一种改进的启动子及其组成的T载体和应用的制作方法
本发明属于基因工程领域,涉及一种改进的启动子及其组成的T载体和应用,具体涉及一种改进的启动子、带有所述改进启动子的T载体,带有所述T载体的宿主细胞及其应用。
背景技术:
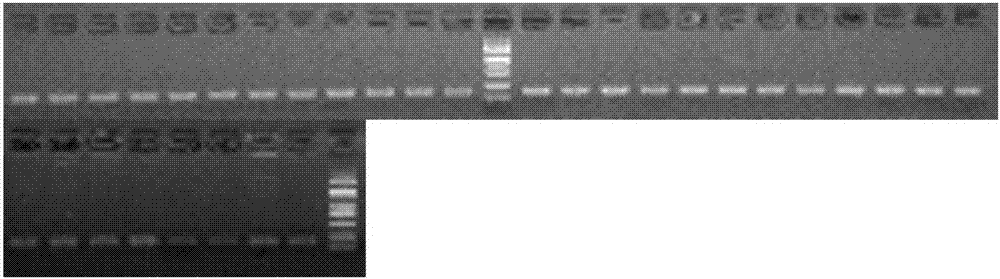
:PCR技术的发明是分子生物学与基因工程领域的重大突破。PCR技术诞生之后,将PCR产物克隆入载体(通常为质粒)的技术也获得发展。常用且相对简单的克隆方法包括TA克隆和平末端连接。由Taq酶扩增出的PCR产物含有dAMP尾巴,在T4连接酶的作用下,可以和含有T末端的载体(T载体)连接,这就是TA克隆。而高保真DNA聚合酶通常含有3’-5’核酸外切酶活性,它们扩增出的PCR产物为平末端,将这些片段与切成平末端的载体在T4连接酶的作用下连接就是平末端连接。这两种方法的共同特点是不需要事先用特殊的酶对PCR产物进行处理,而是直接连入载体,具有简单易操作的特性。目前商业化的T载体和可以用于平末端克隆的载体通常是基于蓝白斑筛选的原理,蓝白斑筛选是最常用的将空载体和有插入片段的载体相分开的一种筛选方案。在这种方法中,报告基因LacZα作为蓝白斑筛选的标记基因,然而基于蓝白斑筛选原理的载体在克隆时有以下问题:(1)由于使用强启动子,能够大量启动外源基因的转录、翻译,导致了某些结构复杂的外源基因转录或翻译产物对宿主有毒性而无法克隆;(2)酶切载体时由于限制性内切酶残留的外切酶活性、酶切载体的反复冻融以及酶切线性化载体的长期保存等因素使得制备的载体在酶切位点缺失1-2碱基,导LacZα基因的移码突变,使得不含外源基因的克隆由于LacZα基因的移码突变而显白色,导致产生大量假阳性克隆;(3)在克隆外源DNA小片段并且外源DNA的插入没有改变lacZα基因的读码框时会造成平板都是蓝斑的假阴性现象;(4)载体在平末端克隆大于2kb的外源DNA片段时,白斑会很少,而蓝斑却很多,而且本来就很少的白斑还可能与蓝斑长在一起,使得白斑单克隆极少,挑选足够数量的阳性克隆会很困难。此外,蓝白斑筛选还需要用到X-gal和IPTG等昂贵且有毒性的化学物质。TheuseoftheccdBlethalgeneforconstructingazerobackgroundvectorinordertocloneblunt-endPCRProducts.HuL-L,ZhangS-S,LiX-X,WangB-L..MolecularBiology.2010;44(1):161-4.;和LethalccdBgene-basedzero-backgroundvectorforconstructionofshotgunlibraries.MiyazakiK.Journalofbioscienceandbioengineering.2010Sep;110(3):372-3.公开了应用ccdB致死基因构建不需要蓝白斑筛选的载体,其效果显著,筛选更加简单方便。CN105400809A公开的一种克隆载体及其制备与应用,其公开了一种克隆载体pUC57-ccdB,为在pUC57载体的多克隆位点插入有ccdB基因的改造载体,所述ccdB基因含有平末端限制性内切酶酶切识别位点。利用ccdB蛋白对不含F质粒的大肠杆菌的致死作用,通过分子生物学技术在pUC57质粒上插入ccdB(含限制性内切酶SmaI酶切位点)基因,获得载体pUC57-ccdB。通过SmaI酶切产生平末端,再连接需要克隆的基因,可以实现需克隆基因的插入,同时避免含空载体菌落的出现。然而上述的技术方案仍然存在一定的问题:(1)由于使用强启动子,能够大量启动外源基因的转录、翻译,导致了某些结构复杂的外源基因转录或翻译产物对宿主有毒性而无法克隆;(2)由于反复冻融等因素使得T载体在末端缺失1-2碱基,导致ccdB“致死基因”的移码突变,使得不含外源基因的克隆由于ccdB“致死基因”的移码突变而“幸存”下来,导致产生大量假阳性克隆;(3)在克隆外源DNA小片段并且外源DNA的插入没有改变ccdB“致死基因”的读码框时会造成平板没有菌落的假阴性现象。任何一段能够独立与转录因子结合并起始转录的DNA序列都可以被称为启动子。在启动子中,能够被σ因子识别的区域具有非常保守的序列特征。其中,在转录起始位点(+1)上游大约10nt以及35nt处的两段序列(称为-10区与-35区)对于σ因子的识别具有决定性的作用,因此这两段序列也被称作狭义启动子或核心启动子。除这段核心启动子区域之外,-35区的上游序列也可能对转录的强度产生影响,这些序列被称作UP元件(UPelement)。在启动子位置进行改造,从而能够提高筛选的效率,避免筛选时的假阳性和假阴性,是一个关键的难点。技术实现要素:因此,本发明要解决的技术问题在于克服现有技术中制备的T载体无法克隆,或产生大量假阳性或假阴性克隆,从而提供一种改进的启动子及其组成的T载体和应用。为达此目的,本发明采用以下技术方案:一方面,本发明提供一种改进的启动子,所述改进的启动子为将启动子区域中的-35区至-10区之间的核酸序列突变为核酸内切酶识别位点。本发明中,原核生物中-35区至-10区之间核酸数目的变动会影响基因转录活性的高低,将启动子区域的-35区至-10区之间的核酸序列进行突变,从而能够被核酸内切酶识别,克隆时,先将载体制备为线性化载体,再将外源基因与线性化载体连接,使得启动子调控表达的基因活性下降,表达量下降,进而发挥功能。本发明中,通过在-35区至-10区之间进行突变,可以避免假阳性和假阴性的出现,可以避免载体在酶切位点缺失1-2bp导致基因移码突变产生假阳性克隆的缺陷,可以消除由于外源DNA片段较小并且外源DNA的插入没有改变基因的读码框,造成平板都是蓝斑的假阴性现象。所述核酸内切酶识别位点指的是能够被核酸内切酶酶切后形成平末端的核酸内切酶识别的位点都可行,其对核酸内切酶不作限制,其核酸内切酶的选择主要是基于本领域技术人员实验操作的方便性,突变1个或几个碱基就能够突变成功即可。根据本发明,所述改进的启动子为将β-半乳糖苷酶的启动子区域中的-35区至-10区之间的核酸序列突变为核酸内切酶酶切形成平末端序列的识别位点。本发明中,对于β-半乳糖苷酶的启动子,将其强启动子区域-35区至-10区之间的核酸序列突变为能够被识别的核酸内切酶识别位点,但其被剪切成线性化载体,插入外源片段后,使得β-半乳糖苷酶强启动子由于外源DNA片段的插入使得活性明显下降,进而使得lacZα基因的表达量显著下降,进而使得含有重组质粒的菌落显白色,这样就能够克服由于采用蓝白筛选的载体存在强启动子启动外源基因的转录或翻译产物可能对宿主有毒性而导致无法克隆的问题,可以避免载体在酶切位点缺失1-2bp导致基因移码突变产生假阳性克隆的缺陷,可以消除由于外源DNA片段较小并且外源DNA的插入没有改变基因的读码框,造成平板都是蓝斑的假阴性现象。根据本发明,所述β-半乳糖苷酶的启动子区域中的-35区至-10区之间的核酸序列如SEQIDNO.1-2所示,所述SEQIDNO.1-2所示的核酸序列如下:SEQIDNO.1:5’-TTTACACTTTATGCTTCCGGCTCGTATGTT-3’;SEQIDNO.2:5’-CTTTATGCTTCCGGCTCG-3’;本发明中,RNA聚合酶Ⅱ的结合部位通常在-35区至-10区,这两个部位都很重要:RNA聚合酶能和-35和-10序列中的碱基和DNA主链中的磷酸基相接触;离开共同顺序较远的启动子的活性亦较弱;而发明人发现,通过突变-35区至-10区中的序列,尤其是SEQIDNO.2所示的序列,能够插入外源基因,使得lacZα基因的表达量显著下降。根据本发明,所述核酸内切酶本领域技术人员可以根据需要进行选择,可以根据突变的启动子区域的序列的不同从而选择不同的核酸内切酶识别位点,本申请选自但不限于EcoRV、AfeI、PmlI、AfeI、NruI、PsiI、ScaI、SmaI、SspI或StuI中的任意一种或至少两种的组合。根据本发明,所述改进的启动子的-35区至-10区之间的核酸序列如SEQIDNO.3-10所示,所述SEQIDNO.3-10所示的核酸序列如下:SEQIDNO.3:5’-GATATCGCTTCCGGCTCG-3’;SEQIDNO.4:5’-CTTGATATCTCCGGCTCG-3’;SEQIDNO.5:5’-CTTTATGATATCGGCTCG-3’;SEQIDNO.6:5’-CTTTATGCTGATATCTCG-3’;SEQIDNO.7:5’-CACGTGGCTTCCGGCTCG-3’;SEQIDNO.8:5’-CTTCACGTGTCCGGCTCG-3’;SEQIDNO.9:5’-CTTTATCACGTGGGCTCG-3’;SEQIDNO.10:5’-CTTTATGCTCACGTGTCG-3’.第二方面,本发明提供一种克隆载体,包括如第一方面所述的改进的启动子。根据本发明,所述载体上的lacZα基因替换为对宿主有毒性的基因,所述对宿主有毒性的基因为转录或翻译产物能够导致宿主无法生长或增殖的基因。根据本发明,所述对宿主有毒性的基因为致死基因和/或限制性内切酶基因。根据本发明,所述致死基因为ccdB基因,所述ccdB基因的核酸序列如SEQIDNO.11所示。本发明中,所述载体本领域技术人员可以根据需要进行选择,载体的选择不会对启动子的功能造成影响,所述克隆载体用于克隆所述目的蛋白,所述克隆载体例如可以选择高拷贝克隆载体pUC18、pUC19、pUC57,低拷贝克隆载体pCA、pCK、pCC或单拷贝克隆载体pCC1,其都可以带上本申请启动子,从而进行后续试验,对载体本身并不造成影响,带上本申请启动子的载体仍然是高拷贝克隆载体、低拷贝克隆载体或单拷贝克隆载体。根据本发明,所述载体还包括外源基因,所述外源基因可操作地连接在所述载体上。根据本发明,所述外源基因为lacI表达元件,所述lacI表达元件的核酸序列如SEQIDNO.12所示。第三方面,本发明提供一种T载体,所述T载体为第二方面所述的载体制备成线性化载体后,在所述线性化载体的3’端添加1个双脱氧胸腺嘧啶核苷酸得到。根据本发明,所述T载体还包括外源基因,所述外源基因可操作地连接在所述改进的启动子的核酸内切酶位点之间。第四方面,本发明提供一种如第三方面所述T载体的制备方法,包括如下步骤:(1)将对宿主有毒性的基因替换载体上的lacZα基因;(2)根据要突变的核酸内切酶识别位点设计引物,以原启动子及其调控表达的基因为模板,进行PCR扩增,获得带有改进的启动子的产物;(3)用Gibson重组的方法将步骤(2)的产物进行环化,得到带有启动子的载体;(4)将步骤(2)所述载体进行线性化,在所述线性化的载体的3’端添加1个双脱氧胸腺嘧啶核苷酸,得到所述T载体。根据本发明,步骤(2)所述引物的核酸序列如SEQIDNO.13-28所示。本发明中,通过SEQIDNO.13-14所述的核酸序列的引物对对pUC57-ccdB-lacI或pCK-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.3所示的核酸序列;通过SEQIDNO.15-16所述的核酸序列的引物对对pUC57-ccdB-lacI或pCK-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.4所示的核酸序列;通过SEQIDNO.17-18所述的核酸序列的引物对对pUC57-ccdB-lacI或pCK-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.5所示的核酸序列;通过SEQIDNO.19-20所述的核酸序列的引物对对pUC57-ccdB-lacI或pCK-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.6所示的核酸序列;通过SEQIDNO.21-22所述的核酸序列的引物对对pCC1-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.7所示的核酸序列;通过SEQIDNO.23-24所述的核酸序列的引物对对pCC1-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.8所示的核酸序列;通过SEQIDNO.25-26所述的核酸序列的引物对对pCC1-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.9所示的核酸序列;通过SEQIDNO.27-28所述的核酸序列的引物对对pCC1-ccdB进行PCR扩增构建得到的质粒中,SEQIDNO.2所示的核酸序列突变为SEQIDNO.10所示的核酸序列。根据本发明,步骤(4)所述线性化为核酸内切酶酶切和/或PCR扩增获得。根据本发明,步骤(4)所述添加1个双脱氧胸腺嘧啶核苷酸采用末端转移酶和/或TaqDNA聚合酶。根据本发明,步骤(1)之前还包括将对宿主有毒性的基因进行密码子优化。根据本发明,步骤(1)之后还包括向所述载体上插入外源基因。第五方面,本发明提供一种宿主细胞,所述宿主细胞包括如第三方面所述的T载体。根据本发明,所述宿主细胞为野生型大肠杆菌。第六方面,本发明提供一种基因克隆的方法,包括:将所述外源基因的3’端添加1个A碱基,添加A碱基的外源基因与第三方面所述T载体连接,导入宿主细胞中,在合适的条件下,培养所述宿主细胞,以便获得阳性克隆。根据本发明,所述宿主细胞为野生型大肠杆菌。第七方面,本发明提供一种试剂盒,所述试剂盒包括第一方面所述的改进的启动子、第二方面所述的载体、第三方面所述的T载体或第三方面所述的宿主细胞中的任意一种或至少两种的组合。根据本发明,所述试剂盒用于基因克隆。与现有技术相比,本发明具有如下有益效果:(1)本发明所述的T载体在进行T-A克隆时将外源DNA片段克隆至载体的β-半乳糖苷酶启动子区域的-10区与-35区之间,使得即使在IPTG诱导的条件下β-半乳糖苷酶启动子活性也极低,使得对宿主有毒性的基因表达量极低,进而使得含有外源DNA片段的重组载体的宿主可以正常生长,而由于载体末端缺失1-2个碱基未连接外源DNA片段发生自连的空载体,由于其强启动子仍然具有很强的活性,并且筛选基因没有发生移码突变,能够启动对宿主有毒性基因的大量表达,进而使得携带不含外源DNA片段的载体的宿主不能生长,因此,本发明所述的T载体能够避免筛选基因由于移码突变产生假阳性克隆的问题;(2)本发明所述的T载体在进行T-A克隆时,能够克服载体筛选基因的强启动子启动外源基因转录或翻译产物可能对宿主有毒性而导致无法克隆的问题,并且可以避免克隆外源DNA小片段并且外源DNA的插入没有改变筛选基因的读码框时造成的假阴性现象;(3)本发明所述的克隆载体的构建方法简单,易操作,效率高,可以在短时间内完成所述克隆载体的构建;(4)本发明所述的T载体在基因克隆领域的用途,所述T载体摒除强启动子对外源基因的转录,容易克隆外源DNA大片段。附图说明图1为本申请实施例1的菌落PCR鉴定的电泳图,其中,DNAmarker大小为0.1kb、0.25kb、0.5kb、0.75kb、1kb、1.5kb、2kb、3kb、5kb;图2为本申请实施例3的菌落PCR鉴定的电泳图,其中,DNAmarker大小为0.1kb、0.25kb、0.5kb、0.75kb、1kb、1.5kb、2kb、3kb、5kb;图3为本申请实施例4的菌落PCR鉴定的电泳图,其中,DNAmarker大小为0.1kb、0.25kb、0.5kb、0.75kb、1kb、1.5kb、2kb、3kb、5kb。具体实施方式为更进一步阐述本发明所采取的技术手段及其效果,以下通过具体实施方式来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。本发明用到了遗传工程和分子生物学领域使用的常规技术和方法,一般性参考文献提供了本领域技术人员已知的定义和方法。但是,本领域的技术人员可以在本发明所记载的技术方案的基础上,采用本领域其它常规的方法、实验方案和试剂,而不限于本发明具体实施例的限定。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。术语解释:LacZ基因:广泛用于基因表达调控研究中的一种基因,编码的β一半乳糖苷酶(简称β-gal)是由4个亚基组成的四聚体,可催化乳糖的水解.Beta-gal比较稳定,用X-Gal为底物进行染色时,呈蓝色,便于检测和观察,LacZ基因的诸多优点使它成为基因工程实验中的一个常用标记基因,比如常用于转化菌株筛选,β-半乳糖苷酶显色反应选择法,即,蓝白筛选;LacZα基因:编码β-半乳糖苷酶(lacZ)N端α片段,可以通过α-互补形成具有酶活性的β-半乳糖苷酶可以将无色化合物X-gal(5-溴-4-氯-3-吲哚-β-D-半乳糖苷)切割成半乳糖和深蓝色的物质5-溴-4-靛蓝;核酸内切酶:在核酸水解酶中,为可水解分子链内部磷酸二酯键生成寡核苷酸的酶;PCR技术:聚合酶链式反应,是利用DNA在体外摄氏95°高温时变性会变成单链,低温(经常是60℃左右)时引物与单链按碱基互补配对的原则结合,再调温度至DNA聚合酶最适反应温度(72℃左右),DNA聚合酶沿着磷酸到五碳糖(5'-3')的方向合成互补链。基于聚合酶制造的PCR仪实际就是一个温控设备,能在变性温度,复性温度,延伸温度之间很好地进行控制。材料:实施例1:高拷贝克隆载体的构建及功能验证本实施例提供了一种高拷贝克隆载体的构建方法,包括如下具体步骤:I)采用ccdB基因替换pUC57(卡那霉素抗性)的lacZα基因,具体如下:(1)全基因合成ccdB基因(苏州金唯智生物科技有限公司合成),核苷酸序列如SEQIDNO.11所示:ATGCAGTTTAAGGTTTACACCTATAAAAGAGAGAGCCGTTATCGTCTGTTTGTGGATGTACAGAGTGATATTATTGACACGCCCGGGCGACGGATGGTGATCCCCCTGGCCAGTGCACGTCTGCTGTCAGATAAAGTCTCCCGTGAACTTTACCCGGTGGTGCATATCGGGGATGAAAGCTGGCGCATGATGACCACCGATATGGCCAGTGTGCCGGTCTCCGTTATCGGGGAAGAAGTGGCTGATCTCAGCCACCGCGAAAATGACATCAAAAACGCCATTAACCTGATGTTCTGGGGAATATAA;(2)以具有卡那霉素抗性的pUC57质粒为模板,以SEQIDNO.29-30为引物进行PCR扩增反应,具体序列如下:SEQIDNO.29(正向引物):TTATAGGTGTAAACCTTAAACTGCATAGCTGTTTCCTGTGTGAAATTGTTATCC;SEQIDNO.30(反向引物):ATTAACCTGATGTTCTGGGGAATATAATTAAGCCAGCCCCGACACCCGCCAACAC;PCR反应体系如下表1所示:表1其中,一组以水为样本的阴性对照;反应条件如下表2所示:表2(3)将步骤(2)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物;(4)使用GibsonMasterMix试剂盒将步骤(3)所得的PCR纯化产物与ccdB基因进行连接反应,连接反应体系如下表3所示:表3连接反应条件为:50℃连接反1h;(5)将上述步骤(4)中获得的连接产物转化Top10F’感受态细胞,最终涂布卡那霉素抗性的LB平板并37℃培养过夜,次日挑取单克隆并进行Sanger测序,保留测序正确的质粒,并将质粒命名为pUC57-ccdB;II)向pUC57-ccdB质粒插入lacI表达元件,具体如下:(1)以步骤I)构建成功的pUC57-ccdB质粒为模板,以引物F-vector-Insert、R-vector-Insert(SEQIDNO.31-32)为引物进行PCR扩增反应:SEQIDNO.31(正向引物):CAGCTGCATTAATGAATCGGCCAACGCGC;SEQIDNO.32(反向引物):GCACGACAGGTTTCCCGACTGGAAAGCGG;PCR反应体系如表1所示,反应条件如下表2所示;(2)将步骤(1)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物;(3)基因合成lacI表达元件(苏州金唯智生物科技有限公司合成),核苷酸序列如SEQIDNO.12所示:CCCGCTTTCCAGTCGGGAAACCTGTCGTGCTTGACACCATCGAATGGTGCAAAACCTTTCGCGGTATGGCATGATAGCGCCCGGAAGAGAGTCAATTCAGGGTGGTGAATATGAACGTGAAACCAGTAACGTTATACGATGTCGCAGAGTATGCCGGTGTCTCTTATCAGACCGTTTCCCGCGTGGTGAACCAGGCCAGCCACGTTTCTGCGAAAACGCGGGAAAAAGTGGAAGCGGCGATGGCGGAGCTGAATTACATTCCCAACCGCGTGGCACAACAACTGGCGGGCAAACAGTCGTTGCTGATTGGCGTTGCCACCTCCAGTCTGGCCCTGCACGCGCCGTCGCAAATTGTCGCGGCGATTAAATCTCGCGCCGATCAACTGGGTGCCAGCGTGGTGGTGTCGATGGTAGAACGAAGCGGCGTCGAAGCCTGTAAAGCGGCGGTGCACAATCTTCTCGCGCAACGCGTCAGTGGGCTGATCATTAACTATCCGCTGGATGACCAGGATGCCATTGCTGTGGAAGCTGCCTGCACTAATGTTCCGGCGTTATTTCTTGATGTCTCTGACCAGACACCCATCAACAGTATTATTTTCTCCCATGAAGACGGTACGCGACTGGGCGTGGAGCATCTGGTCGCATTGGGTCACCAGCAAATCGCGCTGTTAGCGGGCCCATTAAGTTCTGTCTCGGCGCGTCTGCGTCTGGCTGGCTGGCATAAATATCTCACTCGCAATCAAATTCAGCCGATAGCGGAACGGGAAGGCGACTGGAGTGCCATGTCCGGTTTTCAACAAACCATGCAAATGCTGAATGAGGGCATCGTTCCCACTGCGATGCTGGTTGCCAACGATCAGATGGCGCTGGGCGCAATGCGCGCCATTACCGAGTCCGGGCTGCGCGTTGGTGCGGATATCTCGGTAGTGGGATACGACGATACCGAAGACAGCTCATGTTATATCCCGCCGTTAACCACCATCAAACAGGATTTTCGCCTGCTGGGGCAAACCAGCGTGGACCGCTTGCTGCAACTCTCTCAGGGCCAGGCGGTGAAGGGCAATCAGCTGTTGCCCGTCTCACTGGTGAAAAGAAAAACCACCCTGGCGCCCAATACGCAAACCGCCTCTCCCCGCGCGTTGGCCGATTCATTAATGCAGCTGGCACGACAGGTTTCCCGACTGGAAAGCGGGCAGTGATGCCTGGCGGCAGTAGCGCGGTGGTCCCACCTGACCCCATGCCGAACTCAGAAGTGAAACGCCGTAGCGCCGATGGTAGTGTGGGGTCTCCCCATGCGAGAGTAGGGAACTGCCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTGGATTGCACGCAGGTTCTCCGGCCGCTTGGGTGGAGAGGCGCAGAAAGTCAAAAGCCTCCGACCGGAGGCTTTTGACTATTAGCACAGCTGCATTAATGAATCGGCCAACGCGCG;(4)使用GibsonMasterMix(NEB)试剂盒将步骤(2)所得的PCR纯化产物与步骤(3)基因合成的lacI表达元件进行连接反应,连接反应体系如表4所示:表4连接反应条件:50℃连接反应1小时;(5)将上述步骤(4)中获得的连接产物转化Top10F’感受态细胞,最终涂布卡那霉素抗性的LB平板并37℃培养过夜,次日挑取单克隆并进行Sanger测序,保留测序正确的质粒,并将质粒命名为pUC57-ccdB-lacI。Ⅲ)将pUC57-ccdB-lacI质粒的β-半乳糖苷酶启动子区域的-35区与-10区之间的序列5’-CTTTATGCTTCCGGCTCG-3’突变为能够被核酸内切酶酶切形成平末端的序列,具体如下:(1)以步骤II)构建成功的pUC57-ccdB-lacI质粒为模板,以引物F1-EcoRV、R1-EcoRV、F2-EcoRV、R2-EcoRV、F3-EcoRV、R3-EcoRV、F4-EcoRV、R4-EcoRV(SEQIDNO.13-SEQIDNO.20)为引物进行PCR扩增反应,具体序列如下表5:表5具体的PCR反应体系如表1所示,反应条件如表2所示;(2)将步骤(1)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物,然后分别使用GibsonMasterMix试剂盒进行连接反应,连接反应体系如下表6所示:表6连接反应条件:50℃连接反应1小时;(3)将步骤(2)中获得的连接产物分别转化Top10F’感受态细胞,最终涂布卡那霉素抗性的LB平板并37℃培养过夜,次日挑取单克隆进行Sanger测序,保留测序正确的质粒构建,分别命名为pUC57-ccdB-lacI-Mu-1(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-GATATCGCTTCCGGCTCG-3’,质粒由F1-EcoRV+R1-EcoRV引物构建)、pUC57-ccdB-lacI-Mu-2(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTGATATCTCCGGCTCG-3’,质粒由F2-EcoRV+R2-EcoRV引物构建)、pUC57-ccdB-lacI-Mu-3(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTTATGATATCGGCTCG-3’,质粒由F3-EcoRV+R3-EcoRV引物构建)、pUC57-ccdB-lacI-Mu-4(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTTATGCTGATATCTCG-3’,质粒由F4-EcoRV+R4-EcoRV引物构建)。IV)制备T载体(1)使用EcoRV核酸内切酶酶切步骤III)构建的pUC57-ccdB-lacI-Mu-1、pUC57-ccdB-lacI-Mu-2、pUC57-ccdB-lacI-Mu-3、pUC57-ccdB-lacI-Mu-4质粒,酶切产物经1%琼脂糖凝胶电泳后切胶回收纯化得到平末端线性化载体,酶切反应体系如下表7所示:表7PCR扩增产物约0.9ug,3μLEcoRV/PmlI1μL10×buffer2μL灭菌去离子H2O14μL(2)使用TaqDNA聚合酶将单个双脱氧胸腺嘧啶核苷酸(ddTTP)添加到步骤(1)制备的线性化载体的3’端制备T载体,分别命名为pUC57-ccdB-lacI-Mu-1-T、pUC57-ccdB-lacI-Mu-2-T、pUC57-ccdB-lacI-Mu-3-T、pUC57-ccdB-lacI-Mu-4-T载体,反应体系如表8所示:表8线性化平末端载体5ug,50μL10×buffer10μLddTTP5mM,0.5μLTaqDNA聚合酶5U/μL,1μL灭菌去离子H2O38.5μL反应条件为68℃,1h,反应结束后使用Axygen纯化试剂盒进行回收纯化;V)载体克隆实验(1)合成两条24bp引物,两条引物中有23bp的序列反向互补,退火后能形成3’端突出一个A碱基的dsDNA,两条24bp引物核苷酸序列如SEQIDNO.33-SEQIDNO.34所示,具体如下:SEQIDNO.33:GCACCGGGATAACACGCTCACCAA;SEQIDNO.34:TGGTGAGCGTGTTATCCCGGTGCA;(2)以λDNA为模板,F-λDNA-200bp+R-λDNA-200bp为引物进行PCR扩增,所述引物F-λDNA-200bp、R-λDNA-200bp的核苷酸序列如SEQIDNO.35-SEQIDNO.36所示,具体如下:SEQIDNO.35(F-λDNA-200bp):GTTGAATGGGCGGATGCTAATTACTATCTCCCG;SEQIDNO.36(R-λDNA-200bp):TTATGCTCTATAAAGTAGGCATAAACACCCAGC;PCR反应体系如表1所示,PCR扩增程序如表9所示;表9(3)将步骤(2)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物;(4)将步骤(2)退火形成3’端突出一个A的dsDNA、步骤(3)纯化得到的PCR产物分别与步骤III)制备的pUC57-ccdB-lacI-Mu-1-T、pUC57-ccdB-lacI-Mu-2-T、pUC57-ccdB-lacI-Mu-3-T、pUC57-ccdB-lacI-Mu-4-T载体进行连接反应,连接反应体系如下表10所示:表10外源DNA约90ng,3μL酶切后的载体约30ng,1μL10×缓冲液1μLT4DNA连接酶1μL灭菌去离子H2O4μL连接反应条件:22℃连接反应1小时;(5)将上述步骤(4)中获得的连接产物分别转化Top10F’感受态细胞,最终涂布含IPTG的卡那霉素抗性的LB平板并37℃培养过夜,次日从克隆约200bpDNA片段的平板上分别挑取8个单克隆进行菌落PCR鉴定,PCR反应体系如表11所示:表11PCR反应体系PCR扩增程序如表12所示:表12PCR鉴定结果如图1所示,图1结果显示所有菌落均为阳性克隆。从克隆24、48bp外源DNA片段的平板上分别挑取8个单克隆连同菌检阳性的单克隆,分别进行Sanger测序,测序结果显示所有克隆的序列均正确,实验结果表明本发明的载体可以用于克隆不小于24bp的外源DNA。实施例2本发明所述克隆载体克服假阳性克隆的实验验证构建三种pUC57-ccdB-lacI-Mu-4-T突变质粒(pUC57-ccdB-lacI-Mu-4-T-Mim-1、pUC57-ccdB-lacI-Mu-4-T-Mim-2、pUC57-ccdB-lacI-Mu-4-T-Mim-3)模拟pUC57-ccdB-lacI-Mu-4-T载体在酶切位点两端缺失1-2碱基并发生自连,构建步骤如下:(1)以实施例1中构建的质粒pUC57-ccdB-lacI-Mu-4-T为模板,分别以F1-del+R1-del、F2-del+R2-del、F3-del+R3-del为引物进行PCR扩增反应,所述引物F1-del、R1-del、F2-del、R2-del、F3-del、R3-del的核苷酸序列如SEQIDNo.37-SEQIDNo.42所示,具体如下:SEQIDNO.37(F1-del):ACAACATACGAGATTCAGCATAAAGTGTAAAGCCTGGGGTGC;SEQIDNO.38(R1-del):CTTTATGCTGAATCTCGTATGTTGTGTGGAATTGTGAGC;SEQIDNO.39(F2-del):CACAACATACGAGAATCAGCATAAAGTGTAAAGCCTGGGGTG;SEQIDNO.40(R2-del):CACTTTATGCTGATTCTCGTATGTTGTGTGGAATTGTGAGCG;SEQIDNO.41(F3-del):ACACAACATACGAGATCAGCATAAAGTGTAAAGCCTGGGGTG;SEQIDNO.42(R3-del):ACACTTTATGCTGATCTCGTATGTTGTGTGGAATTGTGAGCGG;PCR反应体系如实施例1中表1所示,PCR扩增程序如实施例1表2所示;(2)将步骤(1)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物,然后分别使用GibsonMasterMix(NEB)试剂盒进行连接反应,连接反应体系如实施例1表4所示,连接反应条件:50℃连接反应1小时;(3)将步骤(2)中获得的连接产物分别转化Top10F’感受态细胞,最终涂布卡那霉素抗性的LB平板并37℃培养过夜,次日从每个平板分别挑取5个单克隆进行Sanger测序,保留测序正确的质粒,分别命名为pUC57-ccdB-lacI-Mu-4-T-Mim-1、pUC57-ccdB-lacI-Mu-4-T-Mim-2、pUC57-ccdB-lacI-Mu-4-T-Mim-3;(4)将步骤(3)正确的pUC57-ccdB-lacI-Mu-4-T-Mim-1、pUC57-ccdB-lacI-Mu-4-T-Mim-2、pUC57-ccdB-lacI-Mu-4-T-Mim-3质粒分别转化Top10F’感受态细胞,最终将复苏的菌液分别均分为两份,分别涂布含IPTG及不含IPTG的卡那霉素抗性的LB平板并37℃培养过夜,次日检查发现含IPTG的平板均没有菌落形成,不含IPTG的平板菌落的形态、数量均正常。实验结果表明:在IPTG诱导的条件下,pUC57-ccdB-lacI-Mu-4-T-Mim-1、pUC57-ccdB-lacI-Mu-4-T-Mim-2、pUC57-ccdB-lacI-Mu-4-T-Mim-3的β-半乳糖苷酶启动子仍有很强的活性,能够大量表达ccdB,导致菌落无法生长,即在IPTG的诱导条件下,本发明的T载体在末端缺失1-2个碱基发生自连的情况下不会产生假阳性克隆。实施例3:低拷贝T载体的构建及功能验证本实施例提供一种低拷贝T载体的构建方法,包括如下步骤:I)采用ccdB基因替换pCK(卡那霉素抗性)的lacZα基因,具体如下:(1)以具有卡那霉素抗性的pCK质粒为模板,以SEQIDNO.43-44为引物进行PCR扩增反应,具体序列如下:SEQIDNO.43(正向引物):TTATAGGTGTAAACCTTAAACTGCATAGCTGTTTCCTGTGTGAAATTGTTATCC;SEQIDNO.44(反向引物):TTAACCTGATGTTCTGGGGAATATAATTAAGCCAGCCCCGAGTAGCTAGACAGG;PCR反应体系如实施例2表1所示,反应条件如实施例2表2所示:(2)将步骤(1)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物;(3)使用GibsonMasterMix试剂盒将步骤(2)所得的PCR纯化产物与实施例1步骤(1)基因合成的ccdB基因进行连接反应,连接反应体系如实施例1表3所示,连接反应条件为:50℃连接反1h;(4)将上述步骤(3)中获得的连接产物转化Top10F’感受态细胞,最终涂布卡那霉素抗性的LB平板并37℃培养过夜,次日挑取单克隆并进行Sanger测序,保留测序正确的质粒,并将质粒命名为pCK-ccdB;II)将pCK-ccdB质粒的β-半乳糖苷酶启动子区域的-35区与-10区之间的序列5’-CTTTATGCTTCCGGCTCG-3’突变为能够被核酸内切酶酶切形成平末端的序列,具体如下:(1)以步骤I)构建成功的pCK-ccdB质粒为模板,以引物F1-EcoRV、R1-EcoRV、F2-EcoRV、R2-EcoRV、F3-EcoRV、R3-EcoRV、F4-EcoRV、R4-EcoRV(SEQIDNO.13-SEQIDNO.20)为引物进行PCR扩增反应,具体序列如实施例1表5所示,具体的PCR反应体系如实施例1表1所示,反应条件如实施例1表2所示;(2)将步骤(1)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物,然后分别使用GibsonMasterMix试剂盒进行连接反应,连接反应体系如实施例1表6所示,连接反应条件:50℃连接反应1小时;(3)将步骤(2)中获得的连接产物分别转化Top10F’感受态细胞,最终涂布卡那霉素抗性的LB平板并37℃培养过夜,次日挑取单克隆进行Sanger测序,保留测序正确的质粒构建,分别命名为pCK-ccdB-Mu-1(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-GATATCGCTTCCGGCTCG-3’,质粒由F1-EcoRV+R1-EcoRV引物构建)、pCK-ccdB-Mu-2(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTGATATCTCCGGCTCG-3’,质粒由F2-EcoRV+R2-EcoRV引物构建)、pCK-ccdB-Mu-3(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTTATGATATCGGCTCG-3’,质粒由F3-EcoRV+R3-EcoRV引物构建)、pCK-ccdB-Mu-4(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTTATGCTGATATCTCG-3’,质粒由F4-EcoRV+R4-EcoRV引物构建)。III)制备T载体(1)使用EcoRV核酸内切酶酶切步骤Ⅱ)构建的pUC57-ccdB-Mu-1、pUC57-ccdB-Mu-2、pUC57-ccdB-Mu-3、pUC57-ccdB-Mu-4质粒,酶切产物经1%琼脂糖凝胶电泳后切胶回收纯化得到平末端线性化载体,酶切反应体系如实施例1表7所示;(2)使用TaqDNA聚合酶将单个双脱氧胸腺嘧啶核苷酸(ddTTP)添加到步骤(1)制备的线性化载体的3’端制备T载体,分别命名为pCK-ccdB-Mu-1-T、pCK-ccdB-Mu-2-T、pCK-ccdB-Mu-3-T、pCK-ccdB-Mu-4-T载体,反应体系如实施例1表8所示,反应条件为68℃,1h,反应结束后使用Axygen纯化试剂盒进行回收纯化;Ⅳ)T载体克隆实验(1)合成两条24bp引物,两条引物中有23bp的序列反向互补,退火后能形成3’端突出一个A碱基的dsDNA,两条24bp引物核苷酸序列如实施例2的SEQIDNO.33-SEQIDNO.34所示;(2)以λDNA为模板,F-λDNA-200bp+R-λDNA-200bp为引物进行PCR扩增,PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物,所述引物F-λDNA-200bp、R-λDNA-200bp的核苷酸序列如实施例2的SEQIDNO.35-SEQIDNO.36所示,PCR反应体系如实施例1表1所示,PCR扩增程序如实施例1表9所示;(3)将步骤(2)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物;(4)将步骤(2)退火形成3’端突出一个A的dsDNA、步骤(3)纯化得到的PCR产物分别与步骤III)制备的pCK-ccdB-Mu-1-T、pCK-ccdB-Mu-2-T、pCK-ccdB-Mu-3-T、pCK-ccdB-Mu-4-T载体进行连接反应,连接反应体系如实施例1表10所示,连接反应条件:22℃连接反应1小时;(5)将上述步骤(4)中获得的连接产物分别转化Top10F’感受态细胞,最终涂布含IPTG的卡那霉素抗性的LB平板并37℃培养过夜,次日从克隆约200bpDNA片段的平板上分别挑取8个单克隆进行菌落PCR鉴定;PCR反应体系如实施例1表11所示,PCR扩增程序如实施例1表12所示。PCR鉴定结果如图2所示,图2结果显示所有菌落均为阳性克隆。从克隆24、48bp外源DNA片段的平板上分别挑取8个单克隆连同菌检阳性的单克隆,分别进行Sanger测序,测序结果显示所有克隆的序列均正确,实验结果表明本发明的载体可以用于克隆不小于24bp的外源DNA。实施例4:单拷贝T载体的构建及功能验证本实施例提供单拷贝T载体的构建方法,包括如下具体步骤:I)采用ccdB基因替换pCC1(氯霉素抗性)的lacZα基因,具体如下:(1)以具有氯霉素抗性的pCC1质粒为模板,以实施例1的SEQIDNO.29-30为引物进行PCR扩增反应,PCR反应体系如实施例1表1所示,反应条件如表13所示:表13(2)将步骤(1)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物;(3)使用GibsonMasterMix试剂盒将步骤(2)所得的PCR纯化产物与实施例1中合成的ccdB基因进行连接反应,连接反应体系如表14所示:表14连接反应条件为:50℃连接反1h;(4)将上述步骤(3)中获得的连接产物转化Top10F’感受态细胞,最终涂布氯霉素抗性的LB平板并37℃培养过夜,次日挑取单克隆并进行Sanger测序,保留测序正确的质粒,并将质粒命名为pCC1-ccdB;II)将pCC1-ccdB质粒的β-半乳糖苷酶启动子区域的-35区与-10区之间的序列5’-CTTTATGCTTCCGGCTCG-3’突变为能够被核酸内切酶酶切形成平末端的序列,具体如下:(1)以步骤I)构建成功的pCC1-ccdB质粒为模板,以引物F1-PmlI+R1-PmlI、F2-PmlI+R2-PmlI、F3-PmlI+R3-PmlI、F4-PmlI+R4-PmlI(SEQIDNO.21-SEQIDNO.28)为引物进行PCR扩增反应,具体序列如表15所示:表15具体的PCR反应体系如实施例1表1所示,反应条件如表13所示;(2)将步骤(1)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物,然后分别使用GibsonMasterMix试剂盒进行连接反应,连接反应体系如表16所示:表16连接反应条件:50℃连接反应1小时;(3)将步骤(2)中获得的连接产物分别转化Top10F’感受态细胞,最终涂布氯霉素抗性的LB平板并37℃培养过夜,次日挑取单克隆进行Sanger测序,保留测序正确的质粒构建,分别命名为pCC1-ccdB-Mu-1(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CACGTGGCTTCCGGCTCG-3’,质粒由F1-PmlI+R1-PmlI引物构建)、pCC1-ccdB-Mu-2(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTCACGTGTCCGGCTCG-3’,质粒由F2-PmlI+R2-PmlI引物构建)、pCC1-ccdB-Mu-3(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTTATCACGTGGGCTCG-3’,质粒由F3-PmlI+R3-PmlI引物构建)、pCC1-ccdB-Mu-4(将5’-CTTTATGCTTCCGGCTCG-3’突变为5’-CTTTATGCTCACGTGTCG-3’,质粒由F4-PmlI+R4-PmlI引物构建)。III)制备T载体(1)使用PmlI核酸内切酶酶切步骤Ⅱ)构建的pCC1-ccdB-Mu-1、pCC1-ccdB-Mu-2、pCC1-ccdB-Mu-3、pCC1-ccdB-Mu-4质粒,酶切产物经1%琼脂糖凝胶电泳后切胶回收纯化得到平末端线性化载体,酶切反应体系如实施例1表7所示;(2)使用TaqDNA聚合酶将单个双脱氧胸腺嘧啶核苷酸(ddTTP)添加到步骤(1)制备的线性化载体的3’端制备T载体,分别命名为pCC1-ccdB-Mu-1-T、pCC1-ccdB-Mu-2-T、pCC1-ccdB-Mu-3-T、pCC1-ccdB-Mu-4-T载体,反应体系如实施例1表8所示,反应条件为68℃,1h,反应结束后使用Axygen纯化试剂盒进行回收纯化;Ⅳ)T载体克隆实验(1)合成两条24bp引物,两条引物中有23bp的序列反向互补,退火后能形成3’端突出一个A碱基的dsDNA,两条24bp引物核苷酸序列如实施例2的SEQIDNO.33-SEQIDNO.34所示;(2)以λDNA为模板,F-λDNA-200bp+R-λDNA-200bp为引物进行PCR扩增,PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物,所述引物F-λDNA-200bp、R-λDNA-200bp的核苷酸序列如实施例2的SEQIDNO.35-SEQIDNO.36所示,PCR反应体系如实施例1表1所示,PCR扩增程序如实施例1表9所示;(3)将步骤(2)获得的PCR反应液经1%琼脂糖凝胶电泳后切胶回收纯化得到PCR扩增产物;(4)将步骤(2)退火形成3’端突出一个A的dsDNA、步骤(3)纯化得到的PCR产物分别与步骤III)制备的pCC1-ccdB-Mu-1-T、pCC1-ccdB-Mu-2-T、pCC1-ccdB-Mu-3-T、pCC1-ccdB-Mu-4-T载体进行连接反应,连接反应体系如实施例1表10所示,连接反应条件:22℃连接反应1小时;(5)将上述步骤(4)中获得的连接产物分别转化Top10F’感受态细胞,最终涂布含IPTG的氯霉素抗性的LB平板并37℃培养过夜,次日从克隆约200bpDNA片段的平板上分别挑取8个单克隆进行菌落PCR鉴定;PCR反应体系如实施例1表11所示,PCR扩增程序如实施例1表12所示。PCR鉴定结果如图3所示,图3结果显示所有菌落均为阳性克隆。从克隆24、48bp外源DNA片段的平板上分别挑取8个单克隆连同菌检阳性的单克隆,分别进行Sanger测序,测序结果显示所有克隆的序列均正确,实验结果表明本发明的载体可以用于克隆不小于24bp的外源DNA。综上所述,本发明所述的T载体在进行T-A克隆时将外源DNA片段克隆至载体的β-半乳糖苷酶启动子区域的-10区与-35区之间,使得即使在IPTG诱导的条件下β-半乳糖苷酶启动子活性也极低,使得对宿主有毒性的基因表达量极低,进而使得含有外源DNA片段的重组载体的宿主可以正常生长,而由于载体末端缺失1-2个碱基未连接外源DNA片段发生自连的空载体,由于其强启动子仍然具有很强的活性,并且筛选基因没有发生移码突变,能够启动对宿主有毒性基因的大量表达,进而使得携带不含外源DNA片段的载体的宿主不能生长,因此,本发明所述的T载体能够避免筛选基因由于移码突变产生假阳性克隆的问题。申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属
技术领域:
的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。序列表<110>中国科学院天津工业生物技术研究所<120>一种流感病毒抗体的新应用<130>2017<141>2017-12-29<160>10<170>SIPOSequenceListing1.0<210>1<211>97<212>PRT<213>人工合成序列()<400>1AspIleValMetThrGlnSerProGlyThrLeuSerLeuSerProGly151015GluArgAlaThrLeuSerCysArgAlaSerGlnSerValSerSerSer202530TyrLeuAlaTrpTyrGlnGlnLysProGlyGlnAlaProArgLeuLeu354045IleTyrArgAlaSerSerArgAlaThrGlyIleProAspArgPheSer505560GlySerGlySerGlyThrAspPheThrLeuThrIleSerArgLeuGlu65707580ProGluAspPheAlaValTyrTyrCysGlnGlnTyrGlySerSerPhe859095Thr<210>2<211>117<212>PRT<213>人工合成序列()<400>2GlnValGlnLeuValGluSerGlyGlyGlyValValGlnProGlyThr151015SerLeuArgLeuSerCysGluAlaSerGlyPheThrSerSerAlaTyr202530AlaMetHisTrpValArgGlnAlaProGlyLysGlyLeuGluTrpVal354045AlaValIleThrPheAspGlyGlyTyrGlnTyrTyrAlaAspSerVal505560LysGlyArgPheThrIleSerArgAspIleSerArgAsnThrLeuHis65707580LeuHisMetAsnSerLeuArgAlaGluAspThrAlaValTyrTyrCys859095AlaArgAspProLeuThrLysLeuLeuProPheAspTrpValSerGly100105110GlyTyrPheAspTyr115<210>3<211>291<212>DNA<213>人工合成序列()<400>3gacatcgtgatgacacagtctccaggcaccctgtctttgtctccaggggaaagagccacc60ctctcctgcagggccagtcagagtgttagcagcagctacttagcctggtaccagcagaaa120cctggccaggctcccaggctcctcatctatcgtgcatccagcagggccactggcatccca180gacaggttcagtggcagtgggtctgggacagacttcactctcaccatcagcagactggag240cctgaagattttgcagtgtattactgtcagcagtatggtagctcgttcact291<210>4<211>351<212>DNA<213>人工合成序列()<400>4caggtgcagctggtggagtctgggggaggcgtggtccagcctgggacgtccctgagactc60tcctgtgaagcctctggattcacctccagtgcctatgctatgcactgggtccgccaggct120ccaggcaagggcctagagtgggtggcagttataacatttgatggaggttatcaatactac180gcagactccgtgaagggccgattcaccatctccagagacatttccaggaacactcttcac240ctgcacatgaacagcctgagagctgaggacacggctgtttattactgtgcgagagatccc300ctaacaaagttactgccttttgactgggtctctggggggtactttgactac351<210>5<211>7<212>PRT<213>人工合成序列()<400>5GlnSerValSerSerSerTyr15<210>6<211>3<212>PRT<213>人工合成序列()<400>6ArgAlaSer1<210>7<211>8<212>PRT<213>人工合成序列()<400>7GlnGlnTyrGlySerSerPheThr15<210>8<211>8<212>PRT<213>人工合成序列()<400>8GlyPheThrSerSerAlaTyrAla15<210>9<211>8<212>PRT<213>人工合成序列()<400>9IleThrPheAspGlyGlyTyrGln15<210>10<211>21<212>PRT<213>人工合成序列()<400>10AlaArgAspProLeuThrLysLeuLeuProPheAspTrpValSerGly151015GlyTyrPheAspTyr20当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1