硒二唑类药物分子印迹聚合物的制备方法及其产品与应用与流程
本发明属于分子印迹聚合物技术领域,具体涉及一种硒二唑类药物分子印迹聚合物的制备方法,还涉及由该制备方法制得的一种硒二唑类药物分子印迹聚合物,以及该硒二唑类药物分子印迹聚合物在制备药物载体中的应用。
背景技术:
纳米药物载体主要利用纳米颗粒的小尺寸效应和高比表面效应改善药物的吸收并控制药物的释放,同时利用载体的屏蔽作用实现对药物的保护,因此,纳米药物载体与传统药物剂型相比,具有明显的优势,主要表现在以下各方面:(1)纳米药物载体的尺寸小,容易进入细胞内而实现高疗效;(2)纳米药物载体的比表面积大,黏附性强,有利于局部用药时滞留性的增加,从而延长药物与消化道壁的接触时间并增大接触面积,最终提高药物口服吸收的生物利用率;(3)水溶性差的药物在纳米药物载体中的溶解度相对增强,从而避免使用常规乳化增溶剂,并因此避免了常规乳化增溶剂的毒副作用;(4)纳米药物载体一般具有多孔、中空、多层等结构特性,易于药物的缓释控制;(5)纳米药物载体可消除特殊生物屏障对药物作用的限制。
分子印迹技术(molecularimprintingtechnology,mit)是指制备对某一特定的目标分子(又称为模板分子或印迹分子)具有选择性的聚合物的技术。1894年,在解释酶作用的专一性的时候,e.fischer等人提出了“锁与钥匙”的学说,即酶和底物结合时,底物的结构和酶的活动中心的结构十分吻合,酶的这种互补形状,使酶只能与对应的化合物匹配,从而排斥了那些形状、大小不适合的化合物。基于酶与底物分子的专一性结合特性,在1949年,dickey等人首次提出了“分子印迹”的概念;dickey以甲基橙作为模板分子,以酸化的硅酸盐作为单体,引发聚合之后,用甲醇作为洗脱液洗脱甲基橙,制得了甲基橙分子的印迹硅胶。这种材料和空白的硅胶相比,对于甲基橙的吸附作用显著增大。1972年,德国的wulff研究小组首次报道了人工合成的分子印迹聚合物后,这项技术才逐渐被人们所认识。特别是在1993年,瑞典科学家mosbach等在《nature》杂志上发表了有关茶碱分子印迹聚合物的研究报告,阐述了分子印迹的“塑料抗体”和仿生免疫分析的应用,使得分子印迹技术得到全世界的瞩目,在此之后,分子印迹技术得到了更加蓬勃的发展。分子印迹技术之所以能够得到科研人员的认可并实现广泛的应用,是因为其主要具有以下优点:(1)预定性:模板分子和功能单体在聚合之前即已进行选择,所以这种预定性就决定了人们可以按照自己的目的制备不同的分子印迹聚合物(mip),以满足各种不同的需要;(2)特异性识别:mip是按照模板分子的构型聚合的,因此它的特异性识别位点和空腔能够专一性识别模板分子;(3)实用性:mip的稳定性好,不易受高温、酸碱的影响;与天然生物分子如酶和底物、抗原和抗体、受体和激素相比,mip能够抵御比较恶劣的环境影响,而且贮存条件温和,能够长时间保留识别特性。
值得注意的是,制备得到纳米级的分子印迹聚合物具有模板分子的“印迹腔”,当分子印迹聚合物应用到有模板分子存在的体系中后,该分子印迹聚合物就能选择性吸附模板分子,从而将模板分子(目标分子)与其它物质分离。
在现有技术中,制备分子印迹聚合物(mip)时至少需要使用以下反应试剂:模板分子(template);功能单体(functionalmonomer);致孔剂(溶剂);交联剂(crosslinking);引发剂(initiator),各反应试剂的功能如下表1所示。
表1各反应试剂的功能
此外,参见图1,现有技术中的分子印迹聚合物的制备工艺主要包括:首先,模板分子与功能单体在致孔剂的作用下通过共价键或非共价键的方式相互作用,形成预聚复合物;然后,在交联剂和引发剂的作用下,使得在模板-功能单体复合物周围进行聚合,直至聚合结束之后,形成一种高分子聚合物;最后,洗脱除去模板分子,如此,在该高分子聚合物中留下了一个与模板分子在空间结构上完全匹配的且含有对模板分子的特异性识别位点的三维空穴。由于该三维空穴对模板分子的记忆特性,mip对模板分子的亲和力大大增强,最终表现出分子识别的能力。
另外,分子印迹聚合物(mip)也被用于制备药物载体,具体地,主要应用于药物缓释和靶向肿瘤这两个方面。在药物缓释方面,通常以药物分子为模板分子,模板分子和功能单体之间通过氢键,疏水作用力等非共价键连接,形成分子印迹聚合物,其利用药物分子和功能单体之间的非共价键作用来达到药物缓释/控释的作用。例如,有文献报道用壳聚糖和甲基丙烯酸甲酯形成的接枝共聚物,能够以5-氟脲嘧啶为模板分子,所生成的共聚物可印迹5-氟脲嘧啶;此外,有研究表明,体外的药物释放实验中,该聚合物的药物释放与ph和时间是相关的。进一步地,分子印迹聚合物可在靶向肿瘤中得到应用;例如,选择在肿瘤表面高表达的短肽作为模板分子,制备出来的分子印迹聚合物能够特异性识别模板分子,由于模板分子是在肿瘤表面高表达的短肽,所以制备的分子印迹聚合物对在肿瘤表面高表达的短肽具有高的亲和力,最终实现对肿瘤组织的靶向性;例如有文献报道,以在肿瘤表面高表达的p32的蛋白,并且对其进行修饰,从而获得具有一定空间结构的短肽作为模板分子;又如,对具有一定空间构象的抗原决定簇进行印迹,所得到的印迹聚合物在动物体内实验中表现出对肿瘤组织的显著靶向性。
技术实现要素:
本发明旨在提供一种硒二唑类药物分子印迹聚合物,并提供一种粒径达纳米级的硒二唑类药物载体,从而获得硒二唑类药物控释制剂,显著提升药物的分散性与稳定性,有利于提升药效。
具体地,本发明的第一方面提供了一种硒二唑类药物分子印迹聚合物的制备方法,包括以下步骤:
s1:准确称取甲基丙烯酸、n-异丙基丙烯酰胺、n,n-二甲基双丙烯酰胺,超声溶于水中;然后加入n-叔丁基丙烯酰胺的无水乙醇溶液,超声脱气;
s2:调节ph=5.6~6.5后,加入作为模板分子的硒二唑类药物;
s3:缓慢地滴加引发剂的水溶液,在32~38℃下聚合反应22~25h;
s4:反应完全后,以乙醇-氯仿为萃取剂,采用索氏提取器进行回流过夜,以洗脱模板分子,制得硒二唑类药物分子印迹聚合物粗品;
s5:将所述硒二唑类药物分子印迹聚合物粗品进行透析以去除未聚合的引发剂或/和甲基丙烯酸或/和n-异丙基丙烯酰胺或/和n-叔丁基丙烯酰胺,即得硒二唑类药物分子印迹聚合物。
优选地,在上述制备方法的s1中,所述甲基丙烯酸与所述n-异丙基丙烯酰胺的摩尔比为1:2,所述甲基丙烯酸与所述n,n-二甲基双丙烯酰胺的摩尔比为1:10,所述甲基丙烯酸与所述n-叔丁基丙烯酰胺的摩尔比为1:1。
优选地,在上述制备方法的s1中,所述超声脱气的持续时间为5~15min。
优选地,在上述制备方法中,所述硒二唑类药物为[1,2,5]硒二唑[3,4-f][1,10]菲咯啉。
优选地,在上述制备方法中,所述甲基丙烯酸与所述硒二唑类药物的摩尔比为1:2~1:16。
优选地,在上述制备方法中,所述引发剂包括过硫酸铵,亚硫酸氢钠和十二烷基硫酸钠。
进一步优选地,在上述制备方法中,所述过硫酸铵的质量为所述硒二唑类药物的质量的6倍,所述亚硫酸氢钠的质量为所述硒二唑类药物的质量的2倍,所述十二烷基硫酸钠的质量为所述硒二唑类药物的质量的4倍。
本发明的第二方面提供了一种硒二唑类药物分子印迹聚合物,其由本发明第一方面所述的制备方法制得。
此外,本发明的第三方面提供了第二方面所述的硒二唑类药物分子印迹聚合物在制备药物载体中的应用,其包括以下步骤:
p1:准确称取甲基丙烯酸、n-异丙基丙烯酰胺、n,n-二甲基双丙烯酰胺,超声溶于水中;然后加入n-叔丁基丙烯酰胺的无水乙醇溶液,超声脱气;
p2:调节ph=5.6~6.5后,加入作为模板分子的硒二唑类药物;
p3:缓慢地滴加引发剂的水溶液,在32~38℃下聚合反应22~25h;
p4:反应完全后,将反应液进行透析以去除未聚合的引发剂或/和甲基丙烯酸或/和n-异丙基丙烯酰胺或/和n-叔丁基丙烯酰胺,即得硒二唑类药物载体。
优选地,在上述应用中,在p1中,所述甲基丙烯酸与所述n-异丙基丙烯酰胺的摩尔比为1:2,所述甲基丙烯酸与所述n,n-二甲基双丙烯酰胺的摩尔比为1:10,所述甲基丙烯酸与所述n-叔丁基丙烯酰胺的摩尔比为1:1。
优选地,在上述应用中,在p1中,所述超声脱气的持续时间为5~15min。
优选地,在上述应用中,所述硒二唑类药物为[1,2,5]硒二唑[3,4-f][1,10]菲咯啉。其中,有报道通过mtt方法证明了[1,2,5]硒二唑[3,4-f][1,10]菲咯啉对各种肿瘤细胞具有较好的抑制作用,尤其是,其对hepg2的ic50值达到了19.2μm。
优选地,在上述应用中,所述甲基丙烯酸与所述硒二唑类药物的摩尔比为1:10。
优选地,在上述应用中,所述引发剂包括过硫酸铵,亚硫酸氢钠和十二烷基硫酸钠。
优选地,在上述应用中,所述过硫酸铵的质量为所述硒二唑类药物的质量的6倍,所述亚硫酸氢钠的质量为所述硒二唑类药物的质量的2倍,所述十二烷基硫酸钠的质量为所述硒二唑类药物的质量的4倍。
另外,值得补充说明的是,在本文中,各步骤所使用的水均为蒸馏水。
与现有技术相比,本发明所提供的硒二唑类药物分子印迹聚合物适用于制备硒二唑类药物载体,主要具备以下有益效果:众所周知,肿瘤组织附近环境与人体正常细胞组织附近的环境相比,肿瘤组织附近的温度更高,通常高达40℃以上,且ph值呈酸性;发明人利用hplc检测证明了负载有硒二唑类药物的分子印迹聚合物在40℃,ph=5.4的条件下能够更加快速地释放药物,而在正常细胞组织附近的环境下,释放药物的速度明显较慢,因此,获得了控释效果与肿瘤靶向性;同时,发明人检测证明了负载有硒二唑类药物的分子印迹聚合物(即硒二唑类药物载体)的粒径≤200nm,并且借助于肿瘤epr效应(实体瘤的高通透性和滞留效应),使得该粒径下的硒二唑类药物载体更趋向于聚集在肿瘤组织附近,从而有利于特异性地杀死肿瘤细胞,而降低对正常细胞的毒性。综上所述,本发明所提供的硒二唑类药物分子印迹聚合物的制备方法与硒二唑类药物分子印迹聚合物,具有广阔的应用前景与良好的市场潜力。
附图说明
图1为分子印迹聚合物的制备原理示意图;
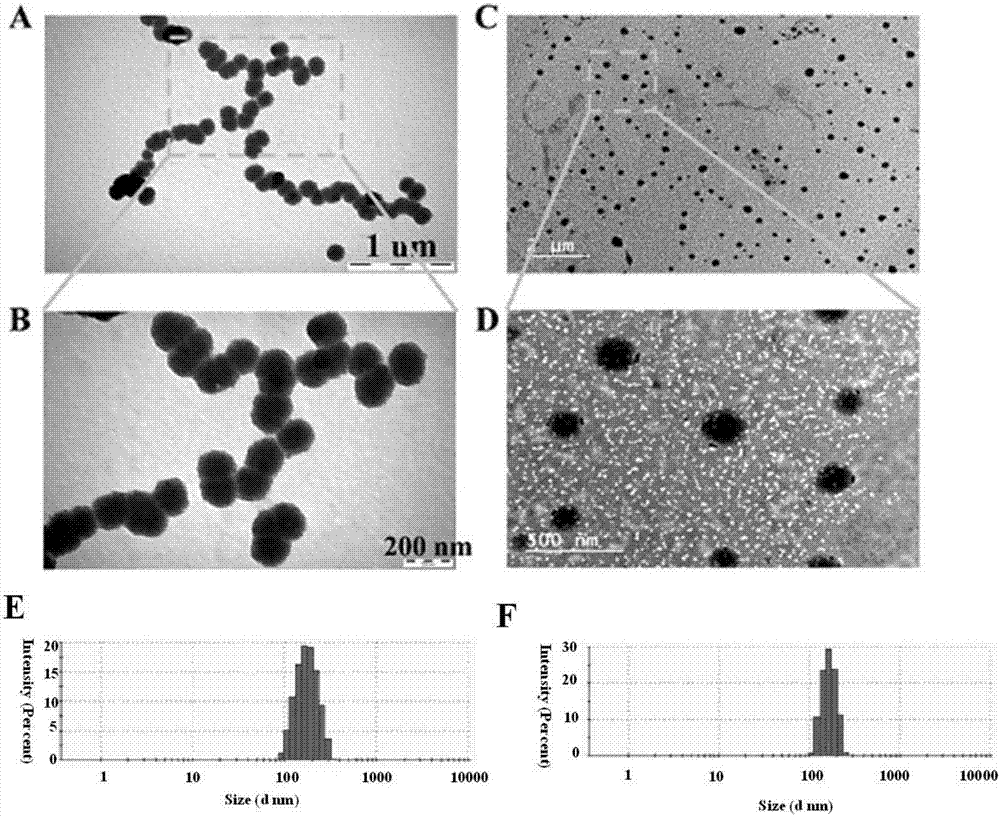
图2(a,b)为nip-3的电镜图;图2(c,d)为nip@seip的电镜图;图2(e)为nip-3的粒径分布图;图2(f)为nip@seip的粒径分布图;
图3为seip(1),nip(2),nip@seip(3)的zeta电位分布图;
图4为seip(1),nip(3),nip@seip(2)的红外光谱图;
图5为nip@seip在水溶液(1),pbs(ph=7.4)(2),含10%fbs的dmem(3)的粒径-时间关系图;
图6(a)为nip@seip在不同温度下的药物释放曲线图;图6(b)为nip@seip在不同ph下的药物释放曲线图。
具体实施方式
下面结合具体实施方式对本发明作进一步阐述,但本发明并不限于以下实施方式。
根据本发明第一方面所述的硒二唑类药物分子印迹聚合物的制备方法,包括以下步骤:
s1:准确称取甲基丙烯酸(0.2-0.3mmol)、n-异丙基丙烯酰胺、n,n-二甲基双丙烯酰胺,超声溶于80ml水中;然后加入n-叔丁基丙烯酰胺的无水乙醇(1ml)溶液,超声脱气10min;
s2:调节ph=5.6~6.5后,加入作为模板分子的硒二唑类药物;
其中,分别按以下摩尔比投加模板分子:
nip-1:(甲基丙烯酸:模板分子=1:2)
nip-2:(甲基丙烯酸:模板分子=1:4)
nip-3:(甲基丙烯酸:模板分子=1:10)
nip-4:(甲基丙烯酸:模板分子=1:16)
s3:缓慢地滴加引发剂的水溶液,在32~38℃下聚合反应22~25h;
s4:反应完全后,以乙醇-氯仿为萃取剂,采用索氏提取器进行回流过夜,以洗脱模板分子,制得硒二唑类药物分子印迹聚合物粗品;
s5:将所述硒二唑类药物分子印迹聚合物粗品进行透析以去除未聚合的引发剂或/和甲基丙烯酸或/和n-异丙基丙烯酰胺或/和n-叔丁基丙烯酰胺,即得硒二唑类药物分子印迹聚合物。
此外,用hplc的方法检测各个分子印迹聚合物对硒二唑类药物的吸附能力,确定吸附量最大的印迹聚合物,结果nip-3的吸附量最大;采用tem和afm对其形貌进行表征;用hplc的方法测定印迹聚合物对靶向分子的吸附平衡曲线。
根据本发明第一方面所述的硒二唑类药物分子印迹聚合物,其由本发明第一方面所述的制备方法制得。
根据本发明第三方面所述的硒二唑类药物分子印迹聚合物在制备药物载体中的应用,其包括以下步骤:
p1:准确称取甲基丙烯酸(0.2-0.3mmol)、n-异丙基丙烯酰胺、n,n-二甲基双丙烯酰胺,超声溶于80ml水中;然后加入n-叔丁基丙烯酰胺的无水乙醇(1ml)溶液,超声脱气10min;
p2:调节ph=5.6~6.5后,加入作为模板分子的[1,2,5]硒二唑[3,4-f][1,10]菲咯啉;
p3:缓慢地滴加引发剂的水溶液,在32~38℃下聚合反应22~25h;
p4:反应完全后,将反应液进行透析以去除未聚合的引发剂或/和甲基丙烯酸或/和n-异丙基丙烯酰胺或/和n-叔丁基丙烯酰胺,即得硒二唑类药物载体。
另外,采用tem和afm对其形貌进行表征;采用纳米粒度仪测定聚合物的稳定性和zeta电位;用hplc的方法测定负载药物的分子印迹聚合物在不同的温度和ph的条件下释放药物的情况。
实施例1:负载药物的分子印迹聚合物的制备及药物释放实验
一、制备负载药物的分子印迹聚合物(nip@seip)
1)准确称取甲基丙烯酸(27.0μl),n-异丙基丙烯酰胺(73.9mg),n,n-二甲基双丙烯酰胺(3.9mg)溶于80ml蒸馏水,n-叔丁基丙烯酰胺(39.2mg)溶于1ml无水乙醇,超声10min;
2)用naoh调节ph=6.0;
3)称取药物分子(甲基丙烯酸:[1,2,5]硒二唑[3,4-f][1,10]菲咯啉的摩尔比=1:10)80.0mg(溶于无水乙醇);
4)超声10min,脱气;
5)通入n230min,逐滴加入引发剂(过硫酸铵(60.0mg),亚硫酸氢钠(20.0mg),十二烷基硫酸钠(40.0mg)溶于20ml的蒸馏水);
6)滴加完毕,立即密封,35℃聚合24h;
7)将反应混合物用蒸馏水进行透析,除去未聚合的功能单体和药物分子。二、采用透射电镜观察制备的负载药物的分子印迹聚合物的形貌
如图2所示,负载药物的分子印迹聚合物显示为粒径小于200nm的纳米球,并且,相较于没有负载药物的nip,nip@seip的分散性更好;如图3所示,zeta电位分布的结果显示,负载有药物的分子印迹聚合物的电位更大,说明体系更加稳定,更加分散,这与透射电镜的观测结果是一致的。进一步地,参见图4,发明人分别测得了seip、nip和nip@seip的红外光谱图,从而证明了药物分子([1,2,5]硒二唑[3,4-f][1,10]菲咯啉)已经成功负载到该分子印迹聚合物之中了。
三、nip@seip稳定性测定
取三份相同质量的nip@seip分别溶于蒸馏水,pbs(ph=7.4),dmem(含10%的fbs)。分别在2,4,8,12,24,48,72,120h的时间点测定其粒径的情况。nip@seip的稳定性测定结果如图5所示,可见nip@seip在这三种溶解介质中的粒径变化都不大,因此表明nip@seip具有优异的稳定性。四、体外药物释放情况
1)在不同温度条件下药物的释放情况
分别称取两份相同质量的nip@seip溶于pbs(ph=7.4)中,分别放在温度为37和℃40℃的水浴锅中。在0,0.5,1.0,2,4,8,12,24h的时间点取2ml溶液,同时补加2ml的pbs,离心(12000rpm,30min),取上清液,用hplc测定溶液中的药物分子([1,2,5]硒二唑[3,4-f][1,10]菲咯啉)的浓度。hplc检测结果显示,在40℃的条件下,药物释放最快(如图6(a)所示)。
2)在不同ph条件下药物的释放情况
分别称取两份相同质量的nip@seip溶于不同ph值的pbs(ph=5.4,6.2,7.4)中,并且分别置于温度为37℃的2个水浴锅中。在0,0.5,1.0,2,4,8,12,24h的时间点取2ml溶液,同时补加2ml的pbs,离心(12000rpm,30min),取上清液,用hplc测定溶液中的药物分子([1,2,5]硒二唑[3,4-f][1,10]菲咯啉)的浓度。hplc检测结果显示,在随着ph值的减小,药物释放的越快,且ph=5.4时药物释放最快(如图6(b)所示)。
经过进一步分析,值得注意的是,在本发明所提供的技术方案中释药机理可能包括:由于在制备硒二唑类药物分子印迹聚合物的方法中,采用了温度敏感的材料n-异丙基丙烯酰胺,从而在温度较低时,该分子印迹聚合物可利用所形成的三维空穴装载足量的药物,而当温度升高后,三维空穴变形,同时在该分子印迹聚合物中生成若干孔隙,由此将负载的药物从孔隙中挤压释放出来;同时,在该制备方法中,还加入了ph敏感的功能单体——甲基丙烯酸,由于甲基丙烯酸含有羧酸基团,各羧酸基团与肿瘤组织之间会产生氢键、范德华力等作用力,当肿瘤组织内的介质ph值降低到一定程度(例如ph=5.4)后,该羧酸基团发生电离,使药物载体的稳定性遭到破坏,引起药物载体的体积溶胀变化,从而促进内部药物的释放。
以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!