一种MI‑2关键中间体及其制备方法与流程
本发明属于有机合成技术领域,具体涉及一种制备mi-2关键中间体的方法。
背景技术:
淋巴瘤(lymphoma)是淋巴结或结外淋巴组织的恶性肿瘤,nhl是成人淋巴瘤的主要类型,占世界上常见恶性肿瘤的第七位,弥漫性大b细胞淋巴瘤(dlbcl)是其中最为常见的一种亚型,其中abc型淋巴瘤是预后最差,治愈率最低,最易产生耐药性的一种类型。因此寻求更加有效的治疗手段尤其是靶向药物用于治疗abc型淋巴瘤成为目前亟待解决的难题。
大量研究数据显示黏膜相关型淋巴组织淋巴瘤易位蛋白(malt1)是abc型淋巴瘤中有效的靶分子,它可以切割多种底物,以促进淋巴细胞增殖,提高淋巴瘤细胞的存活率。malt1的催化活性在正常情况下受抗原受体触发的严格控制,通过其诱导的单泛素化依赖的二聚反应来诱导malt1的激活。通过特异性抑制malt1的活性,可以达到抑制淋巴细胞恶性增值的目的。
mi-2是最近报道的一种骨架新颖、结构简单的malt1抑制剂,该化合物是fontan等人通过高通量筛选(high-throughputscreening,hts)发现的一种具有三氮唑环结构骨架的小分子先导化合物。细胞实验结果表明,mi-2的药效活性可达到纳摩尔级别并对abc型淋巴瘤具有一定的选择性。同时,mi-2(其结构如图1所示)是一种不可逆的malt1抑制剂,其分子结构中的氯甲基酰胺结构部分可以与malt1中的活性位点共价性结合,这可能是其发挥不可逆抑制作用的原因。本课题组前期研究结果显示,在mi-2乙二醇单甲醚支链区域进行合理结构修饰能够大大提高其抑制恶性dlbcl淋巴瘤细胞增殖的能力。
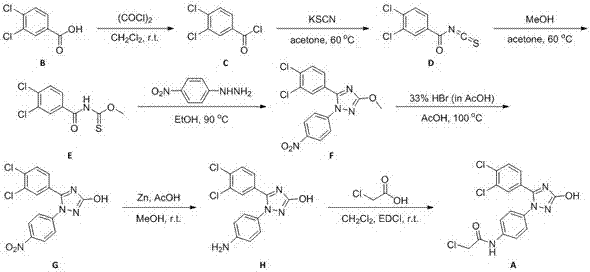
本发明以3,4-二氯苯甲酸为起始原料,经7步常规反应后得到mi-2关键中间体。发明中所涉及的反应操作简单、条件温和、易于处理,所用的试剂及仪器皆为实验室常用易得的,具有很强的操作性。通过该中间体,可快速大量获得mi-2及其在乙二醇单甲醚支链区域改造后的类似物,有助于对mi-2进行系统全面的构效关系研究,具有十分重要的意义。
技术实现要素:
本发明的目的在于提供一种mi-2关键中间体及其制备方法,通过该中间体,可快速大量获得mi-2及其在乙二醇单甲醚支链区域改造后的类似物,有助于对mi-2进行系统全面的构效关系研究,具有十分重要的意义。
为实现上述目的,本发明采用如下技术方案:
一种mi-2关键中间体,其结构式为:
一种制备如上所述的mi-2关键中间体的方法,包括以下步骤:
1)以3,4-二氯苯甲酸为主要起始原料,合成3,4-二氯苯甲酰氯;
2)3,4-二氯苯甲酰氯再与硫氰酸钾反应得到3,4-二氯苯甲酰异硫氰酸酯;
3)3,4-二氯苯甲酰异硫氰酸酯与甲醇反应得到3,4-二氯苯甲酰氨基硫代甲酸甲酯;
4)3,4-二氯苯甲酰氨基硫代甲酸甲酯与对硝基苯肼反应得到3-甲氧基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑;
5)3-甲氧基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑与醋酸反应得到3-羟基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑;
6)3-羟基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑与过量醋酸、锌粉反应得到3-羟基-5-(3,4-二氯苯基)-1-(4-氨基苯基)-1,2,4-三氮唑;
7)将3-羟基-5-(3,4-二氯苯基)-1-(4-氨基苯基)-1,2,4-三氮唑依次与氯乙酸和1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐混合反应得到mi-2关键中间体:3-羟基-5-(3,4-二氯苯基)-1-(4-氯乙酰氨基苯基)-1,2,4-三氮唑。
所述的制备mi-2关键中间体的方法,具体为:
1)将3,4-二氯苯甲酸溶于适量二氯甲烷溶剂中,冰浴条件下依次加入1.1当量草酰氯和催化量的n,n-二甲基甲酰胺,撤去冰浴装置,室温搅拌12h后低温旋干得到3,4-二氯苯甲酰氯;
2)向3,4-二氯苯甲酰氯的丙酮溶液中加入等当量的硫氰酸钾,混合溶液在60℃条件下搅拌反应1小时后,反应液经纯化后得到3,4-二氯苯甲酰异硫氰酸酯;
3)3,4-二氯苯甲酰异硫氰酸酯用丙酮溶解后,向其中加入2.5当量甲醇,混合溶液在60℃条件下搅拌反应3小时后,反应液经纯化后得到3,4-二氯苯甲酰氨基硫代甲酸甲酯;
4)将步骤3)制得的3,4-二氯苯甲酰氨基硫代甲酸甲酯用乙醇溶解后,加入1.2当量对硝基苯肼混合均匀,在90℃条件下搅拌反应12小时后,反应液经纯化后得到3-甲氧基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑;
5)将步骤4)所制得的3-甲氧基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑用醋酸溶解后,加入5当量质量分数为33%hbr醋酸溶液,反应液在100℃条件下搅拌反应6小时,冷却,将反应液加入至50ml冰水中并充分搅拌,抽滤得到3-羟基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑;
6)将步骤5)所得的3-羟基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑用适量甲醇溶剂溶解后,加入锌粉、过量醋酸并混合均匀,室温下搅拌反应过夜后,所得反应液经纯化后得到3-羟基-5-(3,4-二氯苯基)-1-(4-氨基苯基)-1,2,4-三氮唑;
7)将步骤6)制得的3-羟基-5-(3,4-二氯苯基)-1-(4-氨基苯基)-1,2,4-三氮唑用适量的ch2cl2溶剂溶解,依次加入1.5当量氯乙酸和2当量1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐,混合均匀,在室温下搅拌反应2小时后,所得反应液经后处理后得到所述的mi-2关键中间体,即3-羟基-5-(3,4-二氯苯基)-1-(4-氯乙酰氨基苯基)-1,2,4-三氮唑。
一种利用如上所述的mi-2关键中间体制备mi-2及其类似物的方法:将mi-2关键中间体与三苯基膦混合,用四氢呋喃溶剂溶解后,加入乙二醇单甲醚或醇试剂,在冰浴条件下加入偶氮二甲酸二异丙酯,反应液在室温条件下搅拌反应1小时,所得反应液经纯化后即可得到mi-2或其类似物。
所述的纯化具体为:将反应液用乙酸乙酯稀释后,依次加入适量的水、饱和食盐水萃取1~2次,收集有机相并加入无水硫酸钠干燥后,过滤浓缩,得到的粗产物在合适的洗脱极性下经硅胶柱层析纯化后得到mi-2及其类似物。
本发明的显著优点在于:
本发明所设计的合成路线中所涉及的反应操作简单、条件温和、易于处理,所用的试剂及仪器皆为实验室常用易得的,具有很强的操作性。根据本路线可以快速大量获得malt1抑制剂mi-2及其在乙二醇单甲醚支链区域改造后的类似物,有助于对mi-2进行系统全面的构效关系研究,具有十分重要的意义。
附图说明
图1为mi-2的结构式;
图2为mi-2关键中间体的合成路线。
具体实施方式
为了使本发明所述的内容更加便于理解,下面结合具体实施方式对本发明所述的技术方案做进一步的说明,但是本发明不仅限于此。
实施例1
一种制备mi-2关键中间体的方法,具体合成步骤为:
1)3,4二氯苯甲酰氯(c)的制备:
在磁力搅拌反应装置中,往装有磁力搅拌子的200ml圆底烧瓶中加入3,4-二氯苯甲酸(9.55g,50mmol)和100ml二氯甲烷,在0℃冰水浴条件下,逐滴加入草酰氯(4.7ml,55mmol),然后接上尾气吸收装置,末端置于装有氢氧化钠溶液的烧杯中,用保鲜膜将烧杯口包裹(用于吸收反应过程中产生的氯化氢气体)。室温下搅拌,反应12小时,经tlc检测已基本无原料3,4-二氯苯甲酸即可停止反应。用旋转蒸发仪除去二氯甲烷溶剂,无须纯化即可进行下一步的反应;
2)3,4-二氯苯甲酰异硫氰酸酯(d)的制备:
取一个干净干燥的100ml圆底烧瓶,先在室温下,将步骤1)制得的3,4-二氯苯甲酰氯(10.5g,50mmol)溶于20ml丙酮中,并加入kscn(4.9g,50mmol);放入磁力搅拌子,在磁力搅拌反应装置中,加热到60℃,反应1小时。待反应液冷却下来后,经旋转蒸发仪进行旋蒸,除去溶剂后,向粗品中加入20ml乙酸乙酯溶解后转移至分液漏斗中,依次加入水(2×15ml)、饱和食盐水(1×10ml)萃取,收集有机相并加入适量无水硫酸钠干燥后浓缩,得到的粗产物经硅胶柱层析得到10.8g白色固体,洗脱剂为pe/etoac=4:1,反应收率为93%。1hnmr(400mhz,cdcl3)δ8.13(d,j=2.1hz,1h),7.88(dd,j=8.4,2.1hz,1h),7.58(d,j=8.4hz,1h).13cnmr(101mhz,cdcl3)δ160.67,150.23,140.11,133.88,132.27,131.24,130.79,129.36.
3)3,4-二氯苯甲酰氨基硫代甲酸甲酯(e)的合成;
向3,4-二氯苯甲酰异硫氰酸酯(d)(504mg,2.2mmol)的丙酮(5ml)溶液中加入meoh(5ml)。将混合物在60℃下搅拌反应3小时。将反应混合物用etoac(15ml)稀释,加h2o(3×5ml)萃取。有机层用无水硫酸钠干燥,然后浓缩,得到粗产物。残余物通过硅胶柱层析(pe/etoac=2:1)纯化,得到目标产物(481mg,83%),为白色固体。1hnmr(400mhz,cdcl3)δ9.16(s,1h),7.92(d,j=2.1hz,1h),7.68–7.63(m,1h),7.57(d,j=8.3hz,1h),4.18(s,3h).13cnmr(101mhz,cdcl3)δ189.84,160.81,138.07,133.87,132.71,131.18,130.04,126.81,59.74.
4)3-甲氧基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑(f)的合成:
取50ml茄形瓶,加入e(432mg,1.6mmol),对硝基苯肼(250mg,1.6mmol),加入5ml甲醇,固体溶解后放入磁力搅拌子,将反应瓶放在磁力搅拌反应装置中,设置温度为70℃,接上冷凝水回流装置,搅拌反应18小时。经tlc检测已基本无原料e即可停止反应。将反应液倒入60ml分液漏斗中,加入20ml乙酸乙酯和20ml去离子水,震荡萃取,分出水相后,有机相用饱和氯化钠溶液再萃取一次。有机相装于50ml锥形瓶中,加入适量无水硫酸钠,放入磁力搅拌子,进行干燥。干燥后的有机相过滤到100ml茄形瓶中,旋转蒸发仪抽去溶剂。通过tlc检测,确定洗脱剂的极性比为pe:etoac=4:1,粗品经硅胶柱层析纯化得到517mg黄色固体,反应收率为89%。1hnmr(400mhz,cdcl3)δ8.28(d,j=9.0hz,2h),7.69(d,j=2.1hz,1h),7.54(d,j=9.0hz,2h),7.45(d,j=8.4hz,1h),7.23–7.19(m,1h),4.07(s,3h).13cnmr(101mhz,cdcl3)δ169.07,151.90,147.14,142.45,135.62,133.78,131.07,131.01,127.94,127.24,125.25,125.12,57.34.
5)3-羟基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑(g)的合成:
将步骤4)所制得的3-甲氧基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑(f)(109mg,0.30mmol)用5ml醋酸溶解后,加入质量分数为33%的hbr醋酸溶液(736mg,3.0mmol),反应液在100℃条件下搅拌反应6小时;冷却,将反应液加入至50ml冰水中并充分搅拌,抽滤得到104mg浅黄色固体物质即3-羟基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑(g),反应收率为99%。1hnmr(400mhz,dmso-d6)δ11.83(s,1h),8.30(d,j=8.8hz,2h),7.76(d,j=2.0hz,1h),7.68(d,j=8.4hz,1h),7.62(d,j=8.8hz,2h),7.38–7.32(m,1h).13cnmr(101mhz,dmso-d6)δ167.02,150.75,146.48,142.24,133.41,131.73,131.17,130.80,129.04,128.17,125.64,124.99.
6)3-羟基-5-(3,4-二氯苯基)-1-(4-氨基苯基)-1,2,4-三氮唑(h)的合成:
将3-羟基-5-(3,4-二氯苯基)-1-(4-硝基苯基)-1,2,4-三氮唑(g)(105mg,0.3mmol)溶于2ml甲醇溶剂中,依次向反应溶液中加入0.5ml醋酸、锌粉(196mg,3mmol)。放进磁力搅拌子,室温下搅拌过夜。经tlc检测反应液中基本没有原料g时停止反应,旋转除去甲醇。将反应后的产物用乙酸乙酯溶解,用蒸馏水和饱和食盐水萃取若干次,收集有机相,然后用无水硫酸钠进行干燥除去有机相中的水,旋转蒸发仪除去乙酸乙酯。得到的粗产物经硅胶柱层析纯化得到89mg浅黄色固体,洗脱剂极性为ch2cl2/meoh=10:1,反应收率为93%。1hnmr(400mhz,dmso-d6)δ11.30(s,1h),7.66–7.55(m,2h),7.32(d,j=8.4hz,1h),7.00(d,j=8.2hz,2h),6.60(d,j=8.2hz,2h),5.50(s,2h).13cnmr(101mhz,dmso-d6)δ166.19,149.69,149.17,132.47,131.36,130.90,129.95,128.52,128.25,126.98,125.92,113.87.
7)3-羟基-5-(3,4-二氯苯基)-1-(4-氯乙酰氨基苯基)-1,2,4-三氮唑(a)的合成:
取一个25ml茄型瓶,加入3-羟基-5-(3,4-二氯苯基)-1-(4-氨基苯基)-1,2,4-三氮唑(h)(635mg,2.0mmol),氯乙酸(260mg,3.0mmol),edci(760mg,4.0mmol),再加入2ml二氯甲烷,用超声仪使固体溶解,放入磁力搅拌子。常温反应3小时。经tlc检测无原料后,停止反应。将反应后的产物用乙酸乙酯溶解,用蒸馏水和饱和食盐水萃取若干次,收集有机相,然后用无水硫酸钠进行干燥除去有机相中的水,旋转蒸发仪除去乙酸乙酯。得到的粗产物经硅胶柱层析纯化得到725mg浅黄色固体,洗脱剂极性为ch2cl2/meoh=10:1,反应收率为93%。1hnmr(400mhz,dmso-d6)δ11.50(s,1h),10.57(s,1h),7.73–7.61(m,4h),7.36(d,j=8.7hz,2h),7.31–7.26(m,1h),4.27(s,2h).13cnmr(101mhz,dmso-d6)δ166.56,165.17,149.69,139.01,133.01,132.86,131.51,131.04,130.30,128.54,128.29,126.48,119.99,43.65.
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
- 还没有人留言评论。精彩留言会获得点赞!