一种快速建立基因敲除细胞株的重组载体及其方法与流程
本发明涉及分子生物学及遗传学前沿技术领域,具体为一种快速建立基因敲除细胞株的重组载体及其方法。
背景技术:
近年来,随着基因工程技术的发展,由talen技术、crispr/cas9技术引领的针对特定基因、特定位点的精确修饰为人类解码基因功能、探索疾病致病机制提供可靠的技术手段。特别是随着crispr/cas9系统的发现及工程化,使其成为当前基因工程领域功能最为强大的基因修饰工具。与传统的es细胞基因打靶或敲除相比,基于crispr/cas9系统的基因修饰技术,包括基因定点突变、基因敲除和外源基因定点插入等,表现出更具高效、快捷和广泛的应用前景。
crispr/cas9系统针对模式动物受精卵基因组的修饰,使其在短短几年内创造出数以千计的基因工程大鼠和小鼠。但是,在细胞基因修饰方面,由于很难针对单个细胞进行基因修饰并使其扩增建立细胞群,所以唯一可行的方案是针对细胞群进行基因修饰,然后从细胞群中筛选获得基因修饰阳性细胞并建立细胞株。由于进行处理的细胞众多,很难确定哪个细胞为阳性细胞,所以传统的做法是通过大量的单细胞分离培养,使其形成单克隆细胞群,然后进行大量的细胞基因型鉴定。这样的基因敲除细胞株的建立过程费时,费力,获得基因敲除细胞株比较困难。因此,为了满足对不同细胞、不同基因功能的研究,建立一种高效、快速的基因敲除阳性细胞的富集和筛选方法成为当前基因工程领域亟待解决的问题。
技术实现要素:
针对传统基因敲除细胞制作方法的局限,为了使基因敲除细胞株的构建更加高效、快捷,借助crispr/cas9系统,本发明提供了一种快速建立基因敲除细胞株的重组载体及其方法。
为达到上述目的,本发明采取的技术方案包括:
一种快速建立基因敲除细胞株的重组载体,包括目的基因切割载体、donor载体和donor载体的切割载体,其中:
目的基因切割载体为带有针对目的基因设计的grna的grna/cas9表达载体;
donor载体为带有针对目的基因种属相异的非同源基因序列设计的grna/cas9靶序列的egfp和抗性基因表达载体;
donor载体的切割载体为带有针对目的基因种属相异的非同源基因序列设计的grna的grna/cas9表达载体。
具体的,所述的针对目的基因种属相异的非同源基因序列设计的grna/cas9靶序列为gtgaacgtctcctaaagcgtgtgg碱基序列。
更具体的,所述的grna/cas9表达载体可以为px330载体,egfp和抗性基因表达载体可以为pirer2-egfp载体。
详细的,所述的目的基因切割载体为在grna/cas9表达载体的u6启动子下游带有针对目的基因设计的grna的载体;
所述的donor载体为在egfp和抗性基因表达载体ndel酶切位点与多克隆位点之间带有针对目的基因种属相异的非同源基因序列设计的grna/cas9靶序列的载体;
donor载体的切割载体为在grna/cas9表达载体u6启动子下游带有针对目的基因种属相异的非同源基因序列设计的grna的载体。
还有,所述的针对目的基因设计的grna为针对目的基因的第一外显子dna序列或第二外显子dna序列设计的grna。
更详细的,制备方法包括:
(1)目的基因切割载体和donor载体的切割载体的构建:分别针对目的基因和目的基因种属相异的非同源基因序列设计grna,依次命名为grna-a和grna-b,将grna-a连接到grna/cas9表达载体u6启动子下游获得目的基因切割载体,将grna-b连接到grna/cas9表达载体u6启动子下游获得donor载体的切割载体;
(2)donor载体的构建:设计grna-b引物序列得到grna-b正向引物和grna-b反向互补引物,grna-b正向引物和grna-b反向互补引物退火合成得到grna-b双链dna,然后将grna-b双链dna连接到egfp和抗性基因表达载体的ndel酶切位点与多克隆位点之间,即得donor载体。
还有,所述的grna-b的碱基序列为gtgaacgtctcctaaagcgtg。
还有,grna-b的正向引物序列为tatggtgaacgtctcctaaagcgtgtggctgca,grna-b的反向互补引物序列为gccacacgctttaggagacgttcacca。
一种快速建立基因敲除细胞株的方法,包括采用所述的快速建立基因敲除细胞株的重组载体进行细胞目的基因的敲除,再对敲除目标基因的细胞进行克隆富集即得。
该方法的步骤为:
将目的基因切割载体、donor载体和donor载体的切割载体按照质量比为1:1:1的比例与脂质体混合转染目标细胞,再采用遗传霉素g418对转染后的目标细胞进行筛选,待在荧光显微镜下观察到绿色荧光细胞时,将绿色荧光细胞继续扩增培养,进一步通过流式细胞分选富集绿色荧光细胞,将绿色荧光细胞按照有限稀释法或克隆挑取法获得基因敲除细胞株。
使用本发明进行细胞系基因敲除细胞株的构建,与传统方法相比具有以下优点:
(1)高效,本方法借用donor载体上的egfp和neo双标记,首先通过g418快速杀死无基因修饰的野生型细胞,再通过流式细胞技术筛选绿色荧光蛋白表达阳性(gfp+)的细胞,能够高效的富集相同基因型的阳性的细胞;
(2)省时,传统方法至少要用6-12个月甚至更长的时间缩短到2-3个月;
(3)便捷,构建的基因敲除3质粒系统,除了针对目的基因的grna/cas9载体外,其他两个载体均无需重新构建,能够快速进入细胞实验。
附图说明
图1为px330-grna/cas9载体结构示意图;
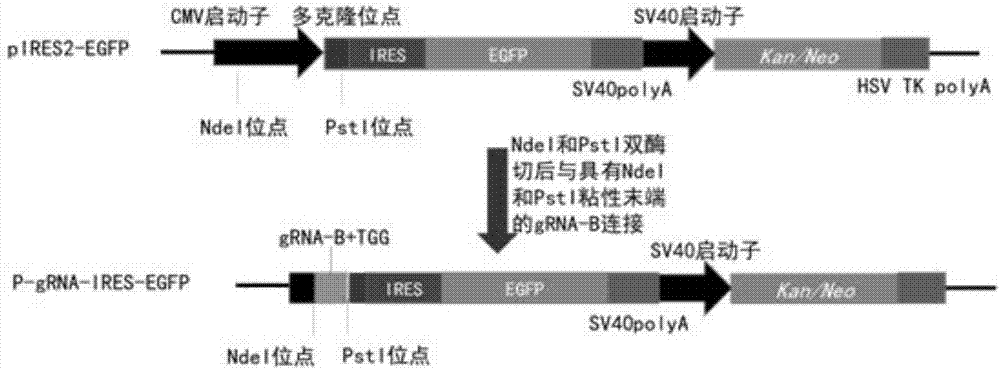
图2为donor载体构建示意图;
图3为目标细胞基因敲除原理示意图;
图4为目标细胞基因敲除细胞株建立流程;
图5为hul-7细胞chop基因敲除细胞经g418初步筛选,基因敲除阳性细胞(gfp+)聚集,荧光显微镜下可见阳性逐渐富集;
图6为hul-7细胞chop基因敲除细胞g418筛选末期流式细胞分析,基因敲除阳性细胞高达85.7%;
图7为hul-7细胞chop基因敲除细胞系经有限稀释法获得单细胞克隆;
图8为c/ebpα基因和c/ebpβ基因敲除细胞基因型鉴定;
图9为c/ebpα基因敲除细胞克隆形成;
图10为c/ebpβ基因敲除细胞克隆形成;
以下结合说明书附图和具体实施方式对本发明做具体说明。
具体实施方式
名词解释:
目的基因指的是待进行基因敲除的基因,包括实施例中的chop、c/ebpa和c/ebpb等。
目的基因种属相异的非同源基因指的是与目的基因种属来源不同,且基因同源性差异较大的基因,以人为例,即人待进行基因敲除基因的种属相异的非同源基因(比如,非同源的小鼠、大鼠基因等,本发明采用的是非同源的小鼠基因)。目的基因切割载体所带的grna的设计序列,与donor切割载体所带的grna的设计序列来自不同种属,同源性差异大,以避免donor切割载体对目的细胞基因组的dna进行切割。
grna指的是guiderna序列,即向导rna序列。
bbsi线性化的px330载体为采用bbsi酶切后的px330载体。
ndel和pstl酶切pirer2-egfp载体骨架为经ndel酶和pstl酶共同酶切后得到的pirer2-egfp载体骨架。
grna/cas9表达载体为grna序列位于u6启动子下游的cas9表达载体,例如可以包括插入grna的pcas-guide载体(origene公司)、pcas9/grna1载体(北京英茂盛业生物技术有限公司)和px330载体等,其中pcas-guide载体grna的插入位点为bamhi和bsmbi酶切位点之间,pcas9/grna1载体grna插入位点ecrorv酶切位点,px330载体grna插入位点为bbsi酶切位点。
egfp和抗性基因表达载体为同时表达egfp和neo抗性基因的载体,通过载体中cmv启动序列中的ndei酶切位点和下游多克隆酶切位点之间插入grna+tgg序列均可构建donor载体,例如可以包括pires-egfp载体(clontech)、pegfpn1载体(clontech)和pires2-egfp载体(clontech)等,其中pires-egfp载体多克隆位点可以选择noti、ecori和bamhi等酶切位点等,pegfpn1和pires2-egfp载体多克隆位点均可以选择psti酶切位点。
下面结合附图和实例对本发明做进一步说明,本文中出现的px330-grna-a/cas9即为目的基因切割载体,px330-grna-b/cas9即为donor载体切割载体,p-grna-ires-egfp即为donor载体。
(a)目的基因切割载体和donor载体的切割载体的构建,(如图1):针对目的基因的第一外显子dna序列或第二外显子dna序列和目的基因种属相异的非同源基因序列设计grna引物,分别命名:grna-a和grna-b,其正向序列为:grna-f,反向互补序列为grna-r,如若grna-f不是以碱基g开始,在grna-f的5′端加上碱基g,再于grna-f和grna-r5′端分别加上cacc和aaac碱基。合成后的grna-f和grna-r引物,分别用1×reactionpcrbuffer按100ul/od稀释,然后按以下组分混合:grna-f,4ul;grna-r,4ul;1×pcrreactionbuffer,12ul共20ul混合液于pcr仪上95℃变性5分钟后取出室温自然冷却退火。将退火合成的grna双链dna与bbsi线性化的px330载体连接分别获得px330-grna-a/cas9和px330-grna-b/cas9载体。grna-b指的是针对目标基因种属相异的非同源基因序列设计的grna,具体的基因序列是gtgaacgtctcctaaagcgtg。
(b)donor载体构建(图2为p-grna-ires-egfp载体构建过程):采用ndel和pstl酶切pirer2-egfp载体,使其上游的cmv启动子元件缺失,具体反应条件如下:pirer2-egfp质粒2ug(4ul),ndel内切酶0.5ul,pstl内切酶0.5ul,neb10×cutsmartbuffer2ul,ddh2o13ul共20ul体系,37℃孵育3小时。酶切反应液琼脂糖凝胶电泳后,割胶回收pirer2-egfp片段,同时合成双链的grna靶序列(即:grna-b+tgg碱基)并在其两端加入ndel和pstl酶切位点,同(a)中双链dna片段退火过程,然后将其连接到pirer2-egfp载体ires元件上游,连接反应条件如下:pirer2-egfp片段0.5ul,grnadna片段1ul,nebquickligase1ul,2×quickligasebuffer10ul,ddh2o7.5ul共20ul体系,室温孵育5分钟。连接产物转化感受态大肠杆菌dh5α,均匀涂布于amp+lb固体平板,过夜37℃培养,挑取阳性克隆,扩增培养,提取质粒获得p-grna-ires-egfp载体。将p-grna-ires-egfp载体转染目标细胞,以pirer2-egfp载体为对照,在荧光显微镜下观察没有荧光细胞出现,即:egfp没有表达。
(c)目标细胞基因敲除细胞株的建立(图3,图4):将生长状态良好的目标细胞以105个细胞接种于6孔板中,待其汇合度达到70-80%时,将px330-grna-a/cas9、px330-grna-b/cas9和p-grna-ires-egfp载体按照1:1:1的比例与lipofectamine2000脂质体混合转染目标细胞(转染步骤按照lipofectamine2000说明书进行),转染48小时之后,将转染细胞连续传代培养(3-5代),采用g418药物筛选1-2周(不同细胞系采用不同筛选浓度),待在荧光显微镜下观察到绿色荧光细胞聚集时,将其继续扩增培养,收集所有细胞做流式细胞分选,进一步富集绿色荧光细胞。将绿色荧光细胞按照有限稀释法或克隆挑取法获得基因敲除细胞株,获得的基因敲除细胞株提取基因组dna,进行pcr基因鉴定,如图2引物primer-f位于基因组上,primer-r位于插入片段上游ires元件上。
px330-grna-a/cas9载体和px330-grna-b/cas9载体的作用:(1)px330-grna-a/cas9靶向目的基因上游外显子序列,使其在靶点处发生dna双链断裂;(2)px330-grna-b/cas9靶向p-grna-ires-egfp载体的grna-b+tgg序列,使其产生dna双链断裂。
p-grna-ires-egfp载体的获得,采用ndel和pstl酶切pirer2-egfp载体,用24bp的grna-b+tgg序列替换cmv序列构建p-grna-ires-egfp载体,其作用是经cas9酶切割后插入到目的基因上游外显子序列中,在目的基因启动子控制下转录egfp基因而终止目的基因下游外显子的转录。
以下结合具体的实施例对本发明的方案做具体说明:
下述实施中的材料来源:grna引物和pcr引物均由华大基因科技有限公司合成;限制性内切酶(bbsi,psti和ndei)和quickligase购于neb公司;lipofectamine2000购于invitrogen公司;10×pcrreactionbuffer购于takara公司;载体px330美国杜克大学免疫学实验室惠赠;载体pirer2-egfp购于clontech公司(catalog#6029-1)。
采用人肝癌huh-7细胞的3个不同的基因(chop基因、c/ebpa基因和c/ebpb基因)验证了这一方法的可靠性,目的基因种属相异的非同源基因序列的设计采用的是小鼠的基因,具体实施例如下:
实施例1:
chop基因,也被称为dna损伤诱导转录基因(dnadamage-inducibletranscript3,ddit3),编码168-169个氨基酸,相对分子质量为29kda,为c/ebp(ccaat/enhancerbindingproteins,c/ebps)家族成员,对其他c/ebp转录因子具有显性抑制作用。近年来,研究发现chop分子与细胞的凋亡、炎症反应以及细胞的癌变有关,使其成为癌症研究重要分子之一。
(1)载体系统的构建:
在实验设计中,针对chop基因的第二外显子,设计grna序列(具体序列为ggctggaaagcagcgcatga),命名为grna-a,以小鼠基因非同源序列设计grna(具体序列为:gtgaacgtctcctaaagcgtg)命名为grna-b。分别合成grna-a和grna-b的正向引物(grna-f)和反向互补引物(grna-r)grna-a-f:caccggctggaaagcagcgcatga/grna-a-r:aaactcatgcgctgctttccagcc;grna-b-f:caccgtgaacgtctcctaaagcgtg/grna-b-r:aaaccacgctttaggagacgttcac,用1×reactionpcrbuffer按100ul/od稀释,然后按以下组分混合:grna-f,4ul;grna-r,4ul;1×pcrreactionbuffer,12ul共20ul混合液于pcr仪上95℃变性5分钟后取出室温自然冷却退火。将退火合成的grna双链dna与bbsi线性化的px330载体连接分别构建px330-grna-a/cas9和px330-grna-b/cas9载体。
采用ndel和pstl酶切pirer2-egfp载体,反应条件如下:pirer2-egfp质粒2ug(4ul),ndel内切酶0.5ul,pstl内切酶0.5ul,neb10×cutsmartbuffer2ul,ddh2o13ul共20ul体系,37℃孵育3小时。酶切反应液琼脂糖凝胶电泳后,割胶回收pirer2-egfp片段。合成grna-b引物序列,并在其两端加入ndel和pstl酶切位点后grna-b的正向序列为tatggtgaacgtctcctaaagcgtgtggctgca,grna-b的反向互补序列为gccacacgctttaggagacgttcacca,如上正向引物与反向互补引物退火条件获得grna-b双链dna序列,然后将grna-b与ndel和pstl酶切后的pirer2-egfp骨架载体连接得到p-grna-ires-egfp载体。
(2)细胞株的构建:
px330-grna-a/cas9载体、px330-grna-b/cas9载体和p-grna-ires-egfp载体按照质量比为1:1:1的比例共转染huh-7细胞,转染细胞48小时后,连续传代培养3代,至细胞状态良好,设置野生型huh-7细胞对照,采用1mg/ml的g418药物筛选7天,野生型细胞已全部凋亡,在荧光显微镜下观察实验组细胞(图5)可见,绿色荧光的阳性细胞停止死亡并开始形成细胞克隆。将药物筛选末期的细胞收集通过流式细胞分选,结果如图(图6)可见,绿色荧光阳性细胞比例已经达到85.7%。有限稀释法获得的阳性克隆细胞如图(7)可见,绿色荧光阳性细胞克隆形成,该克隆细胞扩大培养就可以获得chop基因敲除的huh-7细胞株。
实施例2:
c/ebpα基因转录因子c/ebp家族的重要成员,属于bzip(基本区域亮氨酸拉链)类的转录因子。c/ebpα在正常组织的发育、细胞增殖与分化的调节、磷脂的代谢与合成中发挥重要作用。最近有研究表明,在一些类型的肿瘤中c/ebpα作为抑癌基因而导致细胞分化紊乱和细胞周期停滞。
(1)载体系统的构建:
本实验中,针对c/ebpα基因外显子设计grna序列,序列为:cgccccgacgcgctcgtaca,命名为grna-a,以小鼠基因组非同源序列设计grna,序列为:gtgaacgtctcctaaagcgtg,命名为grna-b,分别合成grna-a和grna-b的正向引物(grna-f)和反向互补引物(grna-r),grna-a-f:cacccgccccgacgcgctcgtaca/grna-a-r:aaactgtacgagcgcgtcggggcg;grna-b-f:caccgtgaacgtctcctaaagcgtg/grna-b-r:aaaccacgctttaggagacgttcac,用1×reactionpcrbuffer按100ul/od稀释,然后按以下组分混合:grna-f,4ul;grna-r,4ul;1×pcrreactionbuffer,12ul共20ul混合液于pcr仪上95℃变性5分钟后取出室温自然冷却退火。将退火合成的grna双链dna与bbsi线性化的px330载体连接分别构建px330-grna-a/cas9和px330-grna-b/cas9载体。
采用ndel和pstl酶切pirer2-egfp载体,反应条件如下:pirer2-egfp质粒2ug(4ul),ndel内切酶0.5ul,pstl内切酶0.5ul,neb10×cutsmartbuffer2ul,ddh2o13ul共20ul体系,37℃孵育3小时。酶切反应液琼脂糖凝胶电泳后,割胶回收pirer2-egfp片段。合成grna-b引物序列,并在其两端加入ndel和pstl酶切位点后grna-b的正向序列为tatggtgaacgtctcctaaagcgtgtggctgca,grna-b的反向互补序列为gccacacgctttaggagacgttcacca,如上正向引物与反向互补引物退火条件获得grna-b双链dna序列,然后将grna-b与ndel和pstl酶切后的pirer2-egfp骨架载体连接得到p-grna-ires-egfp载体。
(2)细胞株的构建:
px330-grna-a/cas9载体、px330-grna-b/cas9载体和p-grna-ires-egfp载体按照质量比为1:1:1的比例共转染huh-7细胞,转染细胞48小时后,连续传代培养3代,至细胞状态良好,设置野生型huh-7细胞对照,采用1mg/ml的g418药物筛选7天,将药物筛选末期的细胞收集通过pcr检测进行基因型鉴定,结果如图(图8)。有限稀释法获得c/ebpα基因敲除的阳性细胞克隆形成(图9)。该克隆细胞扩大培养就可以获得c/ebpα基因敲除细胞株。
实施例3:
c/ebpβ基因(ccaat增强子结合蛋白基因)均为c/ebps家族的重要成员,其c端具有高度保守的dna结合域和二聚化功能域。它们没有内含子,只有一个外显子,其主要功能为通过靶细胞基因转录调节,参与细胞增殖与分化、肿瘤发生与凋亡、细胞周期调控等重要生命活动。
(1)载体系统的构建:
实验中,针对c/ebpβ基因的唯一的外显子设计grna序列(具体序列为:gaggtaagcgcgcagcgcgta),命名为grna,以小鼠基因组非同源序列设计grna(具体序列为:gtgaacgtctcctaaagcgtg)命名为grna-b,分别合成grna-a和grna-b的正向引物(grna-f)和反向互补引物(grna-r),grna-a-f:caccgaggtaagcgcgcagcgcgta/grna-a-r:aaactacgcgctgcgcgcttacctc;grna-b-f:caccgtgaacgtctcctaaagcgtg/grna-b-r:aaaccacgctttaggagacgttcac,用1×reactionpcrbuffer按100ul/od稀释,然后按以下组分混合:grna-f,4ul;grna-r,4ul;1×pcrreactionbuffer,12ul共20ul混合液于pcr仪上95℃变性5分钟后取出室温自然冷却退火。将退火合成的grna双链dna与bbsi线性化的px330载体连接分别构建px330-grna-a/cas9和px330-grna-b/cas9载体。
采用ndel和pstl酶切pirer2-egfp载体,反应条件如下:pirer2-egfp质粒2ug(4ul),ndel内切酶0.5ul,pstl内切酶0.5ul,neb10×cutsmartbuffer2ul,ddh2o13ul共20ul体系,37℃孵育3小时。酶切反应液琼脂糖凝胶电泳后,割胶回收pirer2-egfp片段。合成grna-b引物序列,并在其两端加入ndel和pstl酶切位点后grna-b的正向序列为tatggtgaacgtctcctaaagcgtgtggctgca,grna-b的反向互补序列为gccacacgctttaggagacgttcacca,如上正向引物与反向互补引物退火条件获得grna-b双链dna序列,然后将grna-b与ndel和pstl酶切后的pirer2-egfp骨架载体连接得到p-grna-ires-egfp载体。
(2)细胞株的构建:
px330-grna-a/cas9载体、px330-grna-b/cas9载体和p-grna-ires-egfp载体按照质量比为1:1:1的比例共转染huh-7细胞,转染细胞48小时后,连续传代培养3代,至细胞状态良好,设置野生型huh-7细胞对照,采用1mg/ml的g418药物筛选7周天,将药物筛选末期的细胞收集通过pcr检测进行基因型鉴定,结果如图(图8)。有限稀释法获得c/ebpβ基因敲除的阳性细胞克隆形成(图10)。该克隆细胞扩大培养就可以获得c/ebpβ基因敲除细胞株。
核苷酸序列表电子文件
<110>中国人民解放军第四军医大学
<120>一种快速建立基因敲除细胞株的重组载体及其方法
<210>1
<211>24
<212>rna
<213>人工序列
<220>实施例1中的grna-a
<400>1
ggctggaaagcagcgcatga
<210>2
<211>24
<212>rna
<213>人工序列
<220>实施例1中的grna-a的正向引物序列,即grna-a-f
<400>2
caccggctggaaagcagcgcatga
<210>3
<211>24
<212>rna
<213>人工序列
<220>实施例1中的grna-a的反向引物序列,即grna-a-r
<400>3
aaactcatgcgctgctttccagcc
<210>4
<211>20
<212>rna
<213>人工序列
<220>实施例2中的grna-a
<400>4
cgccccgacgcgctcgtaca
<210>5
<211>24
<212>rna
<213>人工序列
<220>实施例2中的grna-a的正向引物序列,即grna-a-f
<400>5
cacccgccccgacgcgctcgtaca
<210>6
<211>24
<212>rna
<213>人工序列
<220>实施例2中的grna-a的反向引物序列,即grna-a-r
<400>6
aaactgtacgagcgcgtcggggcg
<210>7
<211>21
<212>rna
<213>人工序列
<220>实施例3中的grna-a
<400>7
gaggtaagcgcgcagcgcgta
<210>8
<211>25
<212>rna
<213>人工序列
<220>实施例3中的grna-a的正向引物序列,即grna-a-f
<400>8
caccgaggtaagcgcgcagcgcgta
<210>9
<211>25
<212>rna
<213>人工序列
<220>实施例3中的grna-a的反向引物序列,即grna-a-r
<400>9
aaactacgcgctgcgcgcttacctc
<210>10
<211>21
<212>rna
<213>小鼠
<220>小鼠基因非同源序列设计grna,即grna-b
<400>10
gtgaacgtctcctaaagcgtg
<210>11
<211>25
<212>rna
<213>人工序列
<220>grna-b的正向引物序列,即grna-b-f
<400>11
caccgtgaacgtctcctaaagcgtg
<210>12
<211>25
<212>rna
<213>人工序列
<220>grna-b的反向引物序列,即grna-b-r
<400>12
aaaccacgctttaggagacgttcac
- 还没有人留言评论。精彩留言会获得点赞!