一种PLA-PEG-PLA树枝状聚合物的制备方法及其应用与流程
本发明涉及药物化学技术领域,具体涉及的说,涉及一种pla-peg-pla树枝状聚合物、制备方法及其应用。
背景技术:
聚合物胶束是近年来发展起来的一种主要针对难溶性药物的纳米药物控释系统。聚合物胶束给药系统能显著提高药物的溶解度;减少药物毒副反应,从而提高治疗剂量;药物包裹于核中,可避免药物降解失活;胶束粒径很小,通常在l00nm以下,可以躲避网状内皮系统(res)的吞噬,延长体循环时间,通过增强渗透效应(epr效应)达到对肿瘤被动靶向的效果,从而提高治疗效果。至今为止,形成胶束最重要的材料依然是两亲性嵌段共聚物。胶束中共聚物疏水端向内,亲水端向外,呈典型的核-壳结构,而疏水性的内核主要用来增溶难溶性药物。
对于胶束这种纳米药物控释系统,核心为高分子辅料,载药胶束中高分子辅料和药物之间的相互作用在很大程度上决定了该胶束体内外的性质,同时在很大程度上影响着药效及毒副反应。然而,至今为止,在fda批准上市的药物释放系统中,真正经过人体检验并被证明具有高度安全性的合成高分子辅料只有聚乙二醇(peg)和丙交酯/乙交酯共聚物(plga)两类。这两类高分子及其共聚物也在其他大量进行中的临床实验中作为辅料使用。一般认为peg可经过肾脏排泄,因此不会在体内聚集;而plga最终的降解产物为二氧化碳和水,对人体也没有毒害。目前,用于制备胶束的高分子辅料中研究最广泛,应用最成熟的是甲氧基聚乙二醇和聚消旋丙交酯的嵌段共聚物(mpeg-pla)。
mpeg-pla共聚物不是药典收载成分。根据peg的分子量和mpeg/pla比例的不同,可以形成不同结构和分子量的共聚物。此类共聚物具有两亲性,当在溶液中形成胶束时,胶束外围为亲水性的mpeg链段,内核为疏水性的pla链段,利用共聚物材料的这一性质可以对一些难溶性药物进行增溶。mpeg-pla生物相容性良好,且合成过程简单、易行,其本身可生物降解,体内降解为乳酸和peg,二者均可直接排出体外。研究表明:mpeg-pla无致癌性、无生殖毒性、无致畸、无致突变性,可以顺利以原形从肾脏排泄。
目前,以mpeg-pla为载体的聚合物胶束药物释放系统较为成熟,并在实验室阶段取得了一定的进展,但临床转化的例子依然极少,至今没有fda批准上市的该类胶束制剂。其中最大的原因是mpeg-pla胶束很不稳定(尤其是体内稳定性更差)。例如以mpeg-pla为载体的多西他赛胶束即使在较低的浓度下(2mg/ml),室温下6个小时即发生药物沉淀, 研究发现:这种胶束在进入体内以后迅速解体,药物随即和血液中的蛋白(如白蛋白)结合然后往组织转运,因此无法发挥胶束的增强渗透效应(epr效应)。
虽然胶束作为一种比较有希望的难溶性抗癌药载体受到广泛关注,但以mpeg-pla为辅料的胶束的不稳定性(尤其是体内表现)使得这一纳米制剂的epr效应无法发挥,无法实现靶向给药,因此药效也难以提高。另一方面,在现阶段,主动靶向的胶束前景并不乐观。因此,现阶段对于mpeg-pla胶束比较可行的开发思路是提高胶束的稳定性,尤其是体内稳定性,既保留该高分子优良的生物相容性,同时通过发挥胶束epr效应来提高载体的靶向性,进而提高药效。为此,人们做了大量的努力。如专利201010001047公开了一种在胶束溶液中添加氨基酸提高胶束稳定性的方法,而作为辅助添加剂的氨基酸成分是否在进入体内后经过血液的稀释是否依然能保持胶束的稳定性不得而知,因此体内使用的效果并不明确;添加的氨基酸作为非惰性成分,对药效的发挥是否产生影响同样存在不确定性。另外有研究认为将紫杉醇和多西他赛一起包埋于嵌段共聚物胶束中可显著提高胶束的稳定性,但这种复合药物胶束在临床上使用较难得到认可,体内稳定性目前不得而知。
综上所述,胶束制剂的开发面临较大困难,一方面,是已被临床证明安全的用于药物释放系统的高分子辅料的选择极其有限,而开发一种全新的新型高分子辅料无疑面临巨大风险。其次,已经见诸报道的各类胶束制剂的药效较传统制剂难以显著提升,例如韩国三养社研发的紫杉醇胶束genexolpm虽在美国完成三期临床实验,但由于药效不明显,至今未获得fda上市批准。第三,目前见诸报道的胶束辅料通用性较差,通常一种辅料只能针对一两种药物有效成分形成胶束,影响了其推广应用。
技术实现要素:
为了解决上述问题,本发明以具有公认的安全性的mpeg-pla嵌段共聚物为基础,提供一种新型的pla-peg-pla树枝状聚合物、制备方法及其应用。
本发明提供一种pla-peg-pla树枝状聚合物,分子结构式如下所示:
其中:r4是疏水性基团封端的聚丙交酯链段。
上述树枝状聚合物中,所述r4为疏水性基团封端的聚丙交酯链段,其分子式为
本发明中,端基改性方面所选择的疏水性基团y主要考虑三个方面的作用:第一,能够增加高分子辅料中疏水链段的疏水性,从而提高胶束对药物的包裹能力。由于mpeg-pdlla中末尾端羟基具有一定的亲水性,和疏水性的药物如多西他赛之间难以形成较强的作用力,影响胶束对药物的包核,而通过接枝疏水性端基可以显著提高其疏水性;第二,通过改善药物分子和嵌段共聚物中疏水性链段的相容性,增加药物分子和疏水链的作用力,使药物分子被胶束内核中不易溶出;第三,能够形成分子间作用力,包括氢键、共轭效应,从而有助于将药物包裹在胶束的内核中,进一步增加胶束的稳定性。
上述树枝状聚合物中,所述的疏水性基团y包括下述基团中的任何一种:烷基、叔丁氧羰基、苄基、芴甲氧羰基或者苯环。优选的,为芴甲氧羰基或者苯环。
上述树枝状聚合物中,总数均分子量为2000-60000,其中peg链段的数均分子量为1000-10000。
本发明还提供一种上述的pla-peg-pla树枝状聚合物的制备方法,包括如下步骤如下:
(1)合成端基树枝化聚乙二醇g4-peg-g4
将nhs-peg-nhs和4代树枝化聚酯g4-nh2溶解于有机溶剂后,加入三乙胺tea室温搅拌反应,反应物用乙醚沉淀后的白色粉末状物质为g4-peg-g4;其中:nhs-peg-nhs、g4-nh2和tea的摩尔比为1:(1-5:(0.1-5);
(2)含有32个羟基的树枝化聚乙二醇peg-g4-(oh)32合成
g4-peg-g4溶解于有机醇,分批加入dowexh+交换树脂,室温反应12h后过滤除去树脂,溶液用乙醚沉淀后的白色粉末状固体即为peg-g4-(oh)32;
(3)哑铃型嵌段共聚物plan合成
将dl-丙交酯和peg-g4-(oh)32加入到干燥的聚合瓶中,以丙交酯摩尔比例0.05-0.5%的snoct2作为催化剂,真空熔封聚合瓶后将反应物于130-150℃下聚合6-24h,产物用氯仿溶解,甲醇沉淀后所得白色固体为目标共聚物哑铃型嵌段共聚物plan。
(4)封端反应
将哑铃型嵌段共聚物plan和疏水性的封端剂反应,经过纯化后得到目标pla-peg-pla树枝状聚合物。
进一步的,本发明还提供一种上述的pla-peg-pla树枝状聚合物作为药物载体在制备药物制剂中的应用。
本发明的有益效果在于:首先树枝化的疏水链段由于其特殊的分子结构,胶束核中链段的缠绕程度远高于线型聚合物,药物包埋于其中更不易溶出,由此可提高载药胶束的稳定性;树枝化结构的端基数量明显高于线性聚合物,因此针对端基改性的效果也比线性聚合物明显;树枝化的聚合物形成的胶束结构紧密,粒径较小,更容易发挥epr效应,对肿瘤瘤组织靶向性较好。
附图说明
图1是合成pla-peg-pla树枝状聚合物步骤的化学反应方程式示意图。
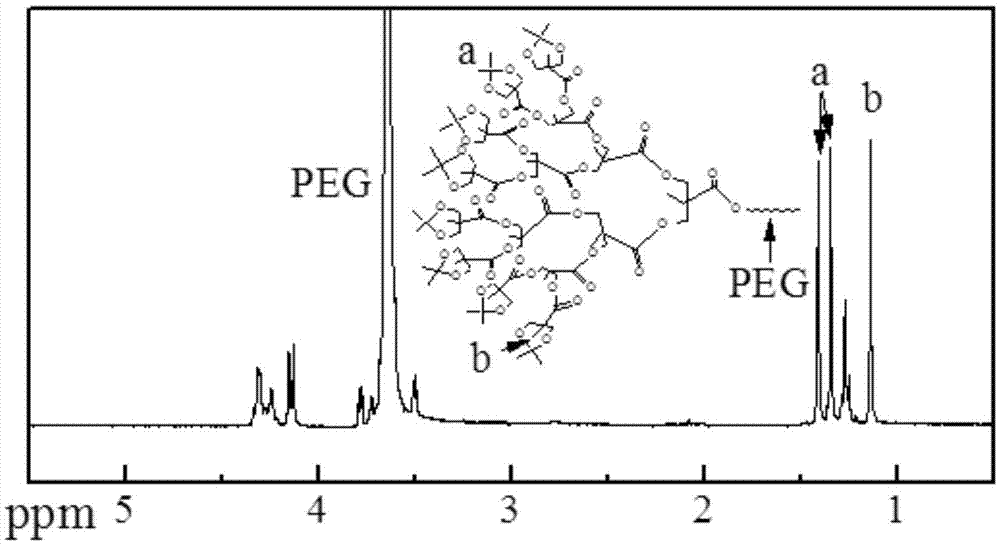
图2是g4-peg-g4的1hnmr谱图。
图3是peg-g4-(oh)32的1hnmr谱图。
图4是哑铃型嵌段共聚物(plan)的1hnmr谱图。
图5是实施例2实测得到的紫杉醇胶束粒径分布图,粒径为25.4±4.5nm
图6是实施例3实测得到的紫杉醇胶束粒径分布图,粒径为25.6±4.6nm。
图7是实施例4实测得到的多西他赛胶束粒径分布图,粒径为24.1±4.4nm。
图8是阿霉素胶束粒径分布图,粒径为26.2±4.5nm。
图9是姜黄素胶束粒径分布图,粒径为24.9±4.6nm。
图10是胶束稳定性对比示意图。
图11是肿瘤体积变化图示。
具体实施方式
本发明的pla-peg-pla树枝状聚合物中,树枝化羟基的数量可以是16个、32个或64 个,反应路径基本相同。
实施例1合成32个羟基的pla-peg-pla树枝状聚合物
其具体反应方式如图1所示。
(1)合成nhs封端聚乙二醇(nhs-peg-nhs)
羧基聚乙二醇(分子量为2000)10g溶解于50ml无水二氯甲烷,加入1.16gn羟基琥珀酰亚胺(nhs,10mmol),2.08g环己基碳二亚胺(dcc),0.12g二羟甲基丙酸(bis-mpa),和1.1g4-二甲胺基吡啶-p-甲苯磺酸盐(dpts),室温下搅拌反应48h得到反应液。将反应液过滤除去不溶物后,倒入1l乙醚中沉淀,将沉淀物重新溶解于有机溶剂,如四氢呋喃,乙酸乙酯,二氯甲烷或氯仿等,随后去除副反应产生的不溶于水的杂质,优选采用离心分离法,将沉淀物除去并将清液重新沉淀于乙醚后,真空干燥得白色粉末状固体即为nhs封端聚乙二醇(nhs-peg-nhs)。
(2)合成端基树枝化聚乙二醇(g4-peg-g4)
3.47g(0.34mmol)nhs-peg-nhs和4.37g(2.04mmol)4代树枝化聚酯(g4-nh2,合成方法参考文献macromolecules2002;35:8307方法合成)溶解于25ml无水二氯甲烷后加入1ml(7.33mmol)三乙胺室温搅拌反应48h。反应物用乙醚沉淀后的白色粉末状物质为g4-peg-g4。将所得到的g4-peg-g4进行核磁共振检测,检测条件为:bruker400m核磁共振仪,氘代氯仿为容液,tms为内标,氢谱。得到的图谱如图2所示,从a峰和b峰可以明显看出成功将树枝化的结构接枝到聚乙二醇的两端。
(3)含有32个羟基的树枝化聚乙二醇(peg-g4-(oh)32)合成
1.05gg4-peg-g4溶解于75ml甲醇,分批加入0.5克-1克dowexh+交换树脂,室温反应12h后过滤除去树脂,溶液用乙醚沉淀后的白色粉末状固体即为peg-g4-(oh)32。
将所得到的peg-g4-(oh)32进行核磁共振检测,检测条件为:bruker400m核磁共振仪,氘代氯仿为容液,tms为内标,氢谱。得到的图谱如图3所示,图3中可以看出树枝化结构表面端羟基脱保护完全。
(4)哑铃型嵌段共聚物(plan)合成
(plan)式中的代表单个丙交酯链段的聚合度。下述步骤得到的产物其中的n=10。
1gdl-丙交酯(6.94mmol)和1gpeg-g4-(oh)32加入到干燥的聚合瓶中,加入5mgsnoct2作为催化剂,真空熔封聚合瓶后将反应物于130℃下聚合12h,产物用氯仿溶解,甲醇沉淀后所得白色固体为目标共聚物,其数均分子量为4000。
将所得到的(plan)进行核磁共振检测,检测条件为:bruker400m核磁共振仪,氘代 氯仿为溶剂,四甲基硅烷(tetramethylsilane,简称tms)为nmr内标,测得其核磁共振氢谱包括以下特征峰,在1.6,5.2ppm处可见表示聚丙交酯的特征峰,在3.6ppm处可见表示聚乙二醇的特征峰,在1.1ppm和4.2-4.4ppm处可见表示树枝化聚酯结构的特征峰。得到的图谱如图4,图4中可以明显看到peg和pla的特征吸收。
(5)封端反应
将反应量的哑铃型嵌段共聚物和疏水性的封端剂反应,经过纯化后得到目标高分子共聚物,共聚物的分子式如下所示:
其中r4为疏水性基团封端的聚丙交酯链段,数据分子量为1000-50000。
实施例2苄基封端共聚物包裹紫杉醇
将第一步所得到的树枝化la-peg-pla树枝状聚合物5g和2g苯甲酰氯加入到圆底烧瓶中,氮气保护下加热至100℃反应6h,产物冷却至室温后用二氯甲烷溶解,乙醚沉淀后得白色粉末状固体既为苄基封端的共聚物辅料bzendcappedmpeg-pla。
将上述步骤所得到的苄基封端的聚合物辅料1g和适量紫杉醇共同溶解于乙酸乙酯,50℃旋转蒸发去除溶剂,加入50ml注射用水溶解后,溶液经0.22μmpvdf膜过滤除去未被包裹的紫杉醇,随后所述溶液冻干得的紫杉醇胶束冻干粉。该冻干粉复溶后测定粒径为25.4nm(见图5粒径分布图)。
通过调节投入紫杉醇的投入数量,可以得到紫杉醇含量不等的胶束冻干粉。每100份共聚物最多可包裹85份紫杉醇,共聚物与紫杉醇的重量比例为100:1~85。
图5为本实施例实测得到的紫杉醇胶束粒径分布图,粒径为25.4±4.5nm
粒径分布均一。
实施例3fmoc基团封端包裹紫杉醇
1.84gfmoc赖氨酸溶解于10ml无水乙酸乙酯后加入三乙胺0.7ml,溶液冷却至-10℃,加入新戊酰氯0.61ml搅拌下升温至0℃反应2h,然后升温至室温再反应1h,过滤去除不溶物,滤液旋转蒸发去除溶剂后得白色固体溶解于5ml干燥二氯甲烷,加入三乙胺0.7ml,4-吡咯烷基吡啶74mg,溶液冷却至0℃后加入所述pla-peg-pla树枝状聚合物1g,反应2h后室温继续反应24h得fmoc封端聚合物fmoc-lysendcappedmpeg-pla。
将上述步骤得到的fmoc封端聚合物1g与适量紫杉醇溶解于乙醇,45℃旋转蒸发去除溶剂,加入50ml注射用水溶解后,液经0.22μmpvdf膜过滤除去未被包裹的紫杉醇,随后冻干得紫杉醇胶束冻干粉。该冻干粉复溶后测定粒径为25.6nm。
通过调节投入紫杉醇的投入数量,可以得到紫杉醇含量不等的胶束冻干粉。每100份共聚物最多可包裹100份紫杉醇,共聚物与紫杉醇的重量比例为100:1~100。
图6为本实施例实测得到的紫杉醇胶束粒径分布图,粒径为25.6±4.6nm。
实施例4,boc-苯丙氨酸封端包裹多西他赛
1.33gboc-苯丙氨酸溶解于10ml无水乙酸乙酯后加入三乙胺0.7ml,溶液冷却至-10℃,加入新戊酰氯0.61ml搅拌下升温至0℃反应2h,然后升温至室温再反应1h,过滤去除不溶物,滤液旋转蒸发去除溶剂后得白色固体溶解于5ml干燥二氯甲烷,加入三乙胺0.7ml,吡咯烷基吡啶74mg,溶液冷却至0℃后加入所述pla-peg-pla树枝状聚合物1g,反应2h后室温继续反应24h得boc-苯丙氨酸封端聚合物boc-pheendcappedmpeg-pla。
将上述步骤得到的boc-苯丙氨酸封端高分子辅料1g和适量多西他赛溶解于乙酸乙酯,40℃旋转蒸发去除溶剂,加入50ml注射用水溶解药物,溶液经0.22μmpvdf膜过滤除去未被包裹的药物,随后冻干得多西他赛胶束冻干粉。图7为本实施例实测得到的多西他赛胶束粒径分布图,粒径为24.1±4.4nm。该冻干粉复溶后测定粒径为24.1nm。通过调节投入紫杉醇的投入数量,可以得到紫杉醇含量不等的胶束冻干粉。每100份共聚物最多可包裹50份多西他赛,共聚物与多西他赛的重量比例为100:1~50。
实施例5:包裹阿霉素
20mg盐酸阿霉素溶解于1ml氯仿,加入0.5ml三乙胺避光室温搅拌12h后加入实施例3所得的高分子共聚物100mg,将上述溶液转移至透析袋中(截留分子量为3000)透析24h后冻干得到胶束阿霉素胶束冻干粉。
通过调节投入阿霉素的投入数量,可以得到阿霉素含量不等的胶束冻干粉。每100份 共聚物最多可包裹33份阿霉素,共聚物与阿霉素的重量比例为100:1~33。
图8为阿霉素胶束粒径分布图,粒径分布均一,粒径为26.2±4.5nm。
实施例6:包裹姜黄素
20mg姜黄素和300mg实施例1所得共聚物溶解于5ml丙酮,45℃旋蒸去除溶剂丙酮后加入5ml超纯水溶解药膜后得到姜黄素胶束溶液,
通过调节投入姜黄素的投入数量,可以得到姜黄素含量不等的胶束冻干粉。每100份共聚物最多可包裹20.5份姜黄素,共聚物与姜黄素的重量比例为100:1~20.5。
该胶束粒径分布如图9所示,粒径分布均一,粒径为24.9±4.6nm。
实验例7,胶束稳定性对比
将实施例2-4得到的载药胶束,采用适量注射用水复溶。另按照专利cn1197396a所述的方法自行制备mpeg-pla紫杉醇胶束。所有溶液的药物浓度统一调整为5mg/ml。然后加入胎牛血清(最后胎牛血清的浓度为50%),用动态光散射记录24h内胶束粒径变化,如图10所示:
如图10所示,实施例1-3制备的三种胶束在高浓度的血清溶液中,24h内粒径几乎没有变化,可见其稳定性非常高。而mpeg-pla胶束在6h内粒径已经显著变大,说明其稳定性较差。
实验例8,抑瘤实验
以a549肺腺癌为模型,采用实施例2和实施例3制备的紫杉醇胶束为药物。采用市售genexolpm作为对比。当肿瘤长到大约为150mm3时分组给药,每组6只裸鼠,共给药4次,每3天一次,测量肿瘤体积并记录如图11:
由图11可见,实施例2和实施例3得到的胶束在等剂量下的抑瘤效果显著强于mpeg-pla-紫杉醇胶束(市售genexolpm),统计学差异p<0.01,说明这两种胶束的靶向性显著高于线性的mpeg-pla紫杉醇胶束,具有较好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!