一种2‑亚氨基噻唑烷‑4‑酮类化合物的制备方法与流程
本发明属于有机中间体合成领域,具体涉及一种2-亚氨基噻唑烷-4-酮类化合物的制备方法。
背景技术:
噻唑烷广泛存在于各种天然产物中,具有较好的生物活性,其中,2-亚氨基噻唑烷-4-酮是很多天然产物、生物活性分子及药物分子的基本结构单元。具有2-亚氨基噻唑烷-4-酮结构单元的化合物具有多种生理活性,如抗癌活性和抗惊厥活性,可用于制备受体抑制剂,以抑制人肝癌细胞生长和抑制人结肠癌细胞增殖等,基于此,2-亚氨基噻唑烷-4-酮类化合物引起人们的广泛关注。
近年来,国内外研究人员已经开发出一些构建2-亚氨基噻唑烷-4-酮类化合物的有效方法,如卢君等以含不同取代基的伯胺化合物为底物,经两步反应,得到含不同取代氨基的硫脲类化合物或缩氨基硫脲类化合物中间体,再经环合反应得到2-亚胺基-4-噻唑烷酮类化合物(参见卢君,《2-亚胺基-4-噻唑烷酮及4-羟甲基-1,3-噁唑啉衍生物的合成研究》,2007年4月),具体反应路线如下:
上述反应路线中,第一步,伯胺、二硫化碳和三乙胺在低于20℃条件下反应3h,然后加入水合肼,在78℃条件下反应24h;第二步,将第一步反应产物与嘧啶醛类化合物在加热回流条件下反应;第三步,将第二步反应产物与溴乙酸乙酯在加热回流条件下反应,得到2-亚氨基噻唑烷-4-酮类化合物。上述路线虽然能成功制备得到2-亚氨基噻唑烷-4-酮类化合物,但反应过程中的三个步骤均需要在在加热条件下完成,反应条件苛刻,不易控制。
技术实现要素:
本发明的目的在于提供一种2-亚氨基噻唑烷-4-酮类化合物的制备方法,本发明提供的制备方法无需加热,在有氧气氛下,仅利用光照即可制备得到2-亚氨基噻唑烷-4-酮类化合物,反应条件温和,易于控制。
为实现以上目的,本发明提供了一种2-亚氨基噻唑烷-4-酮类化合物的制备方法,包括:
将胺类化合物、异硫氰酸酯类化合物、α-卤代羧酸酯和有机溶剂混合,得到原料混合液;
调节所述原料混合液的ph至碱性,得到碱性反应液;
所述碱性反应液在含氧气氛中,进行光照,在常温条件下发生光串联环化反应,得到2-亚氨基噻唑烷-4酮类化合物。
优选地,所述胺类化合物的结构简式为r1-ch2-nh2,所述r1包括芳基、烷基或杂环基;
所述异硫氰酸之类化合物的结构简式为r2n=c=s,所述r2包括芳基或烷基;
所述α-卤代羧酸酯为α-溴代羧酸酯或α-氯代羧酸酯。
优选地,所述胺类化合物、异硫氰酸酯类化合物与α-卤代羧酸酯的物质的量的比为(1~6):1:(2.8~3.2)。
优选地,所述有机溶剂的体积与所述胺类化合物的物质的量的比为1l:0.1~0.16mol。
优选地,所述有机溶剂为极性有机溶剂。
优选地,所述碱性反应液的ph为8~11。
优选地,所述含氧气氛中,o2与胺类化合物的物质的量之比为1~1.5:1。
优选地,所述光照的光源波长为200~1000nm。
优选地,所述光串联环化反应的时间为6~24h。
优选地,所述光串联环化反应的时间为8~20h。
本发明提供的2-亚氨基噻唑烷-4-酮类化合物的制备方法将胺类化合物、异硫氰酸酯类化合物、α-卤代羧酸酯和有机溶剂混合后,再将原料混合液调节为碱性,然后在有氧条件下,进行光照,在常温条件下发生光串联环化反应,得到2-亚氨基噻唑烷-4酮类化合物。在本发明中,胺类化合物亲核进攻异硫氰酸酯,形成活性中间体硫脲,硫脲异构化为硫脲类化合物;在光照条件下,硫脲类化合物氧化生成中间体二硫化合物,二硫化合物通过光促进的均裂反应生成巯基自由基。与此同时,中间体硫脲与α-卤代羧酸酯偶联,在溶液中形成氢键配合物。然后氢键配合物解离生成亚甲基自由基。随后,巯基自由基与中间体亚甲基自由基偶合得到中间体n,n-二取代巯基乙酸乙酯。最后,中间体n,n-二取代巯基乙酸乙酯在碱性条件下氨解环化得到目标产物。本发明提供的制备方法利用光照,为原料提供所需能量,在有氧参与的条件下,即可得到目标产物,反应无需加热,反应条件温和,易于控制。
附图说明
下面结合附图和具体实施方式对本发明作进一步详细的说明。
图1为本发明实施例1所得产品的核磁共振氢谱图;
图2为本发明实施例1所得产品的核磁共振碳谱图;
图3为本发明实施例2所得产品的核磁共振氢谱图;
图4为本发明实施例2所得产品的核磁共振碳谱图;
图5为本发明实施例3所得产品的核磁共振氢谱图;
图6为本发明实施例3所得产品的核磁共振碳谱图;
图7为本发明实施例4所得产品的核磁共振氢谱图;
图8为本发明实施例4所得产品的核磁共振碳谱图;
图9为本发明实施例5所得产品的核磁共振氢谱图;
图10为本发明实施例5所得产品的核磁共振碳谱图;
图11为本发明实施例6所得产品的核磁共振氢谱图;
图12为本发明实施例6所得产品的核磁共振碳谱图;
图13为本发明实施例7所得产品的核磁共振氢谱图;
图14为本发明实施例7所得产品的核磁共振碳谱图;
图15为本发明实施例8所得产品的核磁共振氢谱图;
图16为本发明实施例8所得产品的核磁共振碳谱图;
图17为本发明实施例9所得产品的核磁共振氢谱图;
图18为本发明实施例9所得产品的核磁共振碳谱图;
图19为本发明实施例10所得产品的核磁共振氢谱图;
图20为本发明实施例10所得产品的核磁共振碳谱图;
图21为本发明实施例11所得产品的核磁共振氢谱图;
图22为本发明实施例11所得产品的核磁共振碳谱图;
图23为本发明实施例12所得产品的核磁共振氢谱图;
图24为本发明实施例12所得产品的核磁共振碳谱图;
图25为本发明实施例13所得产品的核磁共振氢谱图;
图26为本发明实施例13所得产品的核磁共振碳谱图;
图27为本发明实施例14所得产品的核磁共振氢谱图;
图28为本发明实施例14所得产品的核磁共振碳谱图。
具体实施方式
本发明提供了一种2-亚氨基噻唑烷-4-酮类化合物的制备方法,包括:
将胺类化合物、异硫氰酸酯类化合物、α-卤代羧酸酯和有机溶剂混合,得到原料混合液;
调节所述原料混合液的ph至碱性,得到碱性反应液;
所述碱性反应液在有氧条件下,进行光照,在常温条件下发生光串联环化反应,得到2-亚氨基噻唑烷-4酮类化合物。
本发明将胺类化合物、异硫氰酸酯类化合物、α-卤代羧酸酯和有机溶剂混合,得到原料混合液。
在本发明中,所述胺类化合物的结构简式为r1-ch2-nh2,所述r1优选为芳基、烷基或杂环基;所述芳基优选为苯基、对腈基取代的苯基、烷基取代的苯基、烷氧基取代的苯基、卤素取代的苯基或萘基。所述烷基取代的苯基优选为1~5个碳原子的烷基取代的苯基,进一步优选为对甲基苯基或间甲基苯基;所述烷氧基取代的苯基优选为甲氧基取代的苯基,进一步优选为对甲氧基苯基、间甲氧基苯基、邻甲氧基苯基或3,4,5-三甲氧基苯基;所述烷基优选为正戊基、环己基或苯乙基;所述卤素取代的苯基优选为f取代的苯基、cl取代的苯基或br取代的苯基,进一步优选为间氟苯基、邻氟苯基、对氟苯基、对氯苯基、间氯苯基、邻氯苯基、对溴苯基、间溴苯基、3,5-二氯苯基或3-氯-4-氟苯基;所述杂环基优选为3-吡啶基、2-噻吩基、2-呋喃基或乙基吗啉基。
在本发明中,所述胺类化合物具体为苄胺、对溴苄胺、对腈基苄胺、对甲氧基苄胺、对甲基苄胺、1-萘基甲胺、2-呋喃基甲胺或正己胺。
在本发明中,所述异硫氰酸酯类化合物的结构简式为r2-n=c=s,所述异硫氰酸酯类化合物的结构简式为r2n=c=s,所述r2优选为芳基或烷基;所述芳基优选为苯基、苄基、萘基、苯甲酰基、烷基取代的苯基、甲氧基取代的苯基、卤素取代的苯基、三氟甲基取代的苯基、对硝基取代的苯基、对腈基取代的苯基、烷基、环己基或甲酸乙酯基;所述烷基取代的苯基优选为对乙基苯基或2,4,6-三甲基苯基;所述甲氧基取代的苯基优选为对甲氧基苯基、2,4-二甲氧基苯基或3,4,5-三甲氧基苯基;所述卤素取代的苯基优选为f取代的苯基、cl取代的苯基或br取代的苯基,进一步优选为对氟苯基、对溴苯基、邻氯苯基、间氯苯基或2,4-二氟苯基;所述三氟甲基取代的苯基优选为对三氟甲基苯基、邻三氟甲基苯基或3,5-双(三氟甲基)苯基;所述烷基优选为1~5个碳原子的烷基,进一步优选为甲基或正丁基。
在本发明中,所述异硫氰酸酯类化合物具体为异硫氰酸苯酯、对氟异硫氰酸苯酯、对甲氧基异硫氰酸苯酯、对乙基异硫氰酸苯酯、2,4-二甲氧基异硫氰酸苯酯、异硫氰酸苄酯、异硫氰酰甲酸乙酯。
在本发明中,所述α-卤代羧酸酯优选为α-溴代羧酸酯或α-氯代羧酸酯。在本发明中,所述α-卤代羧酸酯类化合物的结构简式优选为x(r3)c-coor4;在所述结构简式中,r3优选为甲基、乙基或2-氯苯基,r4优选为甲基或乙基;所述x优选为cl或br。在本发明中,所述α-卤代羧酸酯具体可以为α-溴乙酸乙酯或α-氯乙酸乙酯。
在本发明中,所述有机溶剂优选为极性有机溶剂,进一步优选为二甲基亚砜、n,n-二甲基甲酰胺、甲苯、乙醇、乙腈和二氯甲烷中的一种或几种。
本发明对所述胺类化合物、异硫氰酸酯类化合物、α-卤代羧酸酯和有机溶剂的来源不做任何特殊要求,采用本领域技术人员熟知的市售产品即可。
在本发明中,所述胺类化合物、异硫氰酸酯类化合物与α-卤代羧酸酯的物质的量的比优选为(1~1.6):1:(2.8~3.2);进一步优选为(1.3~1.5):1:(2.9~3.1);更优选为1.4:1:3。
在本发明中,所述有机溶剂的体积与所述胺类化合物的物质的量的比优选为1l:0.1~0.16mol,进一步优选为1l:0.13~0.15mol,更优选为1l:0.14mol。
本发明对所述胺类化合物、异硫氰酸酯类化合物、α-溴代羧酸酯和有机溶剂的混合方式不做任何特殊限定,采用本领域技术人员熟知的混合方式即可。
得到所述原料混合液后,本发明调节原料混合液的ph至碱性,得到碱性反应液。在本发明中,所述碱性反应液的ph优选为8~11,进一步优选为9~10。在本发明中,所述调节优选为向原料反应液中添加碱性物质。在本发明中,所述碱性物质优选为无机碱性物质和/或有机碱性物质;所述无机碱性物质优选为氢氧化钠、氢氧化钾、碳酸钠、碳酸氢钠、碳酸钾和碳酸氢钾中的一种或几种;进一步优选为氢氧化钠或氢氧化钾;所述有机碱性物质优选为有机胺,进一步优选为三乙胺。在本发明中,所述碱性物质为两种或两种以上组分的混合物时,本发明对所述混合物中各组分的用量比不做任何特殊限定。本发明对所述碱性物质的添加方式不做任何特殊限定,采用本领域技术人员熟知的添加方式即可。本发明利用碱性物质将原料反应液的ph调至合适的碱性状态,可促进原料化合物的光串联环化反应的进行,为得到目标产物提供有利条件。
得到碱性反应液后,本发明将所述碱性反应液在含氧气氛中,进行光照,常温条件下发生光串联环化反应,得到2-亚氨基噻唑烷-4酮类化合物。在本发明中,所述含氧气氛中o2与胺类化合物的物质的量的比优选为1~1.5:1,进一步优选为1.2~1.4:1,更优选为1.25:1。在本发明中,所述含氧气氛优选为空气气氛或氧气气氛。在本发明中,所述含氧气氛的提供方式以本领域技术人员熟知的方式即可。在本发明中,所述光照的光源波长优选为200~1000nm,进一步优选为300~800nm。本发明对所述光源不做任何特殊限定,以能提供上述波长的光源即可。在本发明中,所述光串联环化反应的时间优选为6~24h,进一步优选为8~20h。在本发明中,所述光串联环化反应的机理如式i所示,α-卤代羧酸酯以溴乙酸乙酯为例:
式i中,胺类化合物1与异硫酸酯类化合物2混合后,胺类化合物1亲核进攻异硫氰酸酯类化合物2,形成活性中间体硫脲a,活性中间体硫脲a异构化为硫脲类化合物b。在光照条件下,b氧化生成中间体c,c通过光促进的均裂反应生成巯基自由基d。与此同时,中间体a与溴乙酸乙酯3a偶联,在溶液中形成氢键配合物e。然后氢键配合物e解离生成g和f。随后,巯基自由基d与中间体g偶合得到中间体h。最后,在碱性条件下,中间体h氨解环化得到目标产物4。
在本发明中,所述含氧气氛和碱性条件为光串联环化反应提供必要的基础条件,光照为光串联环化反应提供所得的能量,配合原料的选取,在常温条件下,无需加热即可得到2-亚氨基噻唑烷-4-酮类化合物目标产物,整个反应条件温和,易于控制。另外,本发明提供的2-亚氨基噻唑烷-4-酮类化合物的制备方法,对原料的适应性强,可用于不同取代基的2-亚氨基噻唑烷-4-酮化合物的制备。
完成光串联环化反应后,本发明优选对所述光串联环化反应后的混合物进行提纯处理。在本发明中,所述提纯处理优选为柱层析。在本发明中,所述柱层析的洗脱液优选为石油醚和乙酸乙酯的混合溶剂;在本发明中,所述混合溶剂中石油醚和乙酸乙酯的体积比优选为1~30:1,进一步优选为5~20:1,更优选为5:1。本发明对柱层析的具体实施方法不做任何特殊限定,采用本领域技术人员熟知的方式即可。本发明利用石油醚和乙酸乙酯为洗脱液,对光串联环化反应后的混合液进行提纯处理,能够得到纯度较高的2-亚氨基噻唑烷-4-酮类化合物。
在本发明中,所述2-亚氨基噻唑烷-4-酮类化合物的结构式优选如式ii所示:
所述式ii中,r1优选与制备过程中的胺类化合物中r1保持一致,r2优选与制备方法中的异硫氰酸酯类化合物中的r2保持一致,r3与制备过程中的α-卤素羧酸酯中的r3保持一致,在此不再赘述。
在本发明中,所述式ii具体的结构可以为:
在本发明中,所述2-亚氨基噻唑烷-4-酮类化合物的纯度优选为98.5~99.9%,进一步优选为99~99.9%。
为了进一步说明本发明,下面结合实施例对本发明提供的2-亚氨基噻唑烷-4-酮类化合物的制备方法进行详细描述,但不能将它们理解为对本发明保护范围的限定。
实施例1
在反应管中加入0.1mmol苄胺、0.1mmol异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,465nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率93%,纯度为99.9%。
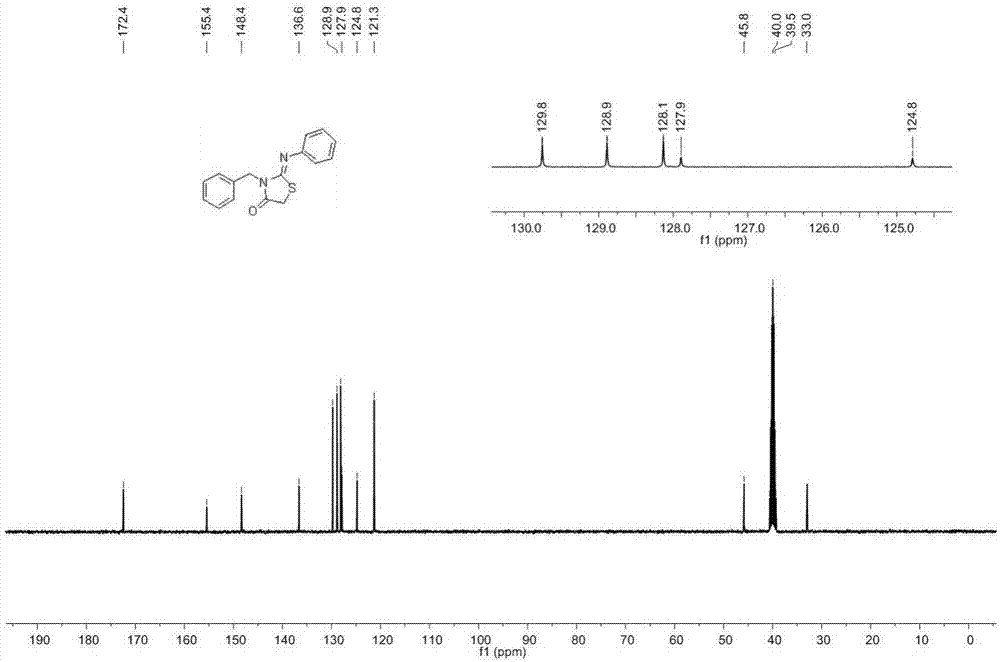
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图1和图2所示,结构表征数据如下所示:
1hnmr(400mhz,dmso):δ=7.39-7.36(m,3h),δ=7.34-7.32(d,j=8.1hz,3h),δ=7.30-7.26(m,1h),δ=7.13-7.10(m,1h),δ=6.90-6.88(t,j=8hz,2h),δ=4.92(s,2h),δ=4.10(s,2h).
13cnmr(100mhz,dmso):δ=172.4,155.5,148.4,136.6,129.8,128.9,128.1,127.9,124.8,121.3,45.8,33.1.
ms(ei,70ev):m/z(%)=282.1(m+),253.1,240.1,207.1,182.1,148.1,136.1,104.1,91.1.
根据以上数据和谱图分析可知,所得产物为目标产物,2-亚氨基噻唑烷-4-酮类化合物其结构如式(1)所示:
实施例2
在反应管中加入0.1mmol的对溴苄胺、0.1mmol异硫氰酸苯酯,0.2mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,365nm光源照射下,搅拌反应6小时,提纯方法同实施例1,产率94%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图3和图4所示,结构表征数据如下所示:
1hnmr(400mhz,dmso):δ=7.55-7.53(d,j=8.4hz,2h),δ=7.36-7.32(t,j=7.8hz,4h),δ=7.13-7.09(t,j=7.4hz,1h),δ=6.90-6.88(d,j=8.2hz,2h),δ=4.88(s,2h),δ=4.09(s,2h).
13cnmr(100mhz,dmso):δ=172.4,155.5,148.4,136.0,131.8,130.5,129.7,124.8,121.3,121.1,45.2,33.0.
ms(ei,70ev):m/z(%)=362.0(m+),320.0,286.0,260.0,227.9,207.1,169.0,136.1,118.1,104.1,90.1,77.1.
根据以上数据推断所得产物的结构如式(2)所示:
实施例3
在反应管中加入0.1mmol的对腈基苄胺、0.1mmol的对腈基异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,425nm光源照射下,搅拌反应12小时,提纯方法同实施例1,产率85%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图5和图6所示,结构表征数据如下所示:
1hnmr(400mhz,dmso):δ=7.83-7.81(d,j=8.3hz,2h),δ=7.55-7.53(d,j=8.4hz,2h),δ=7.38-7.31(m,2h),δ=7.13-7.09(m,1h),δ=6.89-6.86(t,j=1.6hz,2h),δ=5.02(s,2h),δ=4.12(s,2h).
13cnmr(100mhz,dmso):δ=172.5,155.4,148.0,142.2,132.9,129.8,128.8,124.9,121.3,119.1,110.7,45.5,33.1.
ms(ei,70ev):m/z(%)=307.1(m+),278.1,264.1,232.1,207.1,173.0,136.1,116.1,104.1,89.1,77.1.
根据以上数据推断所得产物的结构如式(3)所示:
实施例4
在反应管中加入0.1mmol的对甲氧基苄胺、0.1mmol异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钾、1ml乙腈,420nm光源照射下,搅拌反应12小时,提纯方法同实施例1,得到纯化后的目标产物,产率63%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图7和图8所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.50-7.47(m,2h),δ=7.37-7.32(m,2h),δ=7.16-7.11(m,1h),δ=6.96-6.94(m,2h),δ=6.87-6.83(m,2h),δ=4.96(s,2h),δ=3.79(s,3h),δ=3.77(m,2h).
13cnmr(100mhz,cdcl3):δ=171.6,159.3,154.0,148.0,130.8,129.2,128.3,124.6,121.0,113.8,55.3,45.7,32.7.
ms(ei,70ev):m/z(%)=312.1(m+),270.1,238.1,207.1,178.1,121.1,91.1,77.1.
根据以上数据推断所得产物的结构如式(4)所示:
实施例5
在反应管中加入0.1mmol的对甲基苄胺、0.1mmol异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,415nm光源照射下,搅拌反应12小时,提纯方法同实施例1,产率81%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图9和图10所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.43-7.41(d,j=7.9hz,2h),δ=7.34-7.30(m,2h),δ=7.13-7.12(m,3h),δ=6.95-6.93(m,2h),δ=4.97(s,2h),δ=3.73(s,2h),δ=2.32(s,3h).
13cnmr(100mhz,cdcl3):δ=171.7,154.0,148.0,137.7,133.1,129.3,129.2,129.2,124.6,121.0,46.0,32.8,21.2.
ms(ei,70ev):m/z(%)=296.1(m+),254.1,222.1,162.1,137.1,105.1,77.1.
根据以上数据推断所得产物的结构如式(5)所示:
实施例6
在反应管中加入0.1mmol的1-萘基甲胺、0.1mmol异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钾、1ml乙腈,365nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率72%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图11和图12所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=8.38-8.36(d,j=8.4hz,1h),δ=7.85-7.77(m,2h),δ=7.56-7.40(m,4h),δ=7.33-7.29(t,j=7.6hz,2h),δ=7.13-7.09(t,j=7.4hz,1h),δ=6.93-6.90(t,j=12.0hz,2h),δ=5.50(s,2h),δ=3.79(s,2h).
13cnmr(100mhz,cdcl3):δ=171.8,153.9,147.9,133.8,131.5,130.9,129.3,128.7,128.5,126.7,126.4,125.8,125.2,124.7,123.9,121.0,44.3,32.6.
ms(ei,70ev):m/z(%)=332.1(m+),290.1,257.1,215.1,154.1,141.1,115.1,77.1.
根据以上数据推断所得产物的结构如式(6)所示:
实施例7
在反应管中加入0.1mmol的2-呋喃基甲胺、0.1mmol异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,465nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率73%,纯度为99.7%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图13和图14所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.36-7.31(m,3h),δ=7.15-7.11(t,j=7.4hz,1h),δ=6.97-6.94(dd,j=8.3,1.0hz,2h),δ=6.41-6.40(d,j=3.1hz,1h),δ=6.33-6.31(dd,j=3.1,1.9hz,1h),δ=5.02(s,2h),δ=3.80(s,2h).
13cnmr(100mhz,cdcl3):δ=171.3,153.4,149.1,147.8,142.3,129.2,124.7,121.0,110.5,109.5,39.1,32.6.
ms(ei,70ev):m/z(%)=272.1(m+),243.1,230.1,172.1,138.1,81.2,53.2.
根据以上数据推断所得产物的结构如式(7)所示:
实施例8
在反应管中加入0.1mmol的正己胺、0.1mmol异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,465nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率59%,纯度为99.6%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图15和图16所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.37-7.32(m,2h),δ=7.16-7.11(m,1h),δ=6.96-6.93(m,2h),δ=3.86-3.82(t,j=16.0hz,2h),δ=3.78(s,2h),δ=1.75-1.68(m,2h),δ=1.38-1.29(m,6h),δ=0.91-0.87(t,j=16.0hz,3h).
13cnmr(100mhz,cdcl3):δ=171.8,154.3,148.2,129.2,124.5,121.0,43.3,32.7,31.4,27.2,26.5,22.5,14.0.
ms(ei,70ev):m/z(%)=276.2(m+),247.1,233.1,219.1,205.1,193.1,164,145.1,118.2,104.2,77.2.
根据以上数据推断所得产物的结构如式(8)所示:
实施例9
在反应管中加入0.1mmol苄胺、0.1mmol对氟异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,465nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率84%,纯度为99.7%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图17和图18所示。所得产物的结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.51-7.49(m,2h),δ=7.35-7.26(m,3h),δ=7.05-6.99(m,2h),δ=6.92-6.87(m,2h),δ=5.00(s,2h),δ=3.78(s,2h).
13cnmr(100mhz,cdcl3):δ=171.6,161.2,158.8,154.5,144.0(d,j=2.7hz),135.9,129.1,128.5,128.0,122.4(d,j=8.1hz),116.1-115.9(d,j=22.0hz),46.3,32.7.
ms(ei,70ev):m/z(%)=300.1(m+),271.1,258.1,225.1,200.1,148.1,122.1,104.1,91.1,65.1.
根据以上数据推断所得产物的结构如式(9)所示:
实施例10
在反应管中加入0.1mmol的苄胺、0.1mmol对甲氧基异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,465nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率66%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图19和图20所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.52-7.49(m,2h),δ=7.33-7.25(m,3h),δ=6.92-6.85(m,4h),δ=5.01(s,2h),δ=3.78(s,3h),δ=3.77(s,2h).
13cnmr(100mhz,cdcl3):δ=171.7,156.8,153.6,141.1,136.0,129.1,128.5,127.9,122.1,114.5,55.5,46.2,32.7.
ms(ei,70ev):m/z(%)=312.1(m+),270.1,238.1,223.1,207.1,167.1,148.1,121.1,105.1,91.1,65.1.
根据以上数据推断所得产物的结构如式(12)所示:
实施例11
在反应管中加入0.1mmol的苄胺、0.1mmol对乙基异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,465nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率90%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图21和图22所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.51-7.49(d,j=7.1hz,2h),δ=7.32-7.24(m,3h),δ=7.16-7.14(d,j=8.1hz,2h),δ=6.88-6.86(d,j=7.9hz,2h),δ=4.99(s,2h),δ=3.72(s,2h),δ=2.64-2.59(q,j=7.6hz,2h),δ=1.24-1.20(t,j=7.6hz,3h).
13cnmr(100mhz,cdcl3):δ=171.6,153.5,145.5,140.6,136.1,129.1,128.6,128.5,127.9,120.9,46.2,32.7,28.4,15.6.
ms(ei,70ev):m/z(%)=310.1(m+),268.1,236.2,207.1,165.1,148.1,131.1,105.1,91.1,65.1.
根据以上数据推断所得产物的结构如式(13)所示:
实施例12
在反应管中加入0.1mmol的苄胺、0.1mmol的2,4-二甲氧基异硫氰酸苯酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钾、1ml乙腈,425nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率67%,纯度为99.5%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图23和图24所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.59-7.56(dt,j=8.4,2.1hz,2h),δ=7.34-7.26(m,3h),δ=6.79-6.77(m,j=8.5hz,1h),δ=6.54-6.53(d,j=2.6hz,1h),δ=6.46-6.44(dd,j=8.5,2.6hz,1h),δ=5.05(s,2h),δ=3.79(s,3h),δ=3.78(s,3h),δ=3.75(s,2h).
13cnmr(100mhz,cdcl3):δ=171.8,157.9,155.1,151.9,136.1,130.7,129.2,128.4,127.8,121.5,104.2,100.1,55.9,55.5,46.3,32.8.
ms(ei,70ev):m/z(%)=342.1(m+),311.1,269.1,253.1,225.1,195.1,151.1,91.1,65.1.
根据以上数据推断所得产物的结构如式(12)所示:
实施例13
在反应管中加入0.1mmol的苄胺、0.1mmol的异硫氰酸苄酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,465nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率87%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图25和图26所示,表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.45-7.43(dd,j=7.4,1.7hz,2h),δ=7.37-7.31(m,2h),δ=7.30-7.26(m,6h),δ=4.95(s,2h),δ=4.51(s,2h),δ=3.83(s,2h).
13cnmr(100mhz,cdcl3):δ=171.5,152.7,139.3,136.2,129.0,128.4(d,j=3.6hz),127.8,127.4,126.9,55.3,46.1,32.7.
ms(ei,70ev):m/z(%)=296.2(m+),282.1,253.0,240.1,207.1,182.1,163.0,136.0,104.1,90.1.
根据以上数据推断所得产物的结构如式(13)所示:
实施例14
在反应管中加入0.1mmol的苄胺、0.1mmol的异硫氰酰甲酸乙酯,0.3mmol的2-溴乙酸乙酯、0.4mmol的氢氧化钠、1ml乙腈,410nm光源照射下,搅拌反应12小时,反应结束后通过柱层析分离纯化,柱层析洗脱液中石油醚和乙酸乙酯的体积比为5:1,得到纯化后的目标产物,产率57%,纯度为99.8%。
对所得产物的结构进行表征,核磁共振氢谱图和核磁共振碳谱图分别如图27和图28所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3):δ=7.45-7.42(m,2h),δ=7.34-7.25(m,3h),δ=5.01(s,2h),δ=4.30-4.25(q,j=7.1hz,2h),δ=3.76(s,2h),δ=1.38-1.35(t,j=7.1hz,3h).
13cnmr(100mhz,cdcl3):δ=173.5,172.6,162.3,135.1,128.9,128.6,128.1,62.7,46.9,32.8,14.3.
ms(ei,70ev):m/z(%)=278.1(m+),249.1,236.1,205.0,177.1,133.0,104.1,91.1,65.1.
根据以上数据推断所得产物的结构如式(14)所示:
由实施例1~14的表征结果可知,本发明提供的制备方法能够得到一系列的2-亚氨基噻唑烷-4-酮类化合物;本发明提供的制备方法利用光照,为原料提供所需能量,在有氧参与的条件下,即可得到目标产物,反应无需加热,反应条件温和,易于控制;此外,本发明提供的制备方法原料易得,降低了原料成本;而且制备所得2-亚氨基噻唑烷-4-酮类化合物具有较高的纯度,因此本发明提供的制备方法具有较大的市场推广价值。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对各种取代的胺、异硫氰酸及卤代羧酸酯等化合物做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!