来氟米特晶型化合物、制备方法及其应用与流程
本发明涉及一种来氟米特新晶型化合物/制备方法及其应用,属于医药晶型领域。
背景技术:
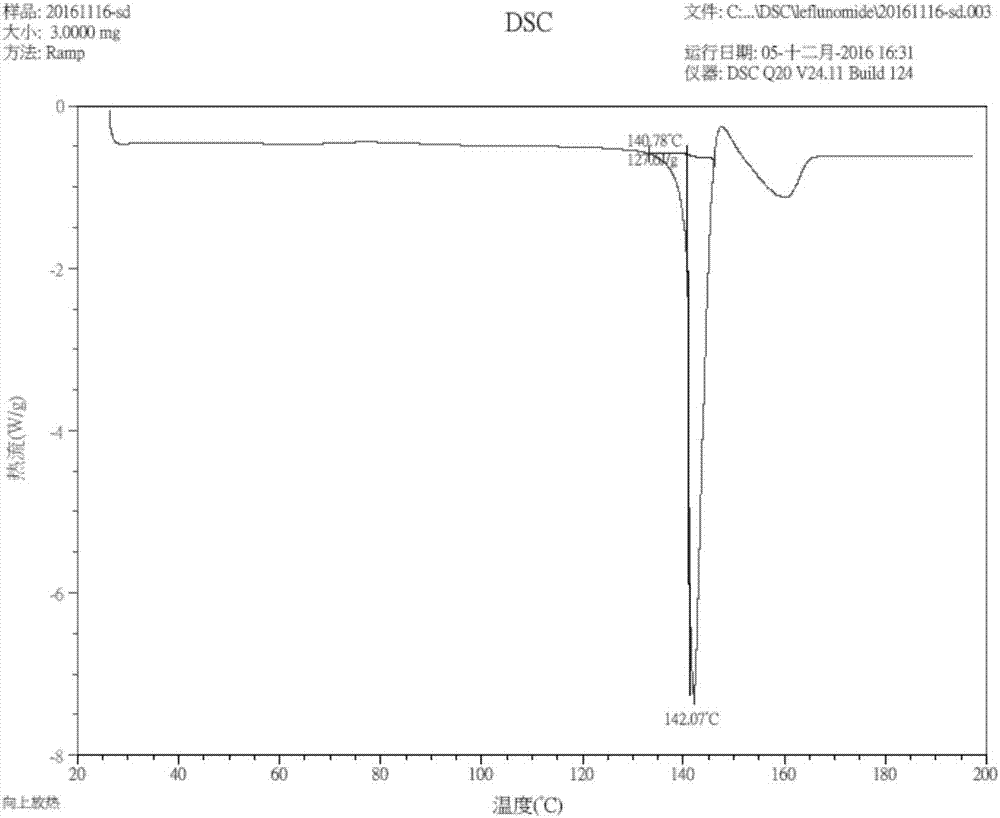
:来氟米特(leflunomide),化学式:c12h9f3n2o2,化学名:n-(4-三氟甲基苯基)-5-甲基异噁唑-4-甲酰胺,结构式如下:是一个具有抗增殖活性的异唑类免疫调节剂,用于治疗风湿性关节炎、系统性红斑狼疮等。其活性代谢物特立唑胺,也被开发用于多发性硬化症。来氟米特目前有三种晶型报道,专利申请cn02104574中报道了晶型i和晶型ii。晶型ⅰ具有特定的x-射线衍射图谱,在2θ角度为16.70°、18.90°、23.00°、23.65°和29.05°处有强的衍射峰,而在2θ为8.25°、12.65°、15.00°、15.30°、18.35°、21.25°、22.15°、24.10°、24.65°、25.45°、26.65°、27.40°、28.00°和28.30°处有弱的衍射峰。晶型ii具有特定的衍射峰,在2θ角为10.65°、14.20°、14.80°、16.10°、21.70°、23.15°、24.40°、24.85°、25.50°、25.85°、26.90°和29.85°有强的衍射峰,在7.40°、9.80°、13.10°、15.45°、16.80°、20.70°、21.45°、22.80°、23.85°、27.25°和28.95°有弱的衍射峰。在10℃—40℃下,晶型i会缓慢转变成晶型ii。在50℃以上,晶型ii转化为晶型i。专利申请cn00817283中的报道了晶型iii,晶型iii为n-甲基吡咯烷酮的溶剂化合物,不适合药用。晶型i在室温情况下会缓慢转变成晶型ii,晶型ii在高温下转化成晶型i。晶型i和晶型ii均不稳定,目前市场上的原料药多数为晶型i与晶型ii的混晶,这不仅影响了原料药和制剂的生产质量,更会影响到制剂的稳定性。基于这种情况,筛选并开发高稳定性的来氟米特新晶型,对于来氟米特药物的开发具有重大意义。技术实现要素:为了找到稳定的来氟米特新晶型化合物,申请人进行了大量的晶型化合物筛选,提出了一种稳定性高的具有式i结构式的来氟米特新晶型化合物,根据发现顺序和行业命名,命名为晶型iv。本发明提供了一种来氟米特晶型化合物,该化合物为新晶型化合物,其中,所述晶型化合物在xrd图谱的2θ角度位置为8.04±0.2°、10.83±0.2°、14.28±0.2°、18.66±0.2°、20.48±0.2°和25.13±0.2°处有特征衍射峰。本发明还提供了一种来氟米特晶型化合物,其中,所述晶型化合物在xrd图谱的2θ角度位置为8.04±0.2°、10.83±0.2°、12.26±0.2°、14.28±0.2°、16.82±0.2°、18.66±0.2°、20.48±0.2°、21.79±0.2°、24.68±0.2°、25.13±0.2°、25.88±0.2°和27.13±0.2°处有特征衍射峰。本发明还提供了一种来氟米特晶型化合物,其中,所述晶型化合物在xrd图谱的2θ角度位置为2θ为:8.04±0.2°、10.83±0.2°、12.26±0.2°、14.28±0.2°、16.08±0.2°、16.82±0.2°、18.66±0.2°、20.48±0.2°、21.79±0.2°、23.11±0.2°、23.71±0.2°、24.68±0.2°、25.13±0.2°、25.88±0.2°、27.13±0.2°、28.99±0.2°、30.12±0.2°、32.13±0.2°、32.61±0.2°、33.72±0.2°、35.49±0.2°、36.65±0.2°和37.99±0.2°处有特征衍射峰。本发明还提供了一种来氟米特晶型化合物,其中,所述晶型化合物具有图1所示xrd图中所示的特征衍射峰。上述晶型化合物的xrd图的特征衍射峰是在以下实验条件下测得的:布鲁克d2phaserx射线衍射仪,电压30kv,电流10ma,扫描范围3°~40°,扫描步长:0.02°,扫描速度19.2s/步。本发明经过系统地晶型筛选实验,其中筛选方法包括,蒸发结晶法(单一溶剂),蒸发结晶法(混合溶剂法),热溶冷析法,反溶剂法等。筛选溶剂包括水、甲苯、戊烷、己烷、环己烷、氯苯、二氯甲烷、甲醇、乙醇、异丙醇、乙醚、环氧丙烷、醋酸甲酯、醋酸乙酯、醋酸丙酯、丙酮、甲基丁酮、甲基异丁酮、乙腈、吡啶、dma、n-甲基吡咯烷酮等,筛选得到的晶型均为晶型ⅰ、晶型ⅱ、晶型ⅲ或者它们的混晶。为了进一步筛选晶型,本发明改变策略,创新性地采用了具有一定粘性的表面活性剂类的溶剂进行筛选。分别以聚乙二醇、吐温、司盘、transcutolp、labrasol、capryol90和transctolhp为溶剂的筛选实验中,从聚乙二醇400中得到了新晶型化合物-晶型ⅳ。具有详细实验报告,由于篇幅问题,全文暂欠奉,如需提供,可以补充提供。本发明还提供了一种来氟米特晶型化合物,其中,该晶型化合物的熔点范围142.07±2℃,所述来氟米特的纯度为99.5%-99.9%(hplc测定)。本发明还进一步提供了一种来氟米特晶型化合物,其中,该晶型化合物具有上述xrd图中所示的特征衍射峰的特征,其由以下方法制得:1)称取来氟米特原料药和聚乙二醇类,混合,加热,搅拌至来氟米特原料药充分溶解;2)降温直至析出晶体;3)过滤得到固体,将固体进行洗涤;4)将洗涤后的固体进行干燥,得到所述新晶型化合物。本发明还提供了一种来氟米特晶型化合物,其中:所述加热优选温度维持在50-100℃。所述来氟米特原料药与聚乙二醇的重量比优选:1:3~10;最优选1:5。步骤3中洗涤溶剂优选水,洗涤次数优选2-5次,更有选3次。干燥优选真空干燥。在本发明还提供了一种所述的氟米特晶型化合物,其中所述来氟米特原料药为来氟米特晶型ⅰ、晶型ⅱ、晶型ⅲ或上述晶型中两种及以上的晶型混合物。所述来氟米特晶型ⅳ制备所使用的聚乙二醇类溶剂为peg200-20000的一种或混合溶剂,,进一步优选peg200-1000,所述聚乙二醇类溶剂进一步优选peg200~400的一种或混合溶剂。本发明还提供了一种来氟米特晶型化合物的制备方法,由以下步骤组成:1)称取来氟米特原料药和聚乙二醇类,混合,加热,搅拌至来氟米特原料药充分溶解;2)降温直至析出晶体;3)过滤得到固体,将固体进行洗涤;4)将洗涤后的固体进行干燥,得到所述晶型。加热优选温度维持在50-100℃。所述来氟米特原料药与聚乙二醇的重量比优选:1:3~10;最优选1:5。步骤3中洗涤溶剂优选水,洗涤次数优选2-5次,更有选3次。干燥优选真空干燥。在所述来氟米特新晶型化合物的制备方法中,所述来氟米特原料药为来氟米特晶型ⅰ、晶型ⅱ、晶型ⅲ或上述晶型中两种及以上的晶型混合物。所述来氟米特晶型ⅳ的制备方法中的聚乙二醇类溶剂为peg200-20000的一种或混合溶剂,优选peg200-1000,所述聚乙二醇类溶剂进一步优选peg200~400的一种或混合溶剂。来氟米特原料药中加入晶型iv晶种可以加速晶型转化。晶种iv使用量为来氟米特原料药的0.1%~1.0%。所述的制备方法,主要溶媒为聚乙二醇类溶媒,也可以加入少许其他溶剂如乙醇、丙酮、水等调节溶媒的溶解度,不影响晶型ⅳ的制备,所述少许为促进所述来氟米特原料药进一步溶解充分为宜。本发明还提供了一种药物组合物,其中,所述药物组合物由所述来氟米特晶型的活性成分和其他药学上可接受的载体组成。其中可接受的载体包括赋形剂、填充剂、润滑剂、粘合剂、助流剂等。所述来氟米特药物组合物可以是片剂,胶囊,分散剂或混悬剂。本发明还提供了一种所述来氟米特晶型化合物用于制备治疗免疫系统异常疾病的药物的应用。所述免疫系统异常疾病包括风湿性关节炎、系统性红斑狼疮和多发性硬化症药物。本发明的组合物包括适于口服和注射等给药途径,优选口服给药途径。剂型包括片剂,胶囊,分散剂和混悬剂,优选片剂和胶囊。本发明晶型稳定性好,在10℃-60℃晶型稳定,目前已报道的晶型ⅰ和晶型ⅱ在10℃-60℃均不稳定。晶型ⅱ在60℃下,放置7小时后转化为晶型ⅰ,如图5所示。晶型ⅰ、晶型ⅱ和晶型ⅳ等量(200mg)混合在5ml水中打浆,16小时后,测定混合物晶型为晶型ⅱ和晶型ⅳ的混晶,如图6所示。由图5和图6说明晶型ⅰ和晶型ⅱ均不稳定。专利cn02104574和专利cn00817283也描述了晶型ⅰ、ⅱ和ⅲ的不稳定性。而本发明来氟米特晶型ⅳ在室温和高湿高温强光照条件下具有很好的稳定性。不同的晶型通常具有不同的溶解度。通过制备了来氟米特晶型ⅰ、晶型ⅱ和晶型ⅳ,并测定其在室温(22℃)在不同ph介质中的溶解度,结果见下表1:表1根据热力学原理,稳定的晶型具有低的溶解度(通常情况下)。同一温度下,溶解度低的晶型稳定性相对较好。尽管来氟米特的晶型ⅰ、晶型ⅱ和晶型ⅳ在各介质中的溶解度均很低,属于难溶性药物,但晶型ⅳ的溶解度最低,表明来氟米特晶型ⅳ在室温下最稳定。本发明制得的来氟米特晶型ⅳ性质非常稳定,在高温高湿强光照条件下以及加速试验条件下(检测报告全文过长,限于篇幅问题,本文暂欠奉,若需要可补充提供)均未发生转晶,且制作工艺简单,适合放大生产,流动性好,具有良好的制剂可加工性,本发明不仅解决了原料药晶型不稳定的问题,而且给制剂工艺提供了更好的选择。本发明所述的晶型ⅳ是一种菱形的薄片状的晶体,如图3的菱片状晶体的对角线长度分别为0.1075mm和0.0449mm,流动性好,有良好的制剂可加工性。本发明所述的晶型ⅳ,其熔点约为142.07℃,如图2。本发明提供了所述的晶型ⅳ的红外图谱,如图4。图6为来氟米特晶型i、晶型ii和晶型iv各200mg,悬浮于5ml水中,打浆16h后xrd图谱,为晶型ⅱ和晶型ⅳ的混晶。本发明中所有xrd图谱中的横坐标的英文意思均为2θ角度。纵坐标为计数。本发明中所述来氟米特晶型iv,具有以下优点:溶解度低,稳定性好,可以通过晶型i、晶型ii和晶型iii通过简易的制备方式得到;在高温高湿强光照条件下,以及加速试验条件下晶型iv稳定性极好。附图说明图1为来氟米特晶型iv的xrd图谱;图1中所述晶型化合物在xrd图谱的2θ角度位置为2θ为:8.04±0.2°、10.83±0.2°、12.26±0.2°、14.28±0.2°、16.08±0.2°、16.82±0.2°、18.66±0.2°、20.48±0.2°、21.79±0.2°、23.11±0.2°、23.71±0.2°、24.68±0.2°、25.13±0.2°、25.88±0.2°、27.13±0.2°、28.99±0.2°、30.12±0.2°、32.13±0.2°、32.61±0.2°、33.72±0.2°、35.49±0.2°、36.65±0.2°和37.99±0.2°处有特征衍射峰。图2为来氟米特晶型iv的dsc图谱;图2中显示,熔点为142.07℃。图3为来氟米特晶型iv的显微照片;图3的菱片状晶体的对角线长度分别为0.1075mm和0.0449mm;图4为来氟米特晶型iv的红外图谱;图5为来氟米特晶型ⅱ在60℃,放置第0小时、第1小时、第3小时和第7小时的xrd对比图谱;图6为来氟米特晶型i、晶型ii和晶型iv混合打浆后xrd图谱。具体实施方式下面的实施例仅用于进一步说明本发明但不限于本发明。凡基于本发明上述内容所实现的技术均属于本发明范围。实验条件:布鲁克d2phaserx射线衍射仪,电压30kv,电流10ma,扫描范围3°~40°,扫描步长:0.02°,扫描速度19.2s/步。实施例1将8g来氟米特原料药(晶型i)加热溶于10ml下列溶剂中,冷却至室温(约22℃),得到的固体测x-射线粉末衍射检测。在peg400中得到晶型ⅳ,如下表2。表2编号溶剂方法晶型1吐温80热溶冷析未收集到固体2司盘20热溶冷析未收集到固体3聚乙二醇400热溶冷析晶型ⅳ4transcutolp热溶冷析晶型ⅰ5labrasol热溶冷析未收集到固体6capryol90热溶冷析未收集到固体7transcutolhp热溶冷析未收集到固体实施例2来氟米特晶型ⅳ的制备将8g的来氟米特原料药(晶型i)悬浮于10ml的peg400,加热至68℃,并使来氟米特完全溶解,搅拌10小时后冷却至室温(22℃),过滤得到固体,用水洗涤2-5次,洗涤后干燥得到晶体,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ,见图1。实施例3来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)悬浮于5g的peg200,加热至68℃,并使来氟米特完全溶解,搅拌36小时后冷却至室温(22℃),过滤得到固体,用水洗涤2-5次,洗涤后干燥得到晶体,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例4来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)悬浮于3g的peg400,加热至50℃,并使来氟米特完全溶解,搅拌72小时后冷却至室温(22℃),过滤得到固体,用水洗涤3次,洗涤后干燥得到晶体,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例5来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)悬浮于5g的peg400,加热至75℃,并使来氟米特完全溶解,搅拌72小时后冷却至室温(22℃),过滤得到固体,用水洗涤3次,洗涤后干燥得到晶体,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例6来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)悬浮于10g的peg400,加热至100℃,并使来氟米特完全溶解,搅拌72小时后冷却至室温(22℃),过滤得到固体,用水洗涤3次,洗涤后干燥得到晶体,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例7来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)与5gpeg6000混合,加热至完全溶解,温度为50℃,若少许未溶,补少许乙醇;冷却至室温得到固体,将固体放入水中打浆16h,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例8来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)与5gpeg10000混合,加热至完全溶解,温度为50℃,若少许未溶,补少许乙醇;冷却至室温得到固体,将固体放入水中打浆16h,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例9来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)与5gpeg12000混合,加热至完全溶解,温度为100℃,冷却至室温,将固体放入水中打浆16小时,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例10来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)与5gpeg20000混合,加热至完全溶解,温度为100℃,冷却至室温,将固体放入水中打浆16小时,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例11来氟米特晶型iv的制备将1g的来氟米特原料药(晶型ii)与5gpeg400混合,加热至68℃,并使来氟米特完全溶解,搅拌10小时后冷却至室温(22℃),过滤得到固体,用水洗涤2-5次,洗涤后干燥得到晶体,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ,实施例12来氟米特晶型ⅳ的制备将1g的来氟米特原料药(晶型i)和0.05g来氟米特iv晶型共同悬浮于5g的peg400,加热至68℃,并使来氟米特完全溶解,搅拌72小时后冷却至室温(22℃),过滤得到固体,用水洗涤2-5次,洗涤后干燥得到晶体,进行x-射线粉末衍射检测和dsc检测,晶体为晶型ⅳ。实施例13高温条件下,来氟米特晶型ⅱ的稳定性研究来氟米特晶型ⅱ在60℃进行放置,于第0小时、第1小时、第3小时和第7小时进行x-射线粉末衍射检测,发现第7小时已经基本转化为晶型ⅰ,如图5,说明晶型ⅱ在高温条件下稳定性差。实施例14来氟米特晶型ⅰ、晶型ⅱ和晶型ⅳ室温下,水中打浆研究在室温下(约22℃),分别称取200mg来氟米特晶型ⅰ、晶型ⅱ和晶型ⅳ,悬浮于5ml水中,打浆16h后,进行x-射线粉末衍射检测,为晶型ⅱ和晶型ⅳ的混晶,如图6,说明晶型ⅰ在室温条件下稳定性差。实施例15晶型ⅳ的稳定性考察取实施例2制得的来氟米特晶型ⅳ进行影响因素试验和加速试验,其中影响因素具体为:取来200mg氟米特晶型ⅳ分别在高温(60℃),高湿(25℃,rh90%),光照(1.2×106lux·hr)条件下放置30天,于0天、5天、10天、30天末取样,对其晶型进行考察,x-射线粉末衍射检测显示放置1个月后来氟米特晶型ⅳ的晶型无变化。加速试验具体为:取200mg来氟米特晶型ⅳ在40±2℃,75%±5%rh条件下放置3个月,于0个月、3个月末取样,对其晶型进行考察,x-射线粉末衍射检测显示加速放置3个月后氟米特晶型ⅳ样品的晶型无变化。实施例16晶型ⅳ的片剂分别称量来氟米特晶型ⅳ0.5g,乳糖6.2g,交联聚维酮0.75g,二氧化硅25mg和硬脂酸镁25mg。在来氟米特晶型ⅳ中,依次加入乳糖,交联聚维酮,再加入二氧化硅,硬脂酸镁,手工混合均匀。调节压片机使片重约为150mg,直接干法直压得到来氟米特晶型ⅳ片剂,其在纯水中30分钟溶出度可达到80%。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制,应当指出的是,对于本领域的技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改造,这些都属于本发明的保护范围,因此,本发明专利的保护范围以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1