一种可注射水凝胶及其制备方法与流程
本发明涉及多糖类高分子材料技术领域,尤其涉及一种可注射水凝胶及其制备方法。
背景技术:
水凝胶材料是一种以物理或化学方式交联形成的三维网状结构,亲水残基与水分子结合,将水分子连接在网状内部,而疏水残基遇水膨胀的交联聚合物,其具有含水量高、保水能力强、柔软、拥有一定的粘弹性和形状、生物相容性良好等优点。水凝胶材料具有独特的性能,是一类新型功能材料,已经成为生物医用高分子材料领域的研究热点。
可注射水凝胶是以溶液状态存在,可以与药物混合;当通过注射的方法以溶液的状态植入体内后,能够迅速发生溶胶-凝胶相转变而原位成胶;同时,药物被包埋于水凝胶内部,然后通过扩散或者水凝胶自身降解缓慢释放,从而达到长效缓释的目的;并且,还具有使用方便,手术创伤微小,能够填充任何形状缺损部位的优势。因而,可注射水凝胶作为抗肿瘤药物载体已经在抗肿瘤领域引起众多研究者极大的兴趣与关注。另外,近年来,可注射水凝胶在伤口敷料和填料、烧伤感染、假体移植和组织工程等领域得到了广泛的研究和应用。
生物相容性水凝胶材料的研究已经引起众多研究者极大的兴趣与关注,也是其应用于实际的关键。其中,生物相容性水凝胶担载蛋白类药物(ChemicalSociety Reviews,2012,41,4860-4883)能够直接注射到病变部位,实现药物的持续释放,延长药物的半衰期,有效减轻病人的痛苦,但是面临制备过程繁琐的问题。随着科研的不断深入,研究人员又制备出了嵌段聚合物基水凝胶(Journal of Controlled Release,2002,85,51-59),这类水凝胶具有温度敏感型,细胞相容性良好,但是,这种嵌段聚合物往往不易生物降解,很难被机体吸收利用或最终排出体外。目前,尽管一些基于合成聚合物的水凝胶也不断的被研制出来,但是很多此类凝胶的生物相容性并不理想,具有引发免疫反应或炎症的潜在风险,并且其制备过程复杂,在一定程度上限制了该类水凝胶的进一步应用。
技术实现要素:
有鉴于此,本发明的目的在于提供一种可注射水凝胶及其制备方法,本发明提供的可注射水凝胶自身具有较高的可调空间,且具有良好的生物相容性和生物降解性。
本发明提供了一种可注射水凝胶,由精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应得到;
所述精氨酸修饰的壳聚糖,具有式(I)所示结构:
其中,n为聚合度,10≤n≤3200;
所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%;
所述交联剂为氧化葡聚糖。
优选的,所述精氨酸修饰的壳聚糖的制备方法包括以下步骤:
a)将壳聚糖、精氨酸衍生物和活化剂混合,进行酰胺化反应,得到式(II)所示化合物;
所述精氨酸衍生物具有式(III)所示结构:
b)将式(II)所示化合物进行脱保护反应,得到精氨酸修饰的壳聚糖。
优选的,步骤a)中所述活化剂选自1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、N-羟基琥珀酰亚胺、N,N-二异丙基碳二亚胺、N,N'-二环己基碳二亚胺和N-羟基硫代琥珀酰亚胺中的一种或多种。
优选的,步骤a)中所述壳聚糖、精氨酸衍生物和活化剂的质量比为1:(0.1~3.2):(0.15~4.8)。
优选的,步骤a)中所述将壳聚糖、精氨酸衍生物和活化剂混合的过程具体为:
a1)将壳聚糖与酸溶液混合,得到第一混合溶液;
a2)将精氨酸衍生物、活化剂与有机溶剂混合,得到第二混合溶液;
a3)将第二混合溶液加入第一混合溶液中,得到壳聚糖、精氨酸衍生物和活化剂的混合溶液;
步骤a1)和a2)没有顺序限制。
优选的,步骤a)中所述酰胺化反应的温度为20℃~40℃,时间为24h~72h。
优选的,步骤b)中脱保护反应的温度为20℃~45℃,时间为2h~8h。
优选的,所述精氨酸修饰的壳聚糖和交联剂的质量比为1:(0.1~20)。
优选的,所述溶剂为水、生理盐水、缓冲溶液、组织培养液或体液。
本发明还提供了一种可注射水凝胶的制备方法,包括以下步骤:
将精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应,得到可注射水凝胶;
所述精氨酸修饰的壳聚糖,具有式(I)所示结构:
其中,n为聚合度,10≤n≤3200;
所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%;
所述交联剂为氧化葡聚糖。
本发明提供了一种可注射水凝胶及其制备方法,所述可注射水凝胶由具有式(I)结构的精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应得到;其中,具有式(I)结构的精氨酸修饰的壳聚糖的聚合度10≤n≤3200;所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%;所述交联剂为氧化葡聚糖。与现有技术相比,本发明提供的可注射水凝胶具有良好的生物相容性,能够在体内原位形成,有利于药物直接作用于病变部位并较长久的发挥药效;而采用氧化葡聚糖作为交联剂安全无毒,环境污染小;同时,所述可注射水凝胶还具有良好的生物降解性,能够在体内降解,而且降解产物为氨基酸和多糖,可通过肾脏直接排除体外,对人体无害。实验结果表明,本发明提供的可注射水凝胶含水量高,不同浓度的多糖衍生物及凝胶浸提液的细胞存活率均大于85%,说明本发明提供的可注射水凝胶没有明显的细胞毒性,细胞相容性良好。
另外,本发明提供的可注射水凝胶具有较高的可调空间,能根据不同需要调节壳聚糖的分子量、壳聚糖侧链上精氨酸的取代度、氧化葡聚糖的分子量、氧化葡聚糖的氧化度等,从而能够制备各种不同物理化学性能的水凝胶材料。
此外,本发明提供的可注射水凝胶的制备方法原料简单易得,成本低;同时操作简单、反应条件温和、制备工艺稳定,易于实现工业化生产,具有较好的应用前景。
附图说明
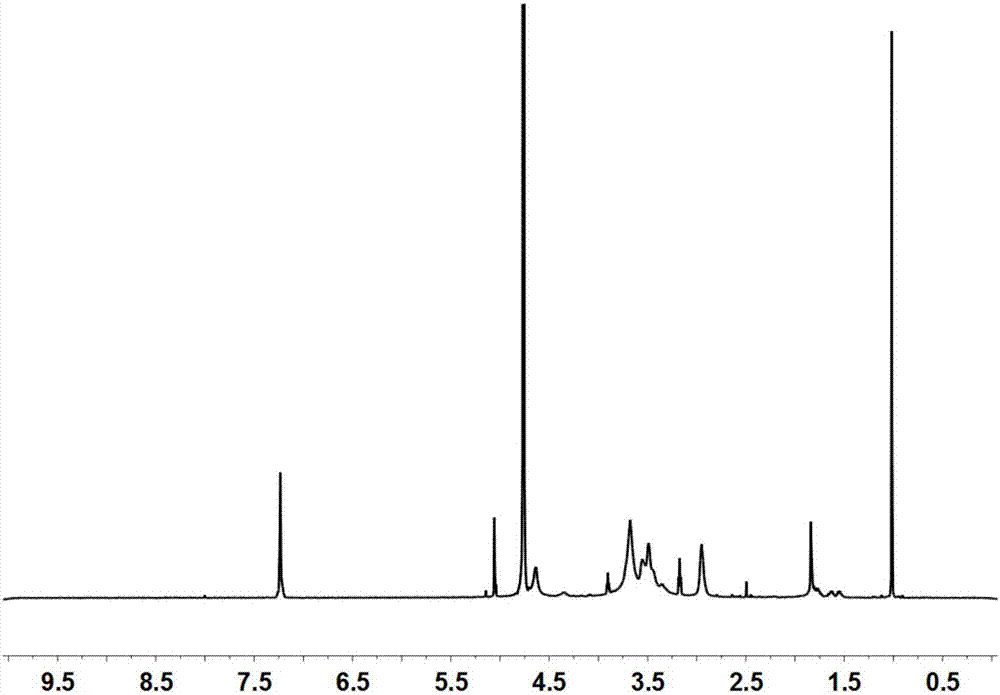
图1为本发明实施例1得到的固体粉末的核磁共振氢谱图;
图2为本发明实施例4得到的固体粉末的核磁共振氢谱图;
图3为本发明实施例5得到的最终产物的核磁共振氢谱图;
图4为本发明实施例45所述的不同浓度的氧化葡聚糖溶液的细胞存活率;
图5为本发明实施例46得到的不同浓度的精氨酸修饰的壳聚糖溶液的细胞存活率;
图6为本发明实施例47得到的不同浓度的凝胶浸提液的细胞存活率;
图7为本发明实施例48得到的不同浓度的凝胶浸提液的细胞存活率;
图8为本发明实施例49得到的不同浓度的凝胶浸提液的细胞存活率;
图9为本发明实施例50得到的不同浓度的凝胶浸提液的细胞存活率。
具体实施方式
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种可注射水凝胶,由精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应得到;
所述精氨酸修饰的壳聚糖,具有式(I)所示结构:
其中,n为聚合度,10≤n≤3200;
所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%;
所述交联剂为氧化葡聚糖。
在本发明中,所述精氨酸修饰的壳聚糖,具有式(I)所示结构:
其中,n为聚合度,10≤n≤3200;
所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%。
在本发明中,具有式(I)结构的精氨酸修饰的壳聚糖,通过引入精氨酸,不仅改善了壳聚糖的水溶性,还提高其生物相容性,从而可赋予水凝胶生物功能性,如细胞粘附性、生物相容性、抗菌性等,也可进行原位凝胶的使用。此外,本发明提供的精氨酸修饰的壳聚糖对生物体无毒,有利于在生物医用材料领域的广泛应用。
在本发明中,所述n为聚合度,10≤n≤3200。在本发明中,所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%,优选为1%~50%,更优选为3%~45%。在本发明中,所述精氨酸修饰的壳聚糖能根据不同需要调节壳聚糖的分子量、壳聚糖侧链上精氨酸的取代度,从而能够用于各种不同物理化学性能的水凝胶材料的制备。
在本发明中,所述精氨酸修饰的壳聚糖的制备方法优选包括以下步骤:
a)将壳聚糖、精氨酸衍生物和活化剂混合,进行酰胺化反应,得到式(II)所示化合物;
所述精氨酸衍生物具有式(III)所示结构:
b)将式(II)所示化合物进行脱保护反应,得到精氨酸修饰的壳聚糖。
本发明首先将壳聚糖、精氨酸衍生物和活化剂混合,进行酰胺化反应,得到式(II)所示化合物。在本发明中,所述壳聚糖具有式(IV)所示结构:
在本发明中,所述壳聚糖的分子量优选为2000Da~500000Da,更优选为8000Da~300000Da,更更优选为10000Da~200000Da;本发明对所述壳聚糖的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
在本发明中,所述精氨酸衍生物具有式(III)所示结构:
本发明对所述精氨酸衍生物的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
在本发明中,所述活化剂优选选自1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)、N-羟基琥珀酰亚胺(NHS)、N,N-二异丙基碳二亚胺(DIC)、N,N'-二环己基碳二亚胺(DCC)和N-羟基硫代琥珀酰亚胺(sulfo-NHS)中的一种或多种,更优选为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)和N-羟基琥珀酰亚胺(NHS)。在本发明中,所述活化剂能够对精氨酸衍生物的羧基起活化作用;本发明对所述活化剂的来源没有特殊限制,采用本领域技术人员熟知的上述1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)、N-羟基琥珀酰亚胺(NHS)、N,N-二异丙基碳二亚胺(DIC)、N,N'-二环己基碳二亚胺(DCC)和N-羟基硫代琥珀酰亚胺(sulfo-NHS)的市售商品即可。
在本发明中,所述壳聚糖、精氨酸衍生物和活化剂的质量比优选为1:(0.1~3.2):(0.15~4.8)。在本发明一个优选的实施例中,所述活化剂选自EDC和NHS;所述壳聚糖、精氨酸衍生物、EDC和NHS的质量比优选为1:0.1:0.1:0.05。在本发明另一个优选的实施例中,所述活化剂选自EDC和NHS;所述壳聚糖、精氨酸衍生物、EDC和NHS的质量比优选为1:0.2:0.2:0.1。在本发明另一个优选的实施例中,所述活化剂选自EDC和NHS;所述壳聚糖、精氨酸衍生物、EDC和NHS的质量比优选为1:0.4:0.4:0.2。在本发明另一个优选的实施例中,所述活化剂选自EDC和NHS;所述壳聚糖、精氨酸衍生物、EDC和NHS的质量比优选为1:0.8:0.8:0.4。在本发明另一个优选的实施例中,所述活化剂选自EDC和NHS;所述壳聚糖、精氨酸衍生物、EDC和NHS的质量比优选为1:1.6:1.6:0.8。在本发明另一个优选的实施例中,所述活化剂选自EDC和NHS;所述壳聚糖、精氨酸衍生物、EDC和NHS的质量比优选为1:3.2:3.2:1.6。
在本发明中,所述将壳聚糖、精氨酸衍生物和活化剂混合的过程优选具体为:
a1)将壳聚糖与酸溶液混合,得到第一混合溶液;
a2)将精氨酸衍生物、活化剂与有机溶剂混合,得到第二混合溶液;
a3)将第二混合溶液加入第一混合溶液中,得到壳聚糖、精氨酸衍生物和活化剂的混合溶液;
步骤a1)和a2)没有顺序限制。
本发明将壳聚糖与酸溶液混合,得到第一混合溶液。在本发明中,所述酸溶液优选为盐酸溶液和/或醋酸溶液,更优选为盐酸溶液。在本发明中,所述酸溶液的作用是促进壳聚糖的溶解,本发明对此没有特殊限制。在本发明中,所述壳聚糖与酸溶液的用量比优选为1g:(25mL~200mL),更优选为1g:(50mL~150mL),更更优选为1g:100mL。
同时,本发明将精氨酸衍生物、活化剂与有机溶剂混合,得到第二混合溶液。在本发明中,所述有机溶剂优选为N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、四氢呋喃(THF)和乙腈中的一种或多种,更优选为N,N-二甲基甲酰胺(DMF)。本发明对所述有机溶剂的来源没有特殊限制,采用本领域技术人员熟知的上述N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、四氢呋喃(THF)和乙腈的市售商品即可。在本发明中,所述精氨酸衍生物与有机溶剂的用量比优选为1g:(1mL~150mL),更优选1g:(3.125mL~100mL)。
分别得到所述第一混合溶液和第二混合溶液后,本发明将第二混合溶液加入第一混合溶液中,得到壳聚糖、精氨酸衍生物和活化剂的混合溶液,即完成混合过程。然后,本发明直接进行酰胺化反应,得到式(II)所示化合物。本发明优选在搅拌的条件下进行酰胺化反应,本发明对此没有特殊限制。在本发明中,所述酰胺化反应的温度优选为20℃~40℃,更优选为30℃;所述酰胺化反应的时间优选为24h~72h,更优选为48h。
完成所述酰胺化反应后,本发明优选还包括将反应产物进行纯化,得到固体粉末。在本发明中,由于反应最终产物为精氨酸修饰的壳聚糖,而多糖核磁难以分析,精氨酸上氢的位移与多糖中氢的位移相近,难以进行取代度的计算,而式(II)所示化合物中苯环结构容易进行核磁分析和取代度的计算,因此本发明对其进行分析表征。
得到式(II)所示化合物后,本发明将式(II)所示化合物进行脱保护反应,得到精氨酸修饰的壳聚糖。在本发明中,所述脱保护反应的过程优选具体为:
将式(II)所示化合物分散在反应介质中,在溴化氢溶液存在下,进行脱保护反应,纯化,得到精氨酸修饰的壳聚糖。在本发明中,所述反应介质优选为三氟乙酸。在本发明中,所述反应介质的用量优选为50mL~200mL,更优选为80mL~150mL;所述溴化氢溶液的用量优选为2mL~25mL,更优选为4mL~20mL。
在本发明中,所述脱保护反应的温度优选为20℃~45℃,更优选为30℃;所述脱保护反应的时间优选为2h~8h,更优选为6h。
在本发明中,所述交联剂为氧化葡聚糖。在本发明中,所述氧化葡聚糖的氧化度优选为10%~100%,更优选为15%~80%,更更优选为20%~50%。本发明采用氧化葡聚糖为交联剂安全无毒,环境污染小。本发明对所述氧化葡聚糖的来源没有特殊限制,采用如下制备方法得到的自制品即可。
在本发明中,所述氧化葡聚糖的制备方法优选具体为:
将葡聚糖与水混合,在高碘酸钠存在下进行避光反应,然后加入异丙醇终止反应,透析冻干,得到氧化葡聚糖。
在本发明中,所述葡聚糖的分子量优选为2000Da~500000Da,更优选为8000Da~300000Da,更更优选为10000Da~200000Da;本发明对所述葡聚糖的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
在本发明中,所述葡聚糖与水的用量比优选为1g:(10mL~50mL),更优选为1g:(20mL~30mL)。在本发明中,所述葡聚糖与高碘酸钠的质量比优选为1:(0.05~1.5),更优选为1:(0.1~1)。
本发明优选在搅拌条件下进行避光反应,本发明对此没有特殊限制。在本发明中,所述避光反应的温度优选为15℃~50℃,更优选为20℃~40℃,更更优选为25℃~35℃;所述避光反应的时间优选为1.5h~6h,更优选为2h~5h。
在本发明中,所述可注射水凝胶由精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应得到。在本发明中,所述精氨酸修饰的壳聚糖和交联剂的质量比优选为1:(0.1~20),更优选为1:(1~10)。
在本发明中,所述溶剂优选为水、生理盐水、缓冲溶液、组织培养液或体液,更优选为缓冲溶液。在本发明一个优选的实施例中,所述溶剂为磷酸的缓冲溶液。
本发明还提供了一种可注射水凝胶的制备方法,包括以下步骤:
将精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应,得到可注射水凝胶;
所述精氨酸修饰的壳聚糖,具有式(I)所示结构:
其中,n为聚合度,10≤n≤3200;
所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%;
所述交联剂为氧化葡聚糖。
在本发明中,所述将精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应的过程优选具体为:
将交联剂、精氨酸修饰的壳聚糖分别与溶剂混合,再进行共混,自交联得到可注射抗菌水凝胶。在本发明中,所述精氨酸修饰的壳聚糖、交联剂、溶剂与上述技术方案所述的相同,在此不再赘述。
在本发明中,交联剂与溶剂混合得到的溶液中交联剂的质量浓度优选为2%~30%,更优选为4%~20%,更更优选为5%~15%。在本发明中,精氨酸修饰的壳聚糖与溶剂混合得到的溶液中精氨酸修饰的壳聚糖的质量浓度优选为2%~30%,更优选为4%~20%,更更优选为5%~15%。
在本发明中,所述自交联的温度优选为10℃~50℃,更优选为15℃~45℃,更更优选为20℃~40℃。
本发明提供了一种可注射水凝胶及其制备方法,所述可注射水凝胶由具有式(I)结构的精氨酸修饰的壳聚糖和交联剂在溶剂中进行自交联反应得到;其中,具有式(I)结构的精氨酸修饰的壳聚糖的聚合度10≤n≤3200;所述精氨酸修饰的壳聚糖中精氨酸的取代度为0.1%~80%;所述交联剂为氧化葡聚糖。与现有技术相比,本发明提供的可注射水凝胶具有良好的生物相容性,能够在体内原位形成,有利于药物直接作用于病变部位并较长久的发挥药效;而采用氧化葡聚糖作为交联剂安全无毒,环境污染小;同时,所述可注射水凝胶还具有良好的生物降解性,能够在体内降解,而且降解产物为氨基酸和多糖,可通过肾脏直接排除体外,对人体无害。实验结果表明,本发明提供的可注射水凝胶含水量高,不同浓度的多糖衍生物及凝胶浸提液的细胞存活率均大于85%,说明本发明提供的可注射水凝胶没有明显的细胞毒性,细胞相容性良好。
另外,本发明提供的可注射水凝胶具有较高的可调空间,能根据不同需要调节壳聚糖的分子量、壳聚糖侧链上精氨酸的取代度、氧化葡聚糖的分子量、氧化葡聚糖的氧化度等,从而能够制备各种不同物理化学性能的水凝胶材料。
此外,本发明提供的可注射水凝胶的制备方法原料简单易得,成本低;同时操作简单、反应条件温和、制备工艺稳定,易于实现工业化生产,具有较好的应用前景。
为了进一步说明本发明,以下结合实施例对本发明提供的可注射水凝胶及其制备方法进行详细描述。本发明以下实施例所用的原料均为市售,其中,精氨酸衍生物由上海吉尔生化有限公司提供的BOC-Arg(Z)-OH,结构式为:
实施例1
(1)称取2g壳聚糖与200mL盐酸溶液混合,得到第一混合溶液;再称取0.2g精氨酸衍生物、0.2g EDC、0.1g NHS于圆底烧瓶,加入20mL DMF使其溶解,得到第二混合溶液;将第二混合溶液加入到第一混合溶液中,30℃下搅拌反应48h,纯化,得到固体粉末;
(2)将步骤(1)得到的固体粉末以100mL三氟乙酸为反应介质分散,加入15mL溴化氢溶液,30℃下反应6h,纯化,得到精氨酸修饰的壳聚糖,产率为80%。
对步骤(1)得到的固体粉末进行核磁共振分析,如图1所示。结果表明,精氨酸成功接到壳聚糖上,同时精氨酸的取代度为3%。
实施例2
(1)称取2g壳聚糖与200mL盐酸溶液混合,得到第一混合溶液;再称取0.4g精氨酸衍生物、0.4g EDC、0.2g NHS于圆底烧瓶,加入20mL DMF使其溶解,得到第二混合溶液;将第二混合溶液加入到第一混合溶液中,30℃下搅拌反应48h,纯化,得到固体粉末;
(2)将步骤(1)得到的固体粉末以100mL三氟乙酸为反应介质分散,加入15mL溴化氢溶液,30℃下反应6h,纯化,得到精氨酸修饰的壳聚糖,产率为85%。
对步骤(1)得到的固体粉末进行核磁共振分析,结果表明,精氨酸成功接到壳聚糖上,同时精氨酸的取代度为6%。
实施例3
(1)称取2g壳聚糖与200mL盐酸溶液混合,得到第一混合溶液;再称取0.8g精氨酸衍生物、0.8g EDC、0.4g NHS于圆底烧瓶,加入20mL DMF使其溶解,得到第二混合溶液;将第二混合溶液加入到第一混合溶液中,30℃下搅拌反应48h,纯化,得到固体粉末;
(2)将步骤(1)得到的固体粉末以100mL三氟乙酸为反应介质分散,加入15mL溴化氢溶液,30℃下反应6h,纯化,得到精氨酸修饰的壳聚糖,产率为83%。
对步骤(1)得到的固体粉末进行核磁共振分析,结果表明,精氨酸成功接到壳聚糖上,同时精氨酸的取代度为10%。
实施例4
(1)称取2g壳聚糖与200mL盐酸溶液混合,得到第一混合溶液;再称取1.6g精氨酸衍生物、1.6g EDC、0.8g NHS于圆底烧瓶,加入20mLDMF使其溶解,得到第二混合溶液;将第二混合溶液加入到第一混合溶液中,30℃下搅拌反应48h,纯化,得到固体粉末;
(2)将步骤(1)得到的固体粉末以100mL三氟乙酸为反应介质分散,加入15mL溴化氢溶液,30℃下反应6h,纯化,得到精氨酸修饰的壳聚糖,产率为81%。
对步骤(1)得到的固体粉末进行核磁共振分析,如图2所示。结果表明,精氨酸成功接到壳聚糖上,同时精氨酸的取代度为23%。
实施例5
(1)称取2g壳聚糖与200mL盐酸溶液混合,得到第一混合溶液;再称取3.2g精氨酸衍生物、3.2g EDC、1.6g NHS于圆底烧瓶,加入20mLDMF使其溶解,得到第二混合溶液;将第二混合溶液加入到第一混合溶液中,30℃下搅拌反应48h,纯化,得到固体粉末;
(2)将步骤(1)得到的固体粉末以100mL三氟乙酸为反应介质分散,加入15mL溴化氢溶液,30℃下反应6h,纯化,得到精氨酸修饰的壳聚糖,产率为84%。
对步骤(1)得到的固体粉末进行核磁共振分析,结果表明,精氨酸成功接到壳聚糖上,精氨酸的取代度为32%。此外,对最终产物进行了核磁共振分析,如图3所示,发现苯环峰消失,说明已成功完成了脱保护过程,生成了精氨酸修饰的壳聚糖。
实施例6
(1)称取2g壳聚糖与200mL盐酸溶液混合,得到第一混合溶液;再称取6.4g精氨酸衍生物、6.4g EDC、3.2g NHS于圆底烧瓶,加入20mL DMF使其溶解,得到第二混合溶液;将第二混合溶液加入到第一混合溶液中,30℃下搅拌反应48h,纯化,得到固体粉末;
(2)将步骤(1)得到的固体粉末以100mL三氟乙酸为反应介质分散,加入15mL溴化氢溶液,30℃下反应6h,纯化,得到精氨酸修饰的壳聚糖,产率为86%。
对步骤(1)得到的固体粉末进行核磁共振分析。结果表明,精氨酸成功接到壳聚糖上,同时精氨酸的取代度为45%。
实施例7
称取2g葡聚糖于烧瓶中,再加入50ml水溶解,得到混合液,然后用锡箔纸包好烧瓶使其避光,再在避光条件下向混合液中加入0.5g高碘酸钠,25℃下搅拌,进行避光反应4h,再加入异丙醇终止反应,透析冻干,得氧化葡聚糖。
实施例8
称取2g葡聚糖于烧瓶中,再加入50ml水溶解,得到混合液,然后用锡箔纸包好烧瓶使其避光,再在避光条件下向混合液中加入1g高碘酸钠,25℃下搅拌,进行避光反应4h,再加入异丙醇终止反应,透析冻干,得氧化葡聚糖。
实施例9
将实施例1中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比10∶1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例10
将实施例1中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1∶1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例11
将实施例1中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1∶10共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例12
将实施例2中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比10∶1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例13
将实施例2中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1∶1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例14
将实施例2中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1∶10共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例15
将实施例3中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比10:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例16
将实施例3中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例17
将实施例3中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:10共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例18
将实施例4中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比8:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例19
将实施例4中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例20
将实施例4中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:8共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例21
将实施例5中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比6:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例22
将实施例5中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例23
将实施例5中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:6共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例24
将实施例6中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比4:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例25
将实施例6中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例26
将实施例6中得到的精氨酸修饰的壳聚糖与实施例7中得到的氧化葡聚糖,分别配制成质量浓度为6%的磷酸缓冲溶液,将制成的两种溶液以体积比1:4共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例27
将实施例1中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为5%的磷酸缓冲溶液,将制成的两种溶液以体积比16:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例28
将实施例1中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为5%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例29
将实施例1中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为5%的磷酸缓冲溶液,将制成的两种溶液以体积比1:16共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例30
将实施例2中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为8%的磷酸缓冲溶液,将制成的两种溶液以体积比14:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例31
将实施例2中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为8%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例32
将实施例2中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为8%的磷酸缓冲溶液,将制成的两种溶液以体积比1:14共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例33
将实施例3中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为10%的磷酸缓冲溶液,将制成的两种溶液以体积比12:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例34
将实施例3中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为10%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例35
将实施例3中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为10%的磷酸缓冲溶液,将制成的两种溶液以体积比1:12共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例36
将实施例4中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为12%的磷酸缓冲溶液,将制成的两种溶液以体积比10:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例37
将实施例4中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为12%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例38
将实施例4中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为12%的磷酸缓冲溶液,将制成的两种溶液以体积比1:10共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例39
将实施例5中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为14%的磷酸缓冲溶液,将制成的两种溶液以体积比9:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例40
将实施例5中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为14%的磷酸缓冲溶液,将制成的两种溶液以体积比1:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例41
将实施例5中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为14%的磷酸缓冲溶液,将制成的两种溶液以体积比1:9共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例42
将实施例6中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为15%的磷酸缓冲溶液,将制成的两种溶液以体积比8:1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例43
将实施例6中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为15%的磷酸缓冲溶液,将制成的两种溶液以体积比1∶1共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例44
将实施例6中得到的精氨酸修饰的壳聚糖与实施例8中得到的氧化葡聚糖,分别配制成质量浓度为15%的磷酸缓冲溶液,将制成的两种溶液以体积比1:8共混,采用小管倒置法观察其在37℃的粘度变化,以小管倒置时,30s内不发生流动为凝胶化,待其自交联得到可注射水凝胶。
实施例45
将实施例7得到的氧化葡聚糖进行体外细胞毒性测试:将细胞种植于96孔板中,细胞密度为1×104个细胞/孔,在37℃,5%CO2的无菌培养箱中培养24h;移除培养基,加入不同浓度倍比稀释的多糖衍生物溶液(简称ODex,最大浓度为10mg/mL,200μL/孔),并以PEI25K作为阳性对照,继续培养24h,移除培养液后采用MTT方法测试。
结果参见图4,图4为本发明实施例45所述的不同浓度的氧化葡聚糖溶液的细胞存活率。由图4可知,不同浓度溶液的细胞存活率均大于85%,说明本发明所述的氧化葡聚糖溶液没有明显的细胞毒性,且氧化葡聚糖中含有醛基可作为大分子交联剂,因此可在生物医用材料领域进一步应用。
实施例46
将实施例4所述的精氨酸修饰的壳聚糖进行体外细胞毒性测试:将细胞种植于96孔板中,细胞密度为1×104个细胞/孔,在37℃,5%CO2的无菌培养箱中培养24h;移除培养基,加入不同浓度倍比稀释的多糖衍生物溶液(简称CA,最大浓度为10mg/mL,200μL/孔),并以PEI25K作为阳性对照,继续培养24h,移除培养液后采用MTT方法测试。
结果参见图5,图5为本发明实施例46得到的不同浓度的精氨酸修饰的壳聚糖溶液的细胞存活率。由图5可知,不同浓度溶液的细胞存活率均大于85%,说明该精氨酸修饰的壳聚糖溶液没有明显的细胞毒性,因此可在生物医用材料领域进一步应用。
实施例47
将实施例12制备的可注射水凝胶进行体外细胞毒性测试:用DMEM浸提实施例12中制备的可注射水凝胶,从而得到凝胶浸提液;将细胞种植于96孔板中,细胞密度为1×104个细胞/孔,在37℃,5%CO2的无菌培养箱中培养24h;移除培养基,加入不同浓度倍比稀释的凝胶浸提液,继续培养不同时间(24h,48h),移除培养液后采用MTT方法测试。
结果参见图6,图6为本发明实施例47得到的不同浓度的凝胶浸提液的细胞存活率。在图6中,不同浓度凝胶浸提液的细胞存活率均大于85%,说明此种可注射水凝胶没有明显的细胞毒性。此外,混合溶液在37℃条件下能够形成水凝胶,即可在体温下形成水凝胶,因此能够作为可注射性型水凝胶作为药物载体或支架材料应用。
实施例48
将实施例19制备的可注射水凝胶进行体外细胞毒性测试:用DMEM浸提实施例19中制备的可注射水凝胶,从而得到凝胶浸提液;将细胞种植于96孔板中,细胞密度为1×104个细胞/孔,在37℃,5%CO2的无菌培养箱中培养24h;移除培养基,加入不同浓度倍比稀释的凝胶浸提液,继续培养不同时间(24h,48h),移除培养液后采用MTT方法测试。
结果参见图7,图7为本发明实施例48得到的不同浓度的凝胶浸提液的细胞存活率。在图7中,不同浓度凝胶浸提液的细胞存活率均大于85%,说明此种可注射水凝胶没有明显的细胞毒性。此外,混合溶液在37℃条件下能够形成水凝胶,即可在体温下形成水凝胶,因此能够作为可注射性型水凝胶作为药物载体或支架材料应用。
实施例49
将实施例15制备的可注射水凝胶进行体外细胞毒性测试:用DMEM浸提实施例15中制备的可注射水凝胶,从而得到凝胶浸提液;将细胞种植于96孔板中,细胞密度为1×104个细胞/孔,在37℃,5%CO2的无菌培养箱中培养24h;移除培养基,加入不同浓度倍比稀释的凝胶浸提液,继续培养不同时间(24h,48h),移除培养液后采用MTT方法测试。
结果参见图8,图8为本发明实施例49得到的不同浓度的凝胶浸提液的细胞存活率。在图8中,不同浓度凝胶浸提液的细胞存活率均大于85%,说明此种可注射水凝胶没有明显的细胞毒性。此外,混合溶液在37℃条件下能够形成水凝胶,即可在体温下形成水凝胶,因此能够作为可注射性型水凝胶作为药物载体或支架材料应用。
实施例50
将实施例18制备的可注射水凝胶进行体外细胞毒性测试:用DMEM浸提实施例18中制备的可注射水凝胶,从而得到凝胶浸提液;将细胞种植于96孔板中,细胞密度为1×104个细胞/孔,在37℃,5%CO2的无菌培养箱中培养24h;移除培养基,加入不同浓度倍比稀释的凝胶浸提液,继续培养不同时间(24h,48h),移除培养液后采用MTT方法测试。
结果参见图9,图9为本发明实施例50得到的不同浓度的凝胶浸提液的细胞存活率。在图9中,不同浓度凝胶浸提液的细胞存活率均大于85%,说明此种可注射水凝胶没有明显的细胞毒性。此外,混合溶液在37℃条件下能够形成水凝胶,即可在体温下形成水凝胶,因此能够作为可注射性型水凝胶作为药物载体或支架材料应用。
由以上实施例可知,本发明提供的可注射水凝胶具有较高的可调空间,能根据不同需要进行调节壳聚糖侧链上精氨酸的取代度、氧化葡聚糖的氧化度、多糖浓度及多糖组分配比等,此外,也能调节壳聚糖及葡聚糖的分子量。同时,利用天然多糖衍生物大分子充当交联剂,避免引入额外的化学交联剂,安全无毒,环境污染小;而且所述可注射水凝胶可以制备各种不同成凝胶时间和凝胶强度的水凝胶。再者,本发明所述精氨酸修饰的壳聚糖可赋予水凝胶生物功能性,如细胞粘附性、生物相容性、抗菌性等,也可以进行原位凝胶的使用。更重要的是,本发明提供的可注射水凝胶含水量高,不同浓度的多糖衍生物材料及凝胶浸提液的细胞存活率均大于85%,说明此种可注射水凝胶没有明显的细胞毒性,细胞相容性良好。并且,该水凝胶能够生物降解,降解产物为氨基酸和多糖,能够通过肾脏排出体外,对人体无毒,具备某些特殊的医用功能,具有推广应用的潜力,市场前景广阔。另外,混合溶液在37℃条件下能够形成水凝胶,即可在体温下形成水凝胶,因此能够用作可注射性型凝胶,作为药物载体或者支架材料应用。再者,本发明提供的水凝胶材料制备工艺稳定,易于实现工业化生产,原料简单易得,成本低、反应条件温和。
所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!