一种三唑席夫碱类化合物及其制备方法与应用与流程
本发明涉及化学药物领域,尤其是一种三唑席夫碱类化合物及其制备方法与应用
背景技术:
目前已发现1,2,4-三唑类化合物具有抗菌,抗癌,抗病毒,抗炎,抗惊厥等多种活性,因而备受人们的青睐(1、elsh.ela,elshelt,mohyedaelf,etal.eur.j.med.chem.2013,66,106-113;2、
然而,随着抗生素药物滥用造成的细菌耐药性迅速增强,细菌的感染率和致病率均呈快速上升趋势。因此,研制合成新型高效广谱杀菌剂已成为生物医药领域迫在眉睫的重要课题。
技术实现要素:
本发明所要解决的技术问题在于提供一种三唑席夫碱类化合物。
本发明所要解决的另一技术问题在于提供上述三唑席夫碱类化合物的制备方法。
本发明所要解决的另一技术问题在于提供上述三唑席夫碱类化合物的应用。
为解决上述技术问题,本发明的技术方案是:
一种三唑席夫碱类化合物,为1,3,4-噁二唑修饰的1,2,4-三唑席夫碱类化合物,具有式(ⅰ)所述结构式:
其中,r1为甲基;r2为氢、氟基、氯基、溴基、甲基或甲氧基。
优选的,上述三唑席夫碱类化合物,当r2为氢时,为:5-(4-(2-羟基-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇。
优选的,上述三唑席夫碱类化合物,当r2为氟基时,为:5-(4-(2-羟基-5-氟-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇。
优选的,上述三唑席夫碱类化合物,当r2为氯基时,为:5-(4-(2-羟基-5-氯-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇。
优选的,上述三唑席夫碱类化合物,当r2为溴基时,为:5-(4-(2-羟基-5-溴-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇。
优选的,上述三唑席夫碱类化合物,当r2为甲基时,为:5-(4-(2-羟基-5-甲基-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇。
优选的,上述三唑席夫碱类化合物,当r2为甲氧基时,为:5-(4-(2-羟基-5-甲氧基-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇。
上述三唑席夫碱类化合物的制备方法,具体步骤如下:
(1)将3-甲基-4-氨基-5-乙氧羰基甲基硫基-1,2,4-三唑溶于甲醇中,后加水合肼,0-70℃下反应,用薄层色谱(tlc)跟踪反应,展开剂为体积比为6:1的二氯甲烷与甲醇的混合物,待原料点消失,旋去部分溶剂,析晶,得到中间体3-甲基-4-氨基-5-乙酸酰肼;
(2)将碱溶于乙醇中,后加入3-甲基-4-氨基-5-乙酸酰肼,完全溶解后,加入含有cs2的乙醇溶液,40-80℃下搅拌反应,用薄层色谱(tlc)跟踪反应,展开剂为体积比为3:1的二氯甲烷与甲醇的混合物,待原料点消失,过滤,并用乙醇重结晶,得到中间体5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇;
(3)将5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇和乙醇混合并搅拌均匀,后加入取代水杨醛,用稀盐酸调ph为5-6,40-80℃下搅拌反应,用薄层色谱(tlc)跟踪反应,展开剂为体积比为10:1的二氯甲烷与甲醇的混合物,待原料点消失,将反应液浓缩,硅胶柱层析分离,得到目标化合物,所述取代水杨醛为水杨醛、5-溴水杨醛、5-氯水杨醛、5-氟水杨醛、5-甲基水杨醛或5-甲氧基水杨醛。
优选的,上述三唑席夫碱类化合物的制备方法,所述步骤(1)中3-甲基-4-氨基-5-乙氧羰基甲基硫基-1,2,4-三唑与水合肼的摩尔比为1:2-4;3-甲基-4-氨基-5-乙氧羰基甲基硫基-1,2,4-三唑与甲醇的用量比为1mmol:4-8ml。
优选的,上述三唑席夫碱类化合物的制备方法,所述步骤(2)中3-甲基-4-氨基-5-乙酸酰肼与cs2的摩尔比为1:2-4;3-甲基-4-氨基-5-乙酸酰肼与乙醇的用量比为1mmol:2-5ml;3-甲基-4-氨基-5-乙酸酰肼与碱的摩尔比为1:2-4。
优选的,上述三唑席夫碱类化合物的制备方法,所述步骤(2)中碱为koh。
优选的,上述三唑席夫碱类化合物的制备方法,所述步骤(3)中5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇与水杨醛的摩尔比为1:1-1.5;5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇与乙醇的用量比为1mmol:1-3ml。
一种上述制备方法的中间产物,为3-甲基-4-氨基-5-乙酸酰肼,具有式(ⅱ)所述结构式:
一种上述制备方法的中间产物,为5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇,具有式(ⅲ)所述结构式:
上述三唑席夫碱类化合物在制备抗菌药物方面的应用。
优选的,上述三唑席夫碱类化合物的应用,所述抗菌药物为抗革兰氏阴性菌、抗革兰氏阳性菌或抗真菌药物。
本发明的有益效果是:
所述三唑席夫碱类化合物,在药物分子中同时引入两个或多个杂环活性中心,能显著改善其生物活性,根据生物活性叠加原理,将“邻羟苯基”分子片断与“1,2,4-三唑”、“1,3,4-噁二唑”合理组装,设计合成了一类1,3,4-噁二唑修饰的1,2,4-三唑席夫碱化合物,通过生物活性测试表明,该类化合物对革兰氏阴性、革兰氏阳性和真菌等都有较好的抑制活性,表现出广谱抗菌活性,极具开发潜力,具有很好的应用前景;其制备方法工艺简单、易于实施、收率可达40-60%,适合规模化工业生产的需要。
附图说明
图1为化合物a的氢谱;
图2为化合物a的碳谱;
图3为化合物b的氢谱;
图4为化合物b的碳谱;
图5为化合物c的氢谱;
图6为化合物c的碳谱;
图7为化合物d的氢谱;
图8为化合物d的碳谱;
图9为化合物e的氢谱;
图10为化合物e的碳谱;
图11为化合物f的氢谱;
图12为化合物f的碳谱。
具体实施方式
为了寻找疗效好和对多种细菌均有抑制作用的抗菌药物,结合对抗菌药物的认识,设计并合成了一类1,3,4-噁二唑修饰的1,2,4-三唑席夫碱化合物,并对获得的目标化合物进行体外抗菌活性测试。发现该类化合物具有很强的抗菌活性,具有开发为新的抗菌药物的前景。目标化合物的结构已经由高分辨质谱(hrms)、核磁共振氢谱(1h-nmr)和核磁共振碳谱(13c-nmr)确证。
实施例1
5-(4-(2-羟基-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇的制备,步骤如下:
1)在50ml的三口瓶中加入20ml甲醇,搅拌下加入2.16g(10mmol)的3-甲基-4-氨基-5-乙氧羰基甲基硫基-1,2,4-三唑使其完全溶解,缓慢滴加1.2ml(30mmol)的水合肼,用薄层色谱(tlc)跟踪反应,展开剂为体积比为6:1的二氯甲烷与甲醇的混合物,待反应结束后,旋去部分溶剂,析晶,得到1.6g3-甲基-4-氨基-5-乙酸酰肼,产率78.8%。m.p.146-148℃.1hnmr(400mhz,dmso-d6),9.30(s,1h,nh),5.88(s,2h,nh2),4.31(s,2h,nh2),3.76(s,2h,ch2),2.28(s,3h,ch3).13cnmr(100mhz,dmso-d6),167.20,153.80,151.01,33.87,10.24.esi-ms,m/z:203.0720[m+h]+.
2)在100ml的三口瓶中加入15ml乙醇,后加入1.12g(20mmol)koh搅拌溶解,搅拌下加入2.03g(10mmol)的3-甲基-4-氨基-5-乙酸酰肼,完全溶解后,升温至40℃,缓慢加入含有2ml(30mmol)cs2的乙醇溶液15ml,加毕,升温至回流。薄层色谱法(tlc)跟踪反应,展开剂为体积比为3:1的二氯甲烷与甲醇的混合物,待原料点消失,过滤,乙醇重结晶,得到1.90g5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇,产率77.8%。m.p.192-194℃.1hnmr(400mhz,dmso-d6),5.94(s,2h,nh2),4.45(s,2h,ch2),2.29(s,3h,ch3).13cnmr(100mhz,dmso-d6),178.48,161.07,153.34,149.25,25.38,10.28.esi-ms,m/z:245.0272[m+h]+。
3)在50ml的圆底烧瓶中加入20ml的乙醇,缓慢加入2.44g(10mmol)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇搅拌溶解,再加入1.34g(1.1mmol)水杨醛,用稀盐酸调ph为5-6,78℃下搅拌反应,用薄层色谱跟踪(tlc),展开剂为体积比为10:1的二氯甲烷与甲醇的混合物,待原料点消失,将反应液浓缩,硅胶柱层析分离,得到目标化合物1.49g。产率42.9%。熔点:191-193℃。该制得的产物设编号为化合物a。
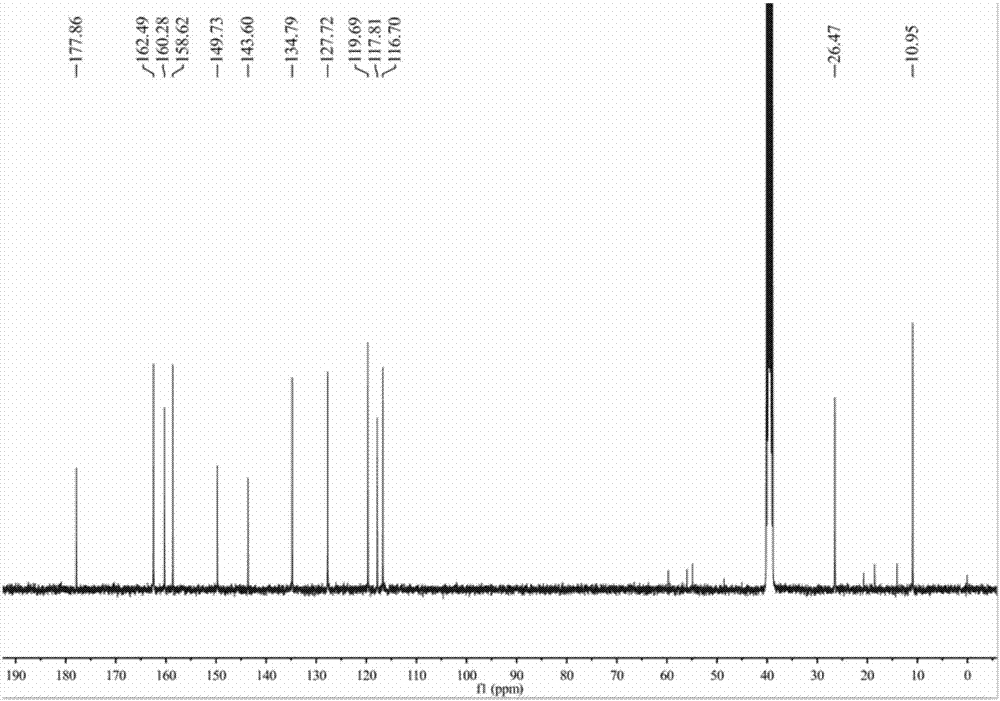
如图1-2所示,通过核磁共振氢谱(1h-nmr)、核磁共振碳谱(13c-nmr)、高分辨质谱(hrms)确证化合物a的结构:1hnmr(400mhz,dmso-d6),14.47(1h,s,sh),10.55(1h,s,oh),8.96(1h,s,n=ch),7.87(1h,dd,j=7.2,1.2hz,arh),7.46(1h,m,arh),7.00(1h,d,j=8.4hz,arh),6.96(1h,t,j=7.6hz,arh),4.42(2h,s,ch2),2.42(3h,s,ch3).13cnmr(100mhz,dmso-d6),177.86,162.49,160.48,158.62,149.73,143.60,134.79,127.72,119.69,117.81,116.70,26.47,10.95.esi-ms,m/z:349.0550[m+h]+。
实施例2
5-(4-(2-羟基-5-溴-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇的制备步骤如下:
1)3-甲基-4-氨基-5-乙酸酰肼制备方法与实施例1相同。
2)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇制备方法与实施例1相同。
3)在50ml的圆底烧瓶中加入20ml的乙醇,缓慢加入2.44g(10mmol)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇搅拌溶解,再加入2.21g(1.1mmol)5-溴水杨醛,用稀盐酸调ph为5-6,78℃下搅拌反应,用薄层色谱跟踪(tlc),展开剂为体积比为10:1的二氯甲烷与甲醇的混合物,待原料点消失,将反应液浓缩,硅胶柱层析分离,得到目标化合物2.15g。产率50.7%。熔点:201-203℃。该制得的产物设编号为化合物b。
如图3-4所示,通过核磁共振氢谱(1h-nmr)、核磁共振碳谱(13c-nmr)、高分辨质谱(hrms)确证化合物b的结构:1hnmr(400mhz,dmso-d6),14.45(1h,s,sh),10.90(1h,s,oh),8.91(1h,s,n=ch),7.97(1h,d,j=2.8hz,arh),7.60(1h,dd,j=8.8,2.4hz,arh),6.97(1h,d,j=8.8hz,arh),4.42(2h,s,ch2),2.43(3h,s,ch3).13cnmr(100mhz,dmso-d6),177.85,160.20,160.07,157.76,149.86,143.71,136.87,129.16,119.97,118.96,110.86,26.56,11.03.esi-ms,m/z:426.9652[m+h]+。
实施例3
5-(4-(2-羟基-5-氯-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇的制备步骤如下:
1)3-甲基-4-氨基-5-乙酸酰肼制备方法与实施例1相同。
2)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇制备方法与实施例1相同。
3)在50ml的圆底烧瓶中加入20ml的乙醇,缓慢加入2.44g(10mmol)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇搅拌溶解,再加入1.71g(1.1mmol)5-氯水杨醛,用稀盐酸调ph为5-6,78℃下搅拌反应,用薄层色谱跟踪(tlc),展开剂为体积比为10:1的二氯甲烷与甲醇的混合物,待原料点消失,将反应液浓缩,硅胶柱层析分离,得到目标化合物1.69g。产率44.2%。熔点:199-201℃。该制得的产物设编号为化合物c。
如图5-6所示,通过核磁共振氢谱(1h-nmr)、核磁共振碳谱(13c-nmr)、高分辨质谱(hrms)确证化合物c的结构:1hnmr(400mhz,dmso-d6),14.46(1h,s,sh),10.88(1h,s,oh),8.92(1h,s,n=ch),7.84(1h,d,j=2.8hz,arh),7.49(1h,dd,j=8.8,2.4hz,arh),7.02(1h,d,j=9.2hz,arh),4.42(2h,s,ch2),2.43(3h,s,ch3).13cnmr(100mhz,dmso-d6),177.85,160.20,160.17,157.38,149.88,143.72,134.10,126.18,123.44,119.39,118.65,26.58,11.02.esi-ms,m/z:383.0162[m+h]+。
实施例4
5-(4-(2-羟基-5-氟-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇的制备步骤如下:
1)3-甲基-4-氨基-5-乙酸酰肼制备方法与实施例1相同。
2)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇制备方法与实施例1相同。
3)在50ml的圆底烧瓶中加入20ml的乙醇,缓慢加入2.44g(10mmol)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇搅拌溶解,再加入1.54g(1.1mmol)5-氟水杨醛,用稀盐酸调ph为5-6,78℃下搅拌反应,用薄层色谱跟踪(tlc),展开剂为体积比为10:1的二氯甲烷与甲醇的混合物,待原料点消失,将反应液浓缩,硅胶柱层析分离,得到目标化合物1.79g。产率48.9%。熔点:198-200℃。该制得的产物设编号为化合物d。
如图7-8所示,通过核磁共振氢谱(1h-nmr)、核磁共振碳谱(13c-nmr)、高分辨质谱(hrms)确证化合物d的结构:1hnmr(400mhz,dmso-d6),14.46(1h,s,sh),10.59(1h,s,oh),8.93(1h,s,n=ch),7.60(1h,dd,j=9.2,3.2hz,arh),7.34(1h,td,j=8.4,3.2hz,arh),7.01(1h,dd,j=9.2,4.4hz,arh),4.42(2h,s,ch2),2.43(3h,s,ch3).13cnmr(100mhz,dmso-d6),177.84,160.37,156.49,155.08,154.15,149.90,143.62,121.69,118.52,118.23,112.18,26.61,10.99.esi-ms,m/z:367.0455[m+h]+
实施例5
5-(4-(2-羟基-5-甲基-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇的制备步骤如下:
1)3-甲基-4-氨基-5-乙酸酰肼制备方法与实施例1相同。
2)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇制备方法与实施例1相同。
3)在50ml的圆底烧瓶中加入20ml的乙醇,缓慢加入2.44g(10mmol)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇搅拌溶解,再加入1.49g(1.1mmol)5-甲基水杨醛,用稀盐酸调ph为5-6,78℃下搅拌反应,用薄层色谱跟踪(tlc),展开剂为体积比为10:1的二氯甲烷与甲醇的混合物,待原料点消失,将反应液浓缩,硅胶柱层析分离,得到目标化合物1.56g。产率43.2%。熔点:192-194℃。该制得的产物设编号为化合物e。
如图9-10所示,通过核磁共振氢谱(1h-nmr)、核磁共振碳谱(13c-nmr)、高分辨质谱(hrms)确证化合物e的结构:1hnmr(400mhz,dmso-d6),14.46(1h,s,sh),10.32(1h,s,oh),8.91(1h,s,n=ch),7.66(1h,s,arh),7.28(1h,dd,j=8.4,2.0hz,arh),6.90(1h,d,j=8.4hz,arh),4.41(2h,s,ch2),2.41(3h,s,ch3),2.27(3h,s,ch3).13cnmr(100mhz,dmso-d6),177.86,162.80,160.25,156.63,149.67,143.53,135.68,128.61,127.29,117.37,116.63,26.55,19.93,10.77.esi-ms,m/z:363.0698[m+h]+。
实施例6
5-(4-(2-羟基-5-甲氧基-苯甲基)氨基-5-甲基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇的制备步骤如下:
1)3-甲基-4-氨基-5-乙酸酰肼制备方法与实施例1相同。
2)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇制备方法与实施例1相同。
3)在50ml的圆底烧瓶中加入20ml的乙醇,缓慢加入2.44g(10mmol)5-(4-氨基-5-苯基-4h-1,2,4-三唑-3-硫烷基)-1,3,4-噁二唑-2-硫醇搅拌溶解,再加入1.67g(1.1mmol)5-甲氧基水杨醛,用稀盐酸调ph为5-6,78℃下搅拌反应,用薄层色谱跟踪(tlc),展开剂为体积比为10:1的二氯甲烷与甲醇的混合物,待原料点消失,将反应液浓缩,硅胶柱层析分离,得到目标化合物1.53g。产率40.5%。熔点:193-195℃。该制得的产物设编号为化合物f。
如图11-12所示,通过核磁共振氢谱(1h-nmr)、核磁共振碳谱(13c-nmr)、高分辨质谱(hrms)确证化合物f的结构:1hnmr(400mhz,dmso-d6),14.46(1h,s,sh),10.14(1h,s,oh),8.92(1h,s,n=ch),7.341h,d,j=3.2hz,arh),7.10(1h,dd,j=9.2,3.2hz,arh),6.94(1h,d,j=8.8hz,arh),4.43(2h,s,ch2),3.75(3h,s,och3),2.42(3h,s,ch3).13cnmr(100mhz,dmso-d6),177.83,161.96,160.20,153.06,152.24,149.74,143.51,122.49,117.93,117.66,109.59,55.52,26.44,10.88.esi-ms,m/z:379.0647[m+h]+。
抑菌实验例
1,3,4-噁二唑修饰的1,2,4-三唑席夫碱类化合物生物活性测试
具体操作如下:
1)抗菌药物贮存液制备
配制浓度为256μg.ml-1的抗菌药物贮存液10ml,用分析天平,称取化合物3.4mg,所用药物为粉剂,其药物的有效力为750μg·ml-1。计算得稀释剂用量为:3.4mg×750μg·ml-1)/256μg·ml-1=10.0ml,用2mldmf和8ml蒸馏水溶解药物。0℃以下贮存抗菌药物贮存液。
2)培养基
配制好相应的培养基:培养细菌用营养肉汤培养基,培养真菌用沙氏葡萄糖液体培养基。
3)稀释抗菌药物的制备
将10支无菌试管(13×100mm)排成一排,第1管除外,其余每管加入1.0ml培养基,在第1管加入抗菌药物贮存液(如256μg·ml-1)2ml混匀,吸取1ml加到第2管,混匀后再吸取1ml至第3管,如此连续至第9管,并从第9管中吸取1ml弃去,第10管为不含药物的阴性对照。第1管至第9管药物浓度分别为256、128、64、32、16、8、4、2、1μg·ml-1。
4)接种物制备
挑取菌落制备0.5麦氏比浊标准的菌悬液,经液体培养基1∶1000稀释后(约含1-2×108cfu·ml-1),向每管加100μl,密封后置37℃恒温培养箱中,细菌培养16-20h,真菌培养72h判断结果。
5)结果判断
待细菌培养16-20h,真菌培养72h后,检查结果并记录数据。首先检查阴性对照管的菌株是否情况良好,并保证没有被污染。然后肉眼观察,最低浓度管无菌株生长者,即为测试菌株的最小抑菌浓度(mic)。
按上述抑菌实验例所述方法,对实施例1-6中的化合物进行抑菌生物活性测试,结果见下表1。
表1化合物的体外最小抑菌浓度(μg·ml-1)
表1检测结果表明:该类化合物对大肠杆菌、金黄色葡萄球菌和白色念珠菌均显示了较好的抑制活性,具有广谱抗菌剂开发价值和应用前景,可以为安全高效抗微生物新药研制提供先导化合物、基础理论和技术支撑。
上述参照实施例对该种满足机体组织生长和修复需求的组合物及其制备方法进行的详细描述,是说明性的而不是限定性的,可按照所限定范围列举出若干个实施例,因此在不脱离本发明总体构思下的变化和修改,应属本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种紫外线吸收剂uv-234的制备方法
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑5-甲基间苯二甲酸锰配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑镉混配配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑间苯二甲酸镉配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑锌混配配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑三维锌配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑对苯二甲酸铜配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-双三唑均苯四酸铜配合物单晶与应用
- 一种4-苯基-3-((4,6-二甲基嘧啶-2-基硫代)甲基)-5-苄硫基三唑类化合物及其应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑二维铜配合物单晶与应用