N‑吡啶类苏氨酸衍生物的手性银配合物的结构及其制备方法与流程
本发明涉及过渡金属配位聚合物技术领域,具体地说涉及一类n-(4-吡啶甲基)-l-苏氨酸银(i)配合物及其制备方法,由于手性配体n-(4-吡啶甲基)-l-苏氨酸银的存在,专利所述银配合物含c、n等多个手性中心,有望在手性拆分、分子识别等领域进行应用。
技术背景
氨基酸是重要的生理活性物质,它们不仅种类繁多,而且具有灵活的配位能力。然而天然氨基酸有限的配位点及其首选与金属离子螯合配位模式,使得氨基酸本身与金属离子配位时很难形成高维度的多孔配位聚合物。基于吡啶基具有较强的配位能力,将氨基酸与吡啶基团有效的结合不仅能增加有机配体配位模式的灵活性,而且更有利于功能性配合物的合成。
氨基酸类吡啶还原schiff碱具有多个氧和氮原子,是一类重要的有机配体,其合成仅需吡啶类醛与氨基酸一步缩合还原反应,因此被广泛应用于过渡金属配合物的合成及相关应用(kuang,x.;ma,y.;zhang,c.;su,h.;zhang,j.;tang,b.chemcommun2015,51,5955;sahoo,s.c.;kundu,t.;banerjee,r.j.am.chem.soc.2011,133,17950.)。而这些配合过渡金属中心多为cu、co、ni、zn以及cd等(中国专利cn103012442a,世界专利wo2013051035a2),d10金属银的氨基酸还原schiff配合物罕见报道。
有鉴于此,本案发明人特提出了一类具有手性的n-(4-吡啶甲基)-l-苏氨酸银(i)配合物的结构及其制备方法,以填补国内外相关领域之空白。
技术实现要素:
本发明的目的是公开一类具有手性的n-(4-吡啶甲基)-l-苏氨酸银(i)配合物,制作方法简单易行,产率高,可重现性好,克服了现有技术中的不足。
本发明中n-(4-吡啶甲基)-l-苏氨酸(l1)银(i)配合物的化学式分别为:{[ag3(l1)2no3](etoh)}n(1),{[ag2(l1)2](h2o)4}n(2),其中l1=n-(4-吡啶甲基)-l-苏氨酸阴离子配体,其分子式如下:
本发明涉及的n-(4-吡啶甲基)-l-苏氨酸阴离子配体:从结构上来看,氨基酸schiff碱配体具有较强的刚性,为了克服氨基酸schiff碱中c=n双键较强的刚性而使其稳定性较差的弱点,所以我们将氨基酸schiff碱配体中的c=n双键用nabh4还原为c–n单键,这样就会使schiff碱配体的结构具有较好的柔韧性,参与配位时有较大的自由度,更有可能通过分子自组装形成多核配合物。
本发明涉及的配合物结构单元分别为:配合物{[ag3(l1)2no3](etoh)}n(1),属于正交晶系,空间群为p212121,晶胞参数为:a=8.9514(19),b=15.932(4),
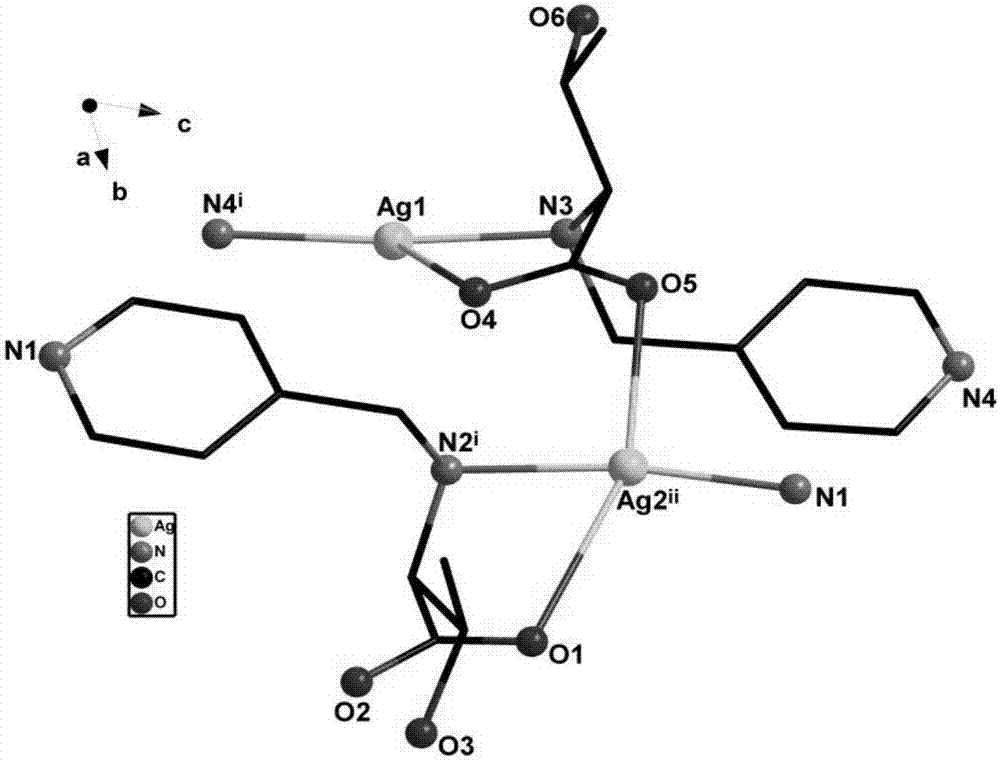
所述的两种ag(i)的配合物均具有手性,配合物1为三核单元,配合物2中含有两个晶体学独立的银,其中,ag(i)的配合物中心银离子与配体中的n,n,o配位,配合物1中还存在ag–ag作用。
n-(4-吡啶甲基)-l-苏氨酸银(i)配合物制备方法,按照下述步骤进行:
向含有n-(4-吡啶甲基)-l-苏氨酸阴离子配体的乙醇与水的混合溶液中,加入弱酸调节ph,除去溶剂得到配体hl1,然后将计量的银盐和配体hl1溶于乙醇和水的混合溶剂中,混合静置一段时间后过滤,将得到的无色澄清溶液常温挥发,3-10天后得到无色晶体,将之过滤收集,然后用乙醇洗涤,干燥,即可得到具有手性ag(i)配合物。
其中所述弱酸为甲酸、乙酸、高氯酸等的弱酸。
其中所述手性配体中引入了苏氨酸手性基团。
其中所述的乙醇和水的体积比为2:1~1:2。
其中所述的银盐和n-(4-吡啶甲基)-l-苏氨酸的摩尔比为2:1~1:2。
其中所述的银盐为硝酸银、氟化银、高氯酸银等可溶性盐。
本发明采用常温挥发法合成了两种具有手性n-(4-吡啶甲基)-l-苏氨酸银(i)配合物,所需的反应设备简单,操作简单易行,可重现性好,产率高等优点,且制备得到的配合物具有手性,有望在手性拆分、分子识别等方面得到广泛应用。
附图说明
图1是n-(4-吡啶甲基)-l-苏氨酸银(i)配合物1的配位环境图;
图2是n-(4-吡啶甲基)-l-苏氨酸银(i)配合物2的配位环境图。
具体实施方式
(1)手性配体的合成及结构
手性吡啶类苏氨酸衍生物配体的结构
实验例1配体的合成:
向含有n-(4-吡啶甲基)-l-苏氨酸阴离子配体的乙醇与水的混合溶液中,加入甲酸调节ph至5-6,将白色沉淀过滤后,再将滤液旋蒸除去溶剂得到配体hl1·甲酸。
实验例2配体的合成:
向含有n-(4-吡啶甲基)-l-苏氨酸阴离子配体的乙醇与水的混合溶液中,加入乙酸调节ph至5-6,将白色沉淀过滤后,再将滤液旋蒸除去溶剂得到配体hl1·乙酸。
实验例3配体的合成:
向含有n-(4-吡啶甲基)-l-苏氨酸阴离子配体的乙醇与水的混合溶液中,加入高氯酸调节ph至5-6,将白色沉淀过滤后,再将滤液旋蒸除去溶剂得到配体hl1·高氯酸。
(2)手性银配合物的合成与表征
实验例1配合物1的合成:
2mmolhl1·乙酸(584mg)溶于5ml水,1mmol硝酸银(169mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色棒状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物1,自然干燥,产率为65%。
实验例2配合物1的合成:
1.5mmolhl1·乙酸(438mg)溶于5ml水,1mmol硝酸银(169mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色棒状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物1,自然干燥,产率为78%。
实验例3配合物1的合成:
2mmolhl1·乙酸(584mg)溶于5ml水,1mmol硝酸银(169mg)溶于10ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色棒状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物1,自然干燥,产率为54%。
实验例4配合物1的合成:
2mmolhl1·乙酸(584mg)溶于10ml水,1mmol硝酸银(169mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色棒状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物1,自然干燥,产率为55%。
实验例5配合物1的合成:
1.5mmolhl1·乙酸(438mg)溶于5ml水,1mmol硝酸银(169mg)溶于10ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色棒状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物1,自然干燥,产率为70%。
实验例6配合物1的合成:
1.5mmolhl1·乙酸(438mg)溶于10ml水,1mmol硝酸银(169mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色棒状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物1,自然干燥,产率为68%。
实验例7配合物2的合成:
1mmolhl1·乙酸(292mg)溶于5ml水,1mmol硝酸银(169mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色针状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物2,自然干燥,产率为75%。
实验例8配合物2的合成:
1mmolhl1·乙酸(292mg)溶于5ml水,2mmol硝酸银(338mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色针状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物2,自然干燥,产率为66%。
实验例9配合物2的合成:
1mmolhl1·乙酸(292mg)溶于5ml水,1mmol硝酸银(169mg)溶于10ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色针状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物2,自然干燥,产率为55%。
实验例10配合物2的合成:
1mmolhl1·乙酸(292mg)溶于10ml水,1mmol硝酸银(169mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色针状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物2,自然干燥,产率为58%。
实验例11配合物2的合成:
1mmolhl1·乙酸(292mg)溶于5ml水,2mmol硝酸银(338mg)溶于10ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色针状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物2,自然干燥,产率为68%。
实验例12配合物2的合成:
1mmolhl1·乙酸(292mg)溶于10ml水,2mmol硝酸银(338mg)溶于5ml乙醇。两溶液混合搅拌后,静置一段时间,常压过滤,将滤液密封扎孔,几天后得到无色针状晶体,将之过滤收集,然后用乙醇洗涤,得到配合物2,自然干燥,产率为64%。
实验例13配合物的结构测定:
晶体结构测定采用brukerapexiiccd衍射仪,于296(2)k下,用经石墨单色化的mokα射线
以上实例仅用于说明本发明的内容,除此之外,本发明还有其它实施方式。但是凡采用等同替换或等效变形方式形成的技术方案均落在本发明的保护范围内。
表1配合物1–2的主要晶体学数据
ar=σ||fo|-|fc||/σ|fo|.
brw=[σ[w(fo2-fc2)2]/σw(fo2)2]1/2.
cgof(f2)={σ[w(fo2-fc2)2]/(n-p)}1/2.
- 还没有人留言评论。精彩留言会获得点赞!