一种芳香族聚酯及其制备方法与流程
本发明涉及一种芳香族聚酯及其制备方法,属于高性能聚合物技术领域。
背景技术:
聚酯类高分子材料包括脂肪族聚酯和芳香族聚酯,脂肪族聚酯由于自身体系的柔性结构,其模量和玻璃化转变温度(Tg≈50oC)远低于具有刚性结构的芳香族聚酯,而后者熔融温度高(Tm≥350oC),加工性较差。为了获得兼具良好加工型和高耐热性的聚酯,人们采用对苯二甲酸二甲酯与二醇类单体进行酯交换反应,制得聚对苯二甲酸乙二醇酯(PET)、聚对苯二甲酸丁二醇酯(PBT)等半芳香族聚酯,并发展成为热塑性聚酯中最主要的品种。这类聚酯的电绝缘性优良,具有优良的物理机械性能,包括抗蠕变性、耐疲劳性、耐摩擦性、尺寸稳定性等,但是缺点也较为明显。虽然耐热性优于脂肪族聚酯,但Tg仍然只能达到80oC;此外,还存在结晶速率慢、成型加工困难、模塑温度高、生产周期长和冲击性能差的问题。因此,长期以来PET、PBT都被归类为通用工程塑料。
近年来人们通过将脂肪族或半芳香族聚酯与热致性液晶聚酯进行共混的方法来发展高性能聚酯。例如,将PET与液晶聚(对氧基苯甲酸酯-co-对氧基萘甲酸酯)共混挤出,制成具有皮芯结构的纤维。采用牵引拉伸的方式驱使作为芯材的液晶聚酯取向,以期达到自增强杨氏模量、拉伸强度的效果。当聚(对氧基苯甲酸酯-co-对氧基萘甲酸酯)含量为15wt%时,纤维样品的拉伸强度提高了120%;然而体系的Tg并未随液晶聚(对氧基苯甲酸酯-co-对氧基萘甲酸酯)的加入而升高,这是因其皮芯结构之间并无任何物理或化学键作用力(参见文献W N Kim and M M Denn. Properties of blends of a thermotropic liquid crystalline polymer with a flexible polymer (Vectra/PET)[J]. Journal of Rheology, 1992, 36: 1477-1498)。因此,如何在确保良好加工性的前提下,制备高拉伸强度、高耐热的聚酯材料是目前高性能热塑性聚合物一个具有重大应用价值的课题。
技术实现要素:
本发明针对现有技术存在的不足,提供一种兼具低粘度、高Tg和高拉伸强度的芳香族聚酯及其制备方法。
实现本发明目的所采用的技术方案是提供一种芳香族聚酯的制备方法,包括如下步骤:
1、将1mol同时含端羧基A和端羟基B的AB型全芳香族单体、1~2.5mol含有端羧基或/和端羟基的AA、BB或AB型半芳香族单体、0.5~2mmol催化剂和1.5~4mol醋酸酐加入反应器中;在氮气气氛、温度为120~140℃的条件下,乙酰化反应30~60min;
2、以0.5~1.5℃/min的速率升温至140~320℃,进行酯交换反应1~5h;
3、在真空度为1~20mbar、温度为 300~320℃的条件下反应10~30min;
4、反应结束后,在氮气气氛中冷却至室温,将产物研磨为精细粉末,再在温度为200~260℃、真空度为1~20mbar的条件下缩聚反应18~36h,即得到一种芳香族聚酯。
上述制备方法中,所述的同时含端羧基A和端羟基B的AB型全芳香族单体为4-羟基苯甲酸、4’-羟基联苯基-4-羧酸、6-羟基-2-萘甲酸中的一种,或它们的任意组合。
所述的含端羧基或/和端羟基的AA、BB或AB型半芳香族单体为HO-(H2C)m-Ar-(CH2)n-OH、HOOC-(H2C)m-Ar-(CH2)n-COOH或HOOC- (H2C)m-Ar-(CH2)n-OH中的一种,或它们的任意组合;其中,Ar为苯基、联苯基或萘基,m、n为2~10。
所述的催化剂为醋酸钠、醋酸钾、醋酸锌、辛酸亚锡、二丁基氧化锡、月桂酸二丁基锡中的一种。
本发明技术方案还包括按上述制备方法得到的一种芳香族聚酯。
与现有技术相比,本发明取得的有益效果是:
1、与传统的聚合物共混改性不同,本发明选用全芳香族和半芳香族单体作为一锅法聚合反应起始物,开辟了一种制备芳香族聚酯的新方法。
2、与传统自增强聚合物制备需要借助外力来实现的方法不同,本发明不需要借助外力作用来达到增强,而是在聚合过程中就地实现自增强;其原理是:体系在熔融加工过程中,由全芳香族单体缩聚形成的刚性分子链,而半芳香族单体缩聚形成柔性分子链,两种分子链相互间存在动态的酯交换反应,有利于建立较强的分子链间化学键作用力;而在冷却过程中,具有规整结构的刚性分子链发生迅速结晶,并且均匀分散在不易结晶的柔性分子链基体中,从而得到一种拉伸强度和Tg自增强的芳香族聚酯。
3、与现有技术不同,本发明提供的芳香族聚酯结构对称性下降,结晶度降低,在成型过程中熔体粘度低,加工性得到改善。此外,得到的制品具有高Tg,这是因为结晶区的存在有效限制了非晶区分子链的运动。
4、由于不需要借助外力,且一锅法简单可控,因此本发明的制备方法具有简便、适用性广的特点。
附图说明
图1是本发明实施例1提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯粉末样品的广角X射线衍射(WXRD)谱图;
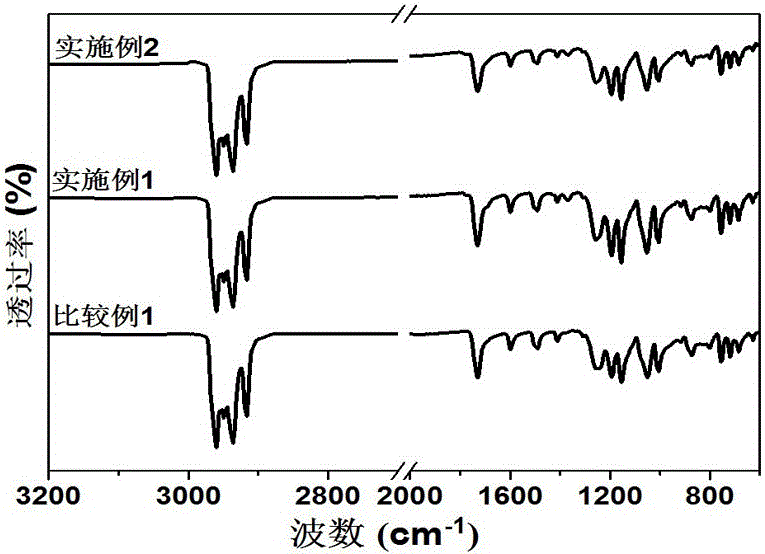
图2是本发明实施例1、2提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯粉末样品的红外光谱(FTIR)图;
图3是本发明实施例1、2、4提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯粉末样品在300℃恒温90min的复合粘度-时间曲线;
图4是本发明实施例1、3、5提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯薄膜样品的动态力学分析(DMA)损耗模量(E”)-温度曲线;
图5是本发明实施例1、6提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯薄膜样品的拉伸强度对比图。
具体实施方式
下面结合附图、实施例和比较例,对本发明技术方案作进一步的描述。
实施例1
含半芳香族单体聚酯的制备:在一个500mL三口圆底烧瓶中加入69.06g 4-羟基苯甲酸、277.85g 4-(3-羟丙基)-苯基正丁酸、300mL醋酸酐和10mg醋酸钾。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度140℃下乙酰化反应30min。在流沙浴中以升温速率1.0℃/min将反应混合物加热,反应温度由140℃上升至310℃。此时,将反应体系缓慢地抽成真空(真空度为10mbar),并在310℃保持30min。不透明的熔体冷却至室温,将产物由烧瓶中移出,而后研磨为精细粉末。在温度250℃和真空度为1mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯,其元素分析、WXRD谱、FTIR谱图和在300℃恒温90min的复合粘度-时间曲线分别参见表1、附图1、2和3。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜,其动态力学分析(DMA)测得的损耗模量(E”)-温度曲线和拉伸强度分别参见附图4和5。
比较例1提供的全芳香族聚酯的制备:在一个500mL三口圆底烧瓶中加入138.12g 4-羟基苯甲酸、200mL醋酸酐和20mg醋酸钾。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度140℃下乙酰化反应30min。在流沙浴中以升温速率1.0℃/min将反应混合物加热,反应温度由140℃上升至310℃。此时,将反应体系缓慢地抽成真空(真空度为10mbar),并在310℃保持30min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在250℃真空度为1mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯,其元素分析、WXRD谱、FTIR谱图和在300℃恒温90min的复合粘度-时间曲线分别参见表1、附图1、2和3。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜,其动态力学分析(DMA)测得的损耗模量(E”)-温度曲线和拉伸强度分别参见附图4和5。
参见附图1,它是本发明中实施例1提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯粉末样品的广角X射线衍射(WXRD)谱。从图中可以看出,比较例1制得的全芳香族聚酯的结晶度高达82%,而实施例1制得的含半芳香族单体聚酯结晶度为48%,这是由于半芳香族链段的加入破坏了分子链的对称性,大大降低了聚合物的结晶度,为体系引入更多的非晶相。这一方面改善了聚合物的加工性,另一方面使得结晶相可以地分散在非晶相内。
实施例2
含半芳香族单体聚酯的制备:在一个1000mL三口圆底烧瓶中加入138.12g 4-羟基苯甲酸、208.25g 4-(3-羟丙基)-苯丙酸、500mL醋酸酐和41mg醋酸钠。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度130℃下乙酰化反应30min。在流沙浴中以升温速率1.1℃/min将反应混合物加热,反应温度由140℃上升至300℃。此时,将反应体系缓慢地抽成真空(真空度为1mbar),并在300℃保持30min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在260℃真空度为15mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯,其元素分析、FTIR谱图和在300℃恒温90min的复合粘度-时间曲线分别参见表1、附图2和3。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜。
不同于溶液缩聚法制得的产物分子量较低,熔融缩聚法制备的芳香族聚酯类高聚物分子量普遍较高,被公认为溶解性差,不仅难溶于普通溶剂,即使采用五氟苯酚、六氟异丙醇或混合溶剂二氯甲烷/六氟异丙醇(v/v=50/50)等鲜见的溶剂仍难以溶解该类聚合物(参见文献:H R Kricheldorf, V Linzer, C Bruhn. Eur. Polym. J. 1994, 30: 549-556)。虽然芳香族聚酯类高聚物可以溶于浓硫酸,但是强酸会导致酯键迅速分解形成低聚物。文献中报道的可以溶解于类似混合溶剂的聚酯类产物通常只有数千(参见文献:① M Gedan-Smolka, D Jehnichen, H Komber, D Voigt, F Böhme, M Rätzsch. Angew. Makromol. Chem. 1995, 229: 159-174; ② A Knijnenberg, E S Weiser, T L StClair, E Mendes, T J Dingemans. Macromolecules 2006, 39: 6936-6943),它们可通过测量特性粘度的方法从侧面来衡量聚合物的分子量,但是不适用于熔融缩聚法制备的芳香族聚酯类高聚物。
另一方面,本文合成的芳香族聚酯类高聚物因大量芳香族结构的存在,使其在诸如固态核磁等谱图中出峰位置相互叠加难以解析;对于分子量高、溶解性差的芳香族聚酯类聚合物,人们不对其结构进行任何表征(参见文献:① T J Dingemans, E S Weiser, T L StClair. U.S. Patent 6,939,940, September 6, 2005;② J Economy. Mol. Cryst. Liq. Cryst. 1989, 169: 1−22;③ Z Yerlikaya, S Aksoy, E Bayramli. J. Polym. Sci., Polym. Chem. 2001, 39: 3263-3277;④ A J East, L F Charbonneau,G W Calundann. Mol. Cryst. Liq. Cryst. Inc. Nonlin. Opt. 1988, 157: 615-637);或者仅能采用红外光谱等基础技术手段来简单表征产物结构(参见文献:① H R Kricheldorf, G Schwarz. Polymer 1990, 31: 481-485;② D Liaw, K Wang, Y Huang, K Lee, J Lai, C Ha, Prog. Polym. Sci. 2012, 37:907-974)。本发明中实施例与比较例提供的芳香族聚酯遇到了相同的难题,因此采用元素分析(表1)和红外光谱(图2)对聚合物的结构进行了表征。
参见表1和附图2,它们分别是本发明中实施例1、2提供的芳香族聚酯与比较例1提供的对位全芳香族聚酯粉末样品的元素分析结果与FTIR谱图。从元素分析表中可以看出,实验值与理论值基本保持一致。由FTIR谱图(图2)可见,2900-2970cm-1为C-H伸缩振动谱带,在1732cm-1处有酯键的C=O对称伸缩振动峰,在1600cm-1和1499cm-1处有明显的芳香族C=C的吸收峰,在1200cm-1附近有C-O基团的伸缩振动谱带。结合三种聚合物的元素分析结果和红外谱图,证明反应是按照预期进行,合成了预期物质。
表1是本发明实施例1、2提供的芳香族聚酯和比较例1提供的对位全芳香族聚酯粉末样品的元素分析结果。
表1:
。
实施例3
含半芳香族单体聚酯的制备:在一个1000mL三口圆底烧瓶中加入138.12g 4-羟基苯甲酸、216.28g 2,6-萘二乙醇、466.65g 4,4’-联苯-二壬酸、380mL醋酸酐和497.9mg二丁基氧化锡。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度140℃下乙酰化反应60min。在流沙浴中以升温速率1.5℃/min将反应混合物加热,反应温度由140℃上升至320℃。此时,将反应体系缓慢地抽成真空(真空度为20mbar),并在320℃保持10min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在250℃真空度为1mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜,其动态力学分析(DMA)测得的损耗模量(E”)-温度曲线参见附图4。
实施例4
含半芳香族单体聚酯的制备:在一个500mL三口圆底烧瓶中加入53.56g 4’-羟基联苯基-4-羧酸、195.32g 1,4-苯基二癸醇、97.09g 1,4-苯基二乙酸、180mL醋酸酐和157.9mg月桂酸二丁基锡。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度130℃下乙酰化反应50min。在流沙浴中以升温速率1.2℃/min将反应混合物加热,反应温度由130℃上升至300℃。此时,将反应体系缓慢地抽成真空(真空度为5mbar),并在300℃保持30min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在250℃真空度为5mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯,其在300℃恒温90min的复合粘度-时间曲线参见附图3。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜。
参见附图3,它是实施例1、2、4提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯粉末样品在300℃恒温90min的复合粘度-时间曲线。由图可见,比较例1制得的全芳香族聚酯在300℃恒温30min,粘度出现小幅下降后出现上升,这是因为其在此温度发生进一步缩聚反应导致分子量增大,这样的短暂的熔融窗口不利于聚合物的加工。实施例1、2和4制得的含半芳香族单体聚酯在300℃恒温分别20min、5min和5min后粘度出现快速下降,且其最小粘度值与比较例1的最小粘度值相比小一个数量级,这主要得益于半芳香族链段的加入和非晶区的存在。虽然实施例1、2和4制得的芳香族聚酯在300℃恒温30min时也同样因为发生进一步缩聚反应导致分子量增大,粘度出现了上升,但是这样的熔融窗口足以完成加工成型。
实施例5
含半芳香族单体聚酯的制备:在一个500mL三口圆底烧瓶中加入94.09g 6-羟基-2-萘甲酸、83.11g 1,4-苯基二乙醇、209.30g 1,4-苯基二癸酸、150mL醋酸酐和124.475mg二丁基氧化锡。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度120℃下乙酰化反应60min。在流沙浴中以升温速率1.0℃/min将反应混合物加热,反应温度由120℃上升至300℃。此时,将反应体系缓慢地抽成真空(真空度为2mbar),并在300℃保持30min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在200℃真空度为1mbar的真空烘箱中后缩聚反应48h,即得到一种芳香族聚酯。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜,其动态力学分析(DMA)测得的损耗模量(E”)-温度曲线参见附图4。
参见附图4,它是本发明中实施例1、3、5提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯薄膜样品的动态力学分析(DMA)损耗模量(E”)-温度曲线。由图可见,实施例1、3、5制得的含半芳香族单体聚酯的玻璃化转变温度为230℃,与比较例1制得的全芳香族聚酯相比提高了约30℃,这是因为实施例1、3、5制得的聚酯的结晶相均匀分散于非晶区,当分子链开始运动时受到了结晶相的限制,使其具有突出的耐热性。
实施例6
含半芳香族单体聚酯的制备:在一个500mL三口圆底烧瓶中加入69.06g 4-羟基苯甲酸、107.11g 4’-羟基联苯基-4-羧酸、104.13g 4-(2-羟乙基)苯基正丁酸、200mL醋酸酐和202.55mg辛酸亚锡。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度120℃下乙酰化反应60min。在流沙浴中以升温速率0.8℃/min将反应混合物加热,反应温度由120℃上升至320℃。此时,将反应体系缓慢地抽成真空(真空度为15mbar),并在320℃保持10min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在230℃真空度为10mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜,其拉伸强度分别参见附图5。
参见附图5,它是本发明中实施例1、6提供的含半芳香族单体聚酯和比较例1提供的全芳香族聚酯薄膜样品的拉伸强度。如图所示,实施例1、6制得的含半芳香族单体聚酯薄膜样品具有较高的拉伸强度,半芳香族链段的加入以及结晶相与非晶相结合的结构确实达到了自增强的效果。
实施例7
含半芳香族单体聚酯的制备:在一个1000mL三口圆底烧瓶中加入53.56g 4’-羟基联苯基-4-羧酸、138.12g 4-羟基苯甲酸、208.25g 4-(3-羟丙基)-苯丙酸、195.32g 1,4-苯基二癸醇、97.09g、1,4-苯基二乙酸、320mL醋酸酐和157.9mg月桂酸二丁基锡。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度135℃下乙酰化反应50min。以升温速率1.2℃/min将反应混合物加热,反应温度由135℃上升至300℃。此时,将反应体系缓慢地抽成真空(真空度为10mbar),并在300℃保持20min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在260℃真空度为20mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯。
将得到的芳香族聚酯粉末平铺于模具中,置于300℃平板硫化仪保压(2bar)30min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜。
实施例8
含半芳香族单体聚酯的制备:在一个500mL三口圆底烧瓶中加入94.09g 6-羟基-2-萘甲酸、83.11g 1,4-苯基二乙醇、209.30g 1,4-苯基二癸酸、150mL醋酸酐和124.475mg二丁基氧化锡。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度120℃下乙酰化反应60min。以升温速率1.0℃/min将反应混合物加热,反应温度由120℃上升至300℃。此时,将反应体系缓慢地抽成真空(真空度为10mbar),并在300℃保持30min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在200℃真空度为1mbar的真空烘箱中后缩聚反应48h,即得到一种芳香族聚酯。
将得到的芳香族聚酯粉末平铺于模具中,置于280℃平板硫化仪保压(2.5bar)60min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜。
实施例9
含半芳香族单体聚酯的制备:在一个1000mL三口圆底烧瓶中加入69.06g 4-羟基苯甲酸、277.85g 4-(3-羟丙基)-苯基正丁酸、94.09g 6-羟基-2-萘甲酸、83.11g 1,4-苯基二乙醇、209.30g 1,4-苯基二癸酸、400mL醋酸酐和10mg醋酸锌。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度125℃下乙酰化反应60min。以升温速率1.0℃/min将反应混合物加热,反应温度由125℃上升至310℃。此时,将反应体系缓慢地抽成真空(真空度为10mbar),并在310℃保持25min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在250℃真空度为1mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯。
将得到的芳香族聚酯粉末平铺于模具中,置于310℃平板硫化仪保压(1bar)20min,自然冷却至40℃脱模,得到一种芳香族聚酯薄膜。
实施例10
含半芳香族单体聚酯的制备:在一个1000mL三口圆底烧瓶中加入138.12g 4-羟基苯甲酸、208.25g 4-(3-羟丙基)-苯丙酸、216.28g 2,6-萘二乙醇、466.65g 4,4’-联苯-二壬酸、480mL醋酸酐和497.9mg二丁基氧化锡。烧瓶配以密封玻璃桨式搅拌器,一个氮气入口管和一个保温蒸馏头。通入适中的氮气流,在温度140℃下乙酰化反应60min。在流沙浴中以升温速率1.2℃/min将反应混合物加热,反应温度由140℃上升至320℃。此时,将反应体系缓慢地抽成真空(真空度为18mbar),并在320℃保持10min。不透明的熔体冷却至室温,将产物由烧瓶中移出,然后研磨为精细粉末。在250℃真空度为5mbar的真空烘箱中后缩聚反应24h,即得到一种芳香族聚酯。
将得到的芳香族聚酯粉末平铺于模具中,置于290℃平板硫化仪保压(1.5bar)40min,自然冷却至30℃脱模,得到一种芳香族聚酯薄膜。
- 还没有人留言评论。精彩留言会获得点赞!