化合物37溶于四氢呋喃和水,在室温下与高碘酸钠进行氧化双键切断反应,再与L-天冬氨酸双叔丁基酯盐酸盐和氰基硼氢化钠进行还原氨化反应,将所得产物溶于甲苯和水,在室温下与苄氧甲酰氯进行氨基保护反应,纯化后,得化合物38;(2)所述化合物38与臭氧进行氧化双键切断反应,再在无臭氧剩余的环境下与琼斯试剂进行醛基氧化反应,纯化后,得化合物39;(3)所述化合物39溶于二氯甲烷,加入苯甲醚和三氟甲磺酸,水解完全脱去保护基,纯化后,得衍生物I-4。本发明所述方法提供了具有特定立体构型的曲霉明素A及其衍生物的合成路线,这种模块化的合成路线可应用于类似结构化合物及相关衍生物的化学合成,为新型抗生素类药物开辟了广阔发展空间。本发明的第三目的是保护所述曲霉明素A及其衍生物在制备药物中的应用。所述曲霉明素A及其衍生物可以与β-内酰胺类抗生素组合使用,用于抑制含有金属β-内酰胺酶的细菌。具体而言,本发明提供了含有所述曲霉明素A及其衍生物的组合物。所述组合物包含本发明所述的曲霉明素A及其衍生物和β-内酰胺类抗生素。所述β-内酰胺类抗生素包括碳青霉烯类抗生素、青霉素类抗生素、头孢菌素类抗生素等;优选为碳青霉烯类抗生素,如美罗培南(Meropenem)。本发明所述组合物优选为所述曲霉明素A与碳青霉烯类抗生素的组合物,该组合物可以实现优异的协 同效果,可以有效提高美罗培南针对含NDM-1细菌的杀菌活性。所述曲霉明素A与碳青霉烯类抗生素优选以摩尔比2~8:1组成,当二者摩尔比为4:1时协同作用最佳。本发明提供的由曲霉明素A及其衍生物与β-内酰胺类抗生素组成的组合物,具有优异的协同效果,可以克服含有金属β-内酰胺酶的细菌对β-内酰胺类抗生素的耐药性。附图说明图1、图2分别为实施例1所述化合物ent-25的1HNMR和13CNMR谱图;图3、图4分别为实施例1所述化合物ent-26的1HNMR和13CNMR谱图;图5、图6分别为实施例1所述化合物32的1HNMR和13CNMR谱图;图7、图8分别为实施例1所述化合物33的1HNMR和13CNMR谱图;图9、图10分别为实施例1所述化合物34的1HNMR和13CNMR谱图;图11、图12分别为实施例1所述化合物(S,S,S)-AMA的1HNMR和13CNMR谱图;图13、图14分别为实施例1所述化合物25的1HNMR和13CNMR谱图;图15、图16分别为实施例1所述化合物26的1HNMR和13CNMR谱图;图17、图18分别为实施例1所述化合物28的1HNMR和13CNMR谱图;图19、图20分别为实施例1所述化合物29的1HNMR和13CNMR谱图;图21、图22分别为实施例1所述化合物30的1HNMR和13CNMR谱图;图23、图24分别为实施例1所述化合物(R,R,S)-AMA的1HNMR和13CNMR谱图;图25为实施例1所述化合物(S,S,S)-AMA和(R,R,S)-AMA对NDM-1蛋白酶活性的抑制效果示意图;图26为实施例2所述衍生物I-2的1HNMR谱图;图27、图28分别为实施例4所述衍生物I-4的1HNMR和13CNMR谱图。具体实施方式以下实施例用于说明本发明,但不用来限制本发明的范围。实施例1:(S,S,S)-AMA及其合成按照以下步骤合成(S,S,S)-AMA:(1)化合物ent-22~ent-25的合成:在一个500mL的圆底烧瓶中加入一个磁子,并加入反应原料ent-21(8.50g,28.79mmol),然后氩气置换气体,向反应瓶中加入二氯甲烷(280mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入DMP(14.6g,34.54mmol),然后升至室温搅拌2.5h;TLC板检测反应完全,在0℃下加入饱和的硫代硫酸钠(300mL)和饱和的碳酸氢钠溶液(300mL),然后升温到室温搅拌1h,用二氯甲烷萃取(200mL×3),将所有萃取液合并,有机相用无水硫酸钠干燥,过滤后,旋干,得到粗品化合物醛ent-22(7.95g),直接用于下一步;在一个250mL的圆底烧瓶中加入一个磁子,并加入反应原料ent-23(936mg,7.99mmol),然后氩气置换气体,向反应瓶中加入甲醇(40mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入溶于(40mL)甲醇的醛ent-22(2.8g,9.59mmol),然后将氰基硼氢化钠(602mg,9.59mmol)和乙酸(0.44mL,7.99mmol)加入反应液中,随后缓慢升至室温搅拌18h;TLC板检测反应完全,旋干,得到粗品二级胺化合物直接用于下一步,将混合物在0℃下溶于乙酸乙酯(80mL)和饱和碳酸氢钠溶液(80mL)中,然后在0℃下加入苄氧甲酰氯(2.25mL,15.98mmol),然后升温到室温搅拌18h,TLC板检测反应完全,用乙酸乙酯萃取(80mL×3),将所有萃取液合并,用饱和食盐水洗(10mL×2),有机相用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=5:1到2:1)得到无色油状二醇ent-25(3.00g,5.66mmol),两步产率71%;所述化合物ent-25的1HNMR和13CNMR谱图分别如图1、图2所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.32-7.25(m,10H),6.04(s,1H),5.29-5.20(m,2H),5.14-5.02(m,5H),4.09-3.93(m,6H),3.63-3.46(m,5H),1.34(s,3H),1.27(s,3H);13CNMR(100MHz,CDCl3):δ156.7,136.1,135.9,131.7,128.5,128.2,128.1,128.0,120.2,109.4,74.6,72.9,67.8,67.1,66.0,64.3,63.5,51.2,49.2,26.0,24.7;IR(neat)νmax3433,2954,2924,1696,1516,1455,1219,1059,771,739,698cm-1;HRMS(ESI):[M+H]+计算值C28H37N2O8:529.2544,测得值:529.2545;[α]23D-40.1(c1.7,MeOH);Rf=0.2(PE:EA=2:1);(2)化合物ent-26的合成:在一个250mL的圆底烧瓶中加入一个磁子,并加入反应原料ent-25(6.49g,12.29mmol),然后氩气置换气体,向反应瓶中加入二氯甲烷(122mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入樟脑磺酸(285.1mg,1.23mmol)和2,2-二甲氧基丙烷(4.5mL,36.87mmol),然后将反应液升到室温反应1h;TLC板检测反应完全,用饱和碳酸氢钠溶液(100mL)淬灭,然后用二氯甲烷(100mL×3)萃取,有机相合并用饱和食盐水水洗(10mL×2),用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=5:1)得到无色油化合物ent-26(6.43g,11.31mmol),产率72%;所述化合物ent-26的1HNMR和13CNMR谱图分别如图3、图4所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.32-7.25(m,10H),5.97(br.s,1H),5.27-5.05(m,7H),5.14-5.02(m,5H),4.42(m,2H),4.09-3.95(m,4H),3.67(m,3H),3.34-3.29(m,1H),1.40-1.30(m,12H);13CNMR(100MHz,CDCl3):δ156.6,156.2,136.4,136.0,132.2,131.8,128.5,128.1,127.9,119.6,119.0,109.8,109.2,76.3,75.6,75.1,67.8,67.7,67.0,66.0,61.1,52.1,51.4,46.9,29.5,29.4,29.2,26.4,26.1,25.1,25.0,22.6,14.0;IR(neat)νmax3353,2984,2918,2850,1699,1512,1215,1125,970,740,697cm-1;HRMS(ESI):[M+H]+计算值C31H41N2O8:569.2857,测得值:569.2860;[α]23D-29.7(c0.5,MeOH);Rf=0.5(PE:EA=5:1);(3)化合物32的合成:在一个250mL的圆底烧瓶中加入一个磁子,并加入反应原料ent-26(5.00g,8.79mmol),然后氩气置换气体,向反应瓶中加入甲醇(88mL),将反应体系置于-78℃的干冰丙酮浴中,然后通入臭氧(40mL)大约1h直到溶液颜色变蓝,TLC板检测反应完全,停止通入臭氧,通入氩气0.5h,并在-78℃加入二甲硫醚(6.5mL,87.9mmol),然后缓慢升至室温搅拌1h,淀粉碘化钾试纸检测无臭氧后,在0℃将L-天冬氨酸双叔丁基酯盐酸盐17(2.6g,9.23mmol)和氰基硼氢化钠(718mg,11.43mmol)加入反应液中,随后缓慢升至室温搅拌18h;TLC板检测反应完全,旋干,得到粗品二级胺化合物直接用于下一步,将混合物在0℃下溶于甲苯(60mL)和水(60mL)中,然后在0℃下加入苄氧甲酰氯(2.49mL,17.58mmol)和碳酸钾(2.43g,17.58mmol)然后升温到室温搅拌5h,TLC板检测反应完全,用乙酸乙酯萃取(100mL×3),将所有萃取液合并,用饱和食盐水洗(10mL×2),有机相用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=10:1到5:1)得到无色油状化合物32(3.94g,4.22mmol),三步产率48%;所述化合物32的1HNMR和13CNMR谱图分别如图5、图6所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.30-7.26(m,15H),5.10-5.01(m,6H),5.27-5.05(m,7H),4.37-4.31(m,3H),4.10-3.56(m,9H),3.29-2.61(m,3H),1.64(s,1H),1.43-1.28(m,30H);13CNMR(100MHz,CDCl3):δ170.7,170.4,169.2,169.1,156.7,156.2,155.7,136.4,136.1,128.4,128.2,128.0,127.8,109.7,109.3,109.2,81.9,80.8,80.6,76.4,76.2,76.0,75.3,67.6,67.5,66.7,66.1,59.3,58.6,51.7,37.6,36.5,28.0,27.8,26.3,26.2,26.0,25.2,24.9;IR(neat)νmax2980,2931,1705,1512,1455,1219,1151,1057,770,739,698cm-1;HRMS(ESI):[M+H]+计算值C50H68N3O14:934.4696,测得值:934.4673;[α]23D-26.9(c1.3,MeOH);Rf=0.5(PE:EA=3:1);(4)化合物33的合成:在一个100mL的圆底烧瓶中加入一个磁子,并加入反应原料32(1.5g,1.61mmol),然后氩气置换气体,向反应瓶中加入四氢呋喃(7.5mL)和水(7.5mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入乙酸(23mL)然后将反应液加热到50℃反应12h;TLC板检测反应完全,旋干溶剂,用饱和碳酸氢钠溶液(30mL)淬灭,然后用二氯甲烷(30mL×3)萃取,有机相合并用用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=5:1到1:1)得到无色油化合物33(1.03g,1.21mmol)产率75%;所述化合物33的1HNMR和13CNMR谱图分别如图7、图8所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.32-7.25(m,15H),5.15-5.00(m,6H),4.60-4.17(m,2H),4.11-4.00(m,3H),3.75-3.45(m,10H),3.00-2.82(m,3H),1.85(br.s,1H),1.43-1.27(m,18H);13CNMR(100MHz,CDCl3):δ170.2,169.0,156.9,156.6,136.2,135.8,129.5,128.8,128.6,128.4,128.2,128.1,128.0,82.3,81.3,77.3,73.7,71.8,70.7,68.2,67.9,67.7,67.0,63.6,63.3,62.1,59.7,59.5,59.2,51.7,37.2, 36.5,28.0,19.1;IR(neat)νmax3425,2976,1696,1498,1455,1392,1247,1148,1054,1001,771,736,697cm-1;HRMS(ESI):[M+H]+计算值C44H60N3O14:854.4070,测得值:854.4053;[α]23D-38.1(c1,MeOH);Rf=0.2(PE:EA=1:2);(5)化合物34的合成:在一个25mL的圆底烧瓶中加入一个磁子,并加入反应原料33(1.03g,1.21mmol),然后氩气置换气体,向反应瓶中加入丙酮(9.6mL)和水(2.4mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入高碘酸钠(1.29g,6.05mmol)然后将反应液升到室温反应3h;TLC板检测反应完全,然后于0℃向反应液中加入琼斯试剂(3.0mL,6.05mmol),然后升到室温反应18h。TLC板检测反应完全,用(15mL)饱和食盐水淬灭,用乙醚(15mL×3)萃取,有机相合并用用无水硫酸钠干燥,过滤后,旋干,用HPLC(70%MeOH/0.1%HCOOHto84%MeOH/0.1%HCOOH)分离纯化得到白色固体化合物34(337mg,0.41mmol)产率34%;所述化合物34的1HNMR和13CNMR谱图分别如图9、图10所示;其表征信息具体为:m.p.=76-78℃;1HNMR(400MHz,CDCl3,50℃):δ9.70(br.s,2H),7.30-7.26(m,15H)6.58(br.s,0.4H),5.98-5.85(m,0.5H),5.28-4.97(m,6H),4.52-4.34(m,3H),4.03-3.46(m,4H),3.04-2.87(m,1H),2.80-2.52(m,1H),1.48-1.32(m,18H);13CNMR(100MHz,CDCl3):δ174.7174.0,173.4,170.8,169.9,168.9,168.7,168.6,168.5,156.7,156.4,156.0,135.9,135.8,135.5,135.3,135.1,128.3,128.0,127.9,82.2,81.9,81.2,68.1,67.5,66.9,61.9,61.2,60.3,59.6,53.2,50.4,37.2,36.8,36.3,36.1,27.8,27.6;IR(neat)νmax2977,1705,1499,1456,1455,1255,1151,1061,1003,843,771,738,698cm-1;HRMS(ESI):[M+H]+计算值C42H52N3O14:822.3444,测得值:822.3451;[α]23D-32.8(c1,MeOH);Rf=0.4(CH2Cl2:MeOH=20:1);(6)化合物(S,S,S)-AMA的合成:在一个10mL的圆底烧瓶中加入一个磁子,并加入反应原料34(41mg,0.05mmol),然后氩气置换气体,向反应瓶中加入二氯甲烷(9.6mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入苯甲醚(27μL,0.30mmol)和三氟甲磺酸(27μL,0.25mmol)然后将反应液置于0℃的冰水混合浴中反应0.5h,然后升到室温反应1h;TLC板检测反应完全,然后于0℃向反应液中加入NaHCO3(27.7mg,0.33mmol)在(8mL)水中,用二氯甲烷(10mL×3)萃取,旋干水相,并用H2O:MeOH:AcOH(1mL:3mL:40μL)重结晶分离纯化得到白色固体化合物I-1(13.7mg,0.0455mmol)产率91%,所述化合物I-1即(S,S,S)-AMA;所述(S,S,S)-AMA的1HNMR和13CNMR谱图分别如图11、图12所示;其表征信息具体为:m.p.=246-256℃;1HNMR(600MHz,D2O):δ3.76-3.73(m,2H),3.34-3.32(dd,J=4.8,9.6Hz,1H)3.21-3.16(m,2H),3.07-3.03(dd,J=9.6,13.2Hz,1H),2.80-2.77(dd,J=4.2,13.2Hz,1H),2.74-2.71(dd,J=4.2,17.6Hz,1H),2.62-2.58(dd,J=9.0,18.0Hz,1H);13CNMR(150MHz,D2O):δ176.6,176.3,172.5,172.4,59.0,58.7,53.8,47.2,46.4,35.0;IR(neat)νmax3386,2362,1592,1392,1069,937,836,690cm-1;HRMS(ESI):[M-H]-计算值C10H16N3O8:306.0932,测得值:306.0929;[α]23D-51.2(c0.8,磷酸盐缓冲液pH=7)。本实施例还合成化合物(R,R,S)-AMA,具体步骤和反应条件与上述(S,S,S)-AMA的合成相同,区别仅在于替换了相应立体构型的原料,以下为各步骤的反应历程及相关产物的表征信息:(1)化合物25的合成:所得化合物25的1HNMR和13CNMR谱图分别如图13、图14所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.32-7.25(m,10H),6.04(s,1H),5.29-5.20(m,2H),5.14-5.02(m,5H),4.09-3.93(m,6H),3.63-3.46(m,5H),1.34(s,3H),1.27(s,3H);13CNMR(100MHz,CDCl3):δ156.7,136.1,135.9,131.7,128.5,128.2,128.1,128.0,120.2,109.4,74.6,72.9,67.8,67.1,66.0,64.3,63.5,51.2,49.2,26.0,24.7;IR(neat)νmax2956,2917,2849,1711,1462,1275,1071,746,702cm-1;HRMS(ESI):[M+H]+计算值C28H37N2O8:529.2544,测得值:529.2551;[α]23D+23.9(c0.2,MeOH);Rf=0.2(PE/EtOAc=2/1);(2)化合物26的合成:所得化合物26的1HNMR和13CNMR谱图分别如图15、图16所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.32-7.25(m,10H),5.97(br.s,1H),5.27-5.05(m,7H),5.14-5.02(m,5H),4.42(m,2H),4.09-3.95(m,4H),3.67(m,3H),3.34-3.29(m,1H),1.40-1.30(m,12H);13CNMR(100MHz,CDCl3):δ156.6,156.2,136.4,136.0,132.2,131.8,128.5,128.1,127.9,119.6,119.0,109.8,109.2,76.3,75.6,75.1,67.8,67.7,67.0,66.0,61.1,52.1,51.4,46.9,29.5,29.4,29.2,26.4,26.1,25.1,25.0,22.6,14.0;IR(neat)νmax2916,2849,1699,1508,1462,1275,1261,1063,801,763,669cm-1;HRMS(ESI):[M+H]+计算值C31H41N2O8:569.2857,测得值:569.2861;[α]23D+33.5(c1.0,MeOH);Rf=0.5(PE/EtOAc=5/1);(3)化合物28的合成:所述化合物28的1HNMR和13CNMR谱图分别如图17、图18所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.30-7.26(m,15H),5.12-4.99(m,6H),4.48-4.30(m,6H),4.48-4.30(m,2H),4.09-3.47(m,9H),3.22-3.04(m,2H),2.63(br.s,1H),1.61(s,1H),1.44-1.27(m,30H);13CNMR(100MHz,CDCl3):δ170.3,169.2,156.7,156.1,155.8,155.7,136.4,136.1,136.0,135.7,128.4,128.1,128.0,127.9,109.2,109.1,82.0,81.0,80.8,80.7,76.3,76.0,75.6,67.5,66.7,66.4,66.0,60.3,59.1,53.4,51.7,48.1,45.3,37.5,28.0,27.8,26.5,26.2,26.0,25.2,25.0,24.3;IR(neat)νmax2916,2848,1712,1513,1461,1422,1370,1263,1153,1059,896,847,801,736,702cm-1;HRMS(ESI):[M+H]+计算值C50H68N3O14:934.4696,测得值:934.4669;[α]23D+0.6(c0.4,MeOH);Rf=0.5(PE/EtOAc=3/1);(4)化合物29的合成:所得化合物29的1HNMR和13CNMR谱图分别如图19、图20所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.31-7.25(m,15H),5.40-4.82(m,6H),4.33-3.85(m,5H),3.75-3.30(m,9H),3.07-2.63(m,5H),1.91-1.84(m,1H),1.44-1.27(m,18H);13CNMR(100MHz,CDCl3):δ170.7,170.4,169.4,169.0,157.1,157.0,156.6,156.5,156.0,136.3,136.1,135.9,135.7,128.9,128.6,128.5,128.4,128.1,128.0,82.5,82.3,82.2,81.2,81.0,77.3,73.3,71.1,68.7,68.2,67.8,67.6,67.4,67.0,63.5,63.3,63.1,62.1,61.9,61.8,59.9,51.3,51.0,49.1,37.6,36.0,28.0,27.8,27.7;IR(neat)νmax3413,2977,2883,1824,1697,1499,1455,1393,1252,1152,1054,1002,772,737,698cm-1;HRMS(ESI):[M+H]+计算值C44H60N3O14:854.4070,测得值:854.4057;[α]23D+8.6(c0.8,MeOH);Rf=0.2(PE/EtOAc=1/2);(5)化合物30的合成:所得化合物30的1HNMR和13CNMR谱图分别如图21、图22所示;其表征信息具体为:1HNMR(400MHz,CDCl3,50℃):δ7.72(br.s,2H),7.29-7.25(m,15H)6.65-6.52(m,0.4H),5.11-4.98(m,6H),4.57-4.28(m,3H),4.11-3.58(m,4H),3.20-2.90(m,1H),2.80-2.59(m,1H),1.40-1.01(m,18H);13CNMR(100MHz,CDCl3):δ174.9174.4,173.4,172.5,170.8,170.4,170.3,169.1,168.9,157.0,156.6,156.4,156.2,156.1,155.7,155.5,155.2,136.2,136.0,135.8,135.5,135.4,135.2,135.1,128.9,128.6,128.4,128.2,128.1,82.3,81.4,81.2,81.0,77.3,68.6,68.5,68.3,68.0,67.9,67.5,67.1,63.1,62.5,60.5,60.1,54.9,53.3,53.0,52.0,50.1,49.9,37.6,37.5,36.4,36.2,29.6,28.0,27.7,27.6,19.1;IR(neat)νmax2978,1697,1499,1420,1394,1368,1249,1060,1001,914,842,771,732,695cm-1;HRMS(ESI):[M+H]+计算值C42H52N3O14:822.3444,测得值:822.3437;[α]23D+9.3(c1.9,MeOH);Rf=0.4(CH2Cl2/MeOH=20/1);(6)化合物(R,R,S)-AMA的合成:所述化合物(R,R,S)-AMA的1HNMR和13CNMR谱图分别如图23、图24所示;其表征信息具体为:1HNMR(400MHz,D2O):δ3.68-3.66(dd,J=4,5.2Hz,1H),3.64-3.61(dd,J=4,8Hz,1H)3.24-3.19(m,2H),3.14-3.10(dd,J=5.6,13.6Hz,1H),2.86-2.80(dd,J=10.8,13.2Hz,1H),2.71-2.61(m,2H),2.57-2.51(dd,J=8,17.6Hz,1H);13CNMR(100MHz,D2O):δ177.5,177.2,173.2,173.0,60.0,59.3,54.4,47.8,47.0,34.8;IR(neat)νmax3280,2340,1634,1152,1063,935,825,668cm-1;HRMS(ESI):[M-H]-计算值C10H16N3O8:306.0932,测得值:306.0933;[α]23D+6.8(c0.3,磷酸盐缓冲液pH=7)。本实施例进一步对上述化合物(S,S,S)-AMA和(R,R,S)-AMA的活性进行检测。具体方法为:将纯化的NDM-1蛋白酶在30μM头孢硝噻吩溶液(含50μMHepes、10μMZnSO4,pH=7.4)中孵育30分钟,分装于96孔板,分别将(S,S,S)-AMA和(R,R,S)-AMA按照浓度梯度加入不同的孔中,在30℃下孵育10~15分钟,使用EnVision2104多标记检测仪(PerkinElmer)在30℃、490nm条件下进行检测,结果如图25所示。由图25所示结果可知,本实施例提供的(S,S,S)-AMA对NDM-1蛋白酶活性呈现剂量依赖性抑制,IC50值为8.1μM;而化合物(R,R,S)-AMA对NDM-1蛋白酶无明显抑制效果。实施例2:衍生物I-2及其合成按照以下步骤合成衍生物I-2:(1)化合物35的合成:在一个10mL的圆底烧瓶中加入一个磁子,并加入反应原料34(44mg,0.054mmol),该反应原料即实施例1步骤(5)合成得到的中间化合物34;然后氩气置换气体,向反应瓶中加入四氢呋喃(1ml),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入N-甲基吗啉(15.4μL,0.14mmol),随后加入氯甲酸乙酯(12.3μL,0.13mmol),然后在0℃搅拌1h;TLC板检测反应完全,在0℃下加入加入新制备的羟胺(0.16mmol)溶于(1ml)甲醇中,然后升温到室温搅拌2h,TLC板检测反应完全,然后用旋转蒸发仪将溶剂旋走,用水(2ml)淬灭反应,用二氯甲烷萃取(3ml×3),将所有萃取液合并,有机相用无水硫酸钠干燥,过滤后,旋干,用LC-MS液相制备仪分离纯化得单羟胺酸化合物35(15mg,0.018mmol,33%)和双羟胺酸化合物(9mg,0.011mmol,20%);所述化合物35的氢谱数据为:1HNMR(400MHz,CDCl3)δ7.28-7.26(m,15H),6.42(br.s,0.3H),5.99-5.91(m,,0.6H),5.29-4.94(m,6H),4.45-4.29(m,4H),4.12-3.83(m,3H),3.66-3.30(m,2H),3.18-2.58(m,3H),1.45-1.25(m,18H);(2)衍生物I-2的合成:在一个10mL的圆底烧瓶中加入一个磁子,并加入所述单羟胺酸化合物35(9mg,0.011mmol),然后氩气置换气体,向反应瓶中加入二氯甲烷(1ml),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入苯甲醚(7.3μL,0.065mmol),随后加入三氟甲磺酸(7.6μL,0.086mmol),然后在0℃搅拌0.5h,然后升温到室温搅拌1h,TLC板检测反应完全,再加入溶于水(5ml)中的NaHCO3(10mg,0.119mmol),用二氯甲烷萃取(5ml×3),将水相旋干,再用sephadexG-10型号的葡聚糖凝胶树脂除盐得到化合物I-2的钠盐(3mg,0.010mmol,71%),经转化即得衍生物I-2的钠盐,经转化,即得衍生物I-2;所述衍生物I-2的氢谱谱图如图26所示,具体数据为:1HNMR(400MHz,D2O)δ3.79-3.71(m,2H),3.30-3.26(dd,J=4.0,8.4Hz,1H),3.22-3.18(dd,J=1.6,5.6Hz,1H),3.08-2.98(m,2H),2.77-2.68(m,2H),2.63-2.57(dd,J=8.4,17.6Hz,1H)。实施例3:衍生物I-3及其合成按照以下步骤合成衍生物I-3:在一个10mL的圆底烧瓶中加入一个磁子,并加入实施例1所得的I-1(15mg,0.049mmol),然后氩气置换气体,向反应瓶中加入水(1mL)和四氢呋喃(1mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入溶在乙醚溶液中的重氮甲烷(0.5M)然后将反应液置于0℃的冰水混合浴中反应0.5h,然后升到室温反应3h;LC-MS检测反应完全,用乙醚(5mL×3)萃取,旋干水相,得到衍生物I-3。实施例4:衍生物I-4及其合成按照以下步骤合成衍生物I-4:(1)化合物37~38的合成:在一个100mL的圆底烧瓶中加入一个磁子,并加入反应原料化合物36(7.99mmol),然后氩气置换气体,在0℃下溶于乙酸乙酯(30mL)和饱和碳酸氢钠溶液(30mL)中,然后在0℃下加入苄氧甲酰氯(3.37mL,23.97mmol),然后升温到室温搅拌18h,TLC板检测反应完全,用乙酸乙酯萃取(30mL×3),将所有萃取液合并,用饱和食盐水洗(5mL×2),有机相用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=5:1到2:1)得到无色油状二醇37(3.3g,6.99mmol),产率80%;在一个50mL的圆底烧瓶中加入一个磁子,加入所述化合物37(0.4g,1.37mmol),然后氩气置换气体,向反应瓶中加入四氢呋喃(4.4mL)和水(8.8mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入高碘酸钠(0.44g,2.06mmol)然后将反应液升到室温反应2h;TLC板检测反应完全,在0℃将L-天冬氨酸双叔丁基酯盐酸盐(0.37g,1.51mmol)和氰基硼氢化钠(129mg,2.06mmol)加入反应液中,随后缓慢升至室温搅拌18h;TLC板检测反应完全,旋干,得到粗品二级胺化合物直接用于下一步,将混合物在0℃下溶于甲苯(6.5mL)和水(6.5mL)中,然后在0℃下加入苄氧甲酰氯(0.23mL,1.64mmol)和碳酸钾(0.24g,1.78mmol)然后升温到室温搅拌5h,TLC板检测反应完全,用乙酸乙酯萃取(20mL×3),将所有萃取液合并,用饱和食盐水洗(5mL×2),有机相用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=10:1到8:1)得到无色油状化合物38(0.3g,0.48mmol),上述三步反应的产率为35%;(2)化合物39的合成:在一个25mL的圆底烧瓶中加入一个磁子,加入所述化合物38(0.3g,0.48mmol),然后氩气置换气体,向反应瓶中加入丙酮(4.8mL),将反应体系置于-78℃的干冰丙酮浴中,然后通入臭氧大约15minh直到溶液颜色变蓝,TLC板检测反应完全,停止通入臭氧,通入氩气0.5h,然后于0℃向反应液中加入琼斯试剂(0.96mL,1.92mmol),然后升到室温反应18h;TLC板检测反应完全,用(10mL)饱和食盐水淬灭,用乙醚(10mL×3)萃取,有机相合并用用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(二氯甲烷/甲醇=50:1到20:1)分离纯化得到无色油状化合物39(125mg,0.19mmol),上述两步反应的产率为40%;(3)衍生物I-4的合成:在一个25mL的圆底烧瓶中加入一个磁子,并加入所述化合物39(35mg,0.05mmol),然后氩气置换气体,向反应瓶中加入二氯甲烷(5mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入苯甲醚(27μL,0.30mmol)和三氟甲磺酸(27μL,0.25mmol)然后将反应液置于0℃的冰水混合浴中反应0.5h,然后升到室温反应1h。TLC板检测反应完全,然后于0℃向反应液中加入NaHCO3(27.7mg,0.33mmol)在(8mL)水中,用二氯甲烷(10mL×3)萃取,旋干水相,并用H2O:MeOH:AcOH(1mL:3mL:40μL)重结晶分离纯化得到白色固体化合物I-4(11.1mg,0.04mmol),产率81%;所述衍生物I-4的氢谱和碳谱谱图分别如图27、图28所示,表征数据为:1HNMR(400MHz,D2O):δ3.64-3.61(m,1H),3.57-3.51(m,1H),3.41-3.39(m,2H),3.17-3.15(d,J=6.4Hz,2H),2.66-2.55(m,1H),2.46-2.40(dd,J=8.4,17.2Hz,1H);13CNMR(100MHz,D2O):δ177.9,175.1,173.4,172.6,60.1,58.6,48.9,45.8,36.8。实施例5:衍生物I-5及其合成按照以下步骤合成衍生物I-5:其中,步骤(1)和(2)与实施例1合成I-1的步骤(1)和(2)相同;(3)化合物45的合成:在一个100mL的圆底烧瓶中加入一个磁子,并加入反应原料ent-26(2.00g,3.52mmol),然后氩气置换气体,向反应瓶中加入甲醇(35mL),将反应体系置于-78℃的干冰丙酮浴中,然后通入臭氧大约0.5h直到溶液颜色变蓝,TLC板检测反应完全,停止通入臭氧,通入氩气0.5h,并在-78℃加入二甲硫醚(2.6mL,35.2mmol),然后缓慢升至室温搅拌1h,淀粉碘化钾试纸检测无臭氧后,在0℃将L-谷氨酸双叔丁基酯盐酸盐44(1.04g,3.69mmol)和氰基硼氢化钠(287mg,4.57mmol)加入反应液中,随后缓慢升至室温搅拌18h;TLC板检测反应完全,旋干,得到粗品二级胺化合物直接用于下一步,将混合物在0℃下溶于甲苯(24mL)和水(24mL)中,然后在0℃下加入苄氧甲酰氯(1mL,7.03mmol)和碳酸钾(0.97g,7.03mmol)然后升温到室温搅拌5h,TLC板检测反应完全,用乙酸乙酯萃取(40mL×3),将所有萃取液合并,用饱和食盐水洗(5mL×2),有机相用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=10:1到5:1)得到无色油状化合物45(1.40g,1.48mmol),上述三步反应的产率为42%;(4)化合物46的合成:在一个50mL的圆底烧瓶中加入一个磁子,加入所述化合物45(1.0g,1.06mmol),然后氩气置换气体,向反应瓶中加入四氢呋喃(4.5mL)和水(4.5mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入乙酸(15mL)然后将反应液加热到50℃反应12h;TLC板检测反应完全,旋干溶剂,用饱和碳酸氢钠溶液(15mL)淬灭,然后用二氯甲烷(20mL×3)萃取,有机相合并用用无水硫酸钠干燥,过滤后,旋干,用硅胶柱色谱分离纯化(石油醚/乙酸乙酯=5:1到1:1)得到无色油化合物46(0.62g,0.72mmol)产率68%;(5)化合物47的合成:在一个25mL的圆底烧瓶中加入一个磁子,加入所述化合物46(0.62g,0.72mmol),然后氩气置换气体,向反应瓶中加入丙酮(6mL)和水(1.5mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入高碘酸钠(0.77g,3.63mmol)然后将反应液升到室温反应3h;TLC板检测反应完全,然后于0℃向反应液中加入琼斯试剂(1.8mL,3.63mmol),然后升到室温反应18h。TLC板检测反应完全,用(10mL)饱和食盐水淬灭,用乙醚(15mL×3)萃取,有机相合并用用无水硫酸钠干燥,过滤后,旋干,用HPLC(70% MeOH/0.1%HCOOHto84%MeOH/0.1%HCOOH)分离纯化得到白色固体化合物47(180mg,0.22mmol)产率30%;(6)衍生物I-5的合成:在一个10mL的圆底烧瓶中加入一个磁子,加入所述化合物47(30mg,0.035mmol),然后氩气置换气体,向反应瓶中加入二氯甲烷(7mL),将反应体系置于0℃的冰水混合浴中,待冷却到0℃后,然后加入苯甲醚(19μL,0.21mmol)和三氟甲磺酸(19μL,0.18mmol)然后将反应液置于0℃的冰水混合浴中反应0.5h,然后升到室温反应1h;TLC板检测反应完全,然后于0℃向反应液中加入NaHCO3(19.4mg,0.23mmol)在(6mL)水中,用二氯甲烷(7mL×3)萃取,旋干水相,并用H2O:MeOH:AcOH(1mL:3mL:40μL)重结晶分离纯化得到衍生物I-5(8.9mg,0.028mmol),产率80%。采用实施例1提供的活性检测方法分别对实施例2~5提供的衍生活性进行检测。由检测结果可知,衍生物I-2、I-3、I-4、I-5均对NDM-1蛋白酶活性呈现剂量依赖性抑制。实施例6:含有(S,S,S)-AMA的组合物本实施例提供的组合物由实施例1所得(S,S,S)-AMA和碳青霉烯类抗生素美罗培南以摩尔比4:1组成。本实施例同时提供了以下对比例:用氨苄青霉素(青霉素类抗生素)、头孢噻肟(头孢菌素类抗生素)、庆大霉素(氨基糖苷类抗生素)、环丙沙星(喹诺酮类抗生素)、四环素(四环素类抗生素)以相同用量比(摩尔比4:1)代替美罗培南,得到组合物。阳性对照:对含有NDM-1酶的肺炎克雷伯菌ATCCBAA-2146具有天然抑菌效果的硫酸多粘菌素B。阴性对照:单独使用氨苄青霉素、头孢噻肟、庆大霉素、环丙沙星、四环素、美罗培南或化合物(S,S,S)-AMA。检测本实施例以及各对比例、阳性对照、阴性对照对含有NDM-1酶的肺炎克雷伯菌ATCCBAA-2146的抑制效果,结果如表1所示;MIC表示最低抑菌浓度。表1:对含有NDM-1酶的肺炎克雷伯菌ATCCBAA-2146的抑菌效果组成MIC(μg/ml)组成MIC(μg/ml)美罗培南>100美罗培南+(S,S,S)-AMA<0.375氨苄青霉素>100氨苄青霉素+(S,S,S)-AMA≈1头孢噻肟>100头孢噻肟+(S,S,S)-AMA≈1庆大霉素>100庆大霉素+(S,S,S)-AMA>10环丙沙星>100环丙沙星+(S,S,S)-AMA>10四环素>100四环素+(S,S,S)-AMA>10(S,S,S)-AMA>100硫酸多粘菌素B0.78由表1可知,实施例1提供的(S,S,S)-AMA与美罗培南的最低抑菌浓度显著低于其它组合物,二者具有显著的增效效果,(S,S,S)-AMA可有效提高美罗培南对含有NDM-1酶的肺炎克雷伯菌的抑菌效果。虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1