一种靶向HDAC的稳定多肽类抑制剂及其用途的制作方法
本发明属于生物工程领域,涉及一种多肽,具体是一种靶向hdac的稳定多肽类抑制剂及其用途。
背景技术:
癌症是世界范围内一个主要的公共卫生问题,在中国,它是首要的疾病致死原因,而且造成严重的疾病负担。据2014年国家中心癌症登记处(nccrc)在中国的癌症统计数据显示,目前每年中国有3,804,000例癌症被诊断,癌症发生率大概为278.07/100,000(男性发病率301.67/100,000,女性发病率253.29/100,000)。肿瘤类型和肿瘤致病原因在不同的性别以及年龄中会不同,肿瘤疾病的复杂性为人类攻克该疾病带来了挑战。关于肿瘤的发生、发展、侵袭、转移和治疗一直是医学研究的热点及难题。肿瘤干细胞学说认为,肿瘤细胞的恶性增殖可能是由肿瘤组织中一小群具有干细胞特性的肿瘤细胞引起,这些细胞具有干细胞的自我更新、快速增殖以及分化的潜能,能导致药物的耐药性以及肿瘤的复发。肿瘤干细胞目前已经在多种肿瘤细胞中发现,比如乳腺癌,结肠癌,卵巢癌和肺癌等,这些肿瘤干细胞的存在为肿瘤的治疗带来了瓶颈,同时也带来了希望。研究表明,表观遗传调控子,尤其是组蛋白去乙酰化酶(hdacs),在肿瘤干细胞的发生发展中起到关键作用。不同于传统的基因突变引起肿瘤的发生或者耐药,hdacs通过催化组蛋白或者非组蛋白的去乙酰化来控制肿瘤细胞的命运,比如组蛋白h4k16位点乙酰化的丢失在多个肿瘤中被发现,而抑癌基因p53的乙酰化水平的改变也会影响其相关的信号通路的作用,从而引起肿瘤性质的改变。因此,hdac一直是药物研发中非常热门的靶点。
自2006年伏立诺他(saha)药物被美国fda批准上市以后,hdac抑制剂被广泛研发,截止2018年,总共有4种hdac抑制剂已经获批上市,主要用于血液癌的治疗,而且大量的药物处于临床研究的各个时期,为该靶点的研发积蓄力量。目前已经上市的hdac抑制剂都属于广谱型的抑制剂,能抑制多个hdac亚型的活性,在临床使用中出现类似的毒副作用,人们试图通过提高hdac抑制剂的选择性来降低其临床使用的毒性,但是收效甚微,最根本的原因是,hdac家族有11个亚型,该亚型的酶活口袋具有高度同源性,而且不同的亚型之间常常形成复合物发挥生物功能,使得研发高效低毒的hdac抑制剂带来了挑战。多肽类药物作为新的治疗工具,能有效抑制蛋白-蛋白相互作用,在抗肿瘤治疗中发挥着越来越重要的作用。与生物大分子类似,多肽类分子对于靶点也有较高的结合力与选择性,相对于小分子类药物具有更小的脱靶效应。而多肽在体内的代谢产物为氨基酸,最大限度地降低了毒性。因而,多肽药物具有良好的生物活性以及生物相容性。
目前已经上市的hdac多肽抑制剂为罗米地辛(fk228),它是一种环肽,对一类hdac家族的成员具有选择性的抑制效果,但是其在临床使用中仍然表现出与其他hdac家族类似的毒副作用,没有显著改善其临床使用的安全性。在2016年,watson等人研发了基于hdac1底物h4k16衍生的多肽抑制剂,该抑制剂偶联了能靶向hdac酶活口袋的基团羟肟酸从而表现出显著的hdac1酶活抑制效果。受限于该线性多肽的稳定性以及细胞穿膜能力,该多肽并没有进行后续的细胞生物学研究。
技术实现要素:
针对现有技术中的上述技术问题,本发明提供了一种靶向hdac的稳定多肽类抑制剂及其用途,所述的这种靶向hdac的稳定多肽类抑制剂及其用途要解决现有技术中的hdac多肽抑制剂治疗癌症的效果不佳,而且毒副作用比较大的技术问题。
本发明提供了一种靶向hdac的稳定多肽类抑制剂,其氨基酸序列结构如下:
上述的氨基酸序列结构式中,r代表精氨酸、a代表丙氨酸、e代表谷氨酸、i代表异亮氨酸、q代表谷氨酰胺、y代表酪氨酸、v代表颉氨酸。
本发明还提供了上述的一种靶向hdac的稳定多肽类抑制剂在制备抑制hdac家族酶活活性的药物中的用途。
本发明还提供了上述的一种靶向hdac的稳定多肽类抑制剂在制备抑制hdac1高表达的肿瘤干细胞的药物中的用途。
本发明还提供了上述的一种靶向hdac的稳定多肽类抑制剂在制备治疗人类卵巢畸胎瘤或者睾丸胚胎癌的药物中的用途。
在本发明中,我们基于hdac1的另一个特异性的底物h3k56设计多肽类的hdac抑制剂,因为该多肽底物为一段螺旋片段,能用稳定螺旋的方法进行稳定,进而提高其细胞穿膜能力、稳定性以及生物学功能。为了进一步提高该多肽的酶活抑制效果,我们将靶向酶活口袋的小分子羟肟酸与多肽共价偶联,本发明设计的多肽在细胞水平和动物水平均表现出非常良好的肿瘤抑制效果。本发明的这种多肽抑制剂能有效改善hdac小分子抑制剂在临床使用中的毒副作用,在小鼠动物模型中,该药物展现出显著的抗肿瘤效果以及良好的生物相容性。
本发明通过hdac酶活抑制实验、免疫印迹分析、荧光定量pcr和人源肿瘤异种移植模型等实验,证实该多肽可以很好的结合hdac蛋白,并能有限抑制肿瘤干细胞的增殖并且能显著影响hdac1相关的信号通路。此外该多肽可以抑制小鼠卵巢畸胎瘤和睾丸胚胎癌肿瘤的生长。本发明有益于解决传统hdac抑制剂的毒副作用,增加其临床使用的治疗窗口。
本发明采用了末端侧链-尾端连接手性二酸修饰多肽的方法来稳定靶向hdac的螺旋多肽。采用之前文献报道(natcommun.2016,7:11262.)的方法,将非天然氨基酸(携带有羟肟酸结构)引入该多肽序列的碳端,通过免疫印迹和免疫共沉淀分析证明该多肽可以有效与hdac家族蛋白结合,而且影响其生物活性。通过荧光定量pcr和对肿瘤细胞的总rna测序实验证明该多肽可以抑制hdac下游基因的表达。
本发明通过一系列免疫印迹的方法较为全面的研究该多肽对hdac相关信号通路的影响,通过细胞增殖实验、凋亡实验和细胞周期阻滞实验进一步证明本发明具有对hdac1和hdac6抑制作用和对肿瘤干细胞生长的抑制作用。两种肿瘤模型证明该多肽具有非常显著的抗肿瘤作用以及体内生物相容性。
本发明和已有技术相比,其技术进步是显著的。通过一系列的酶活抑制实验证明我们的多肽可以有效抑制hdac家族的不同成员,属于广谱类hdac抑制剂。通过细胞增殖、细胞凋亡、细胞周期阻滞实验证明我们的多肽可以有效抑制肿瘤干细胞的增殖。通过荧光定量pcr实验以及westernblot实验证明本发明的多肽可以抑制hdac相关信号通路。两种肿瘤干细胞的小鼠动物模型证明,我们的多肽具有较显著的体内抗肿瘤效果,而且该多肽在高剂量下对小鼠没有明显的组织损伤或者毒性,该研究成果为未来开发新颖的、更广安全治疗窗口的hdac抑制剂提供一种思路。
附图说明
图1为多肽抑制剂的研发设计图。
图2为多肽抑制剂的合成路线图。
图3为多肽16cyc-hxa的hplc分离图谱。
图4为多肽16cyc-hxa的质谱鉴定图谱。
图5为不同多肽抑制剂的圆二色谱(cd)图。
图6为本发明设计的不同多肽对hdac家族的酶活抑制效果图。图6a:稳定多肽抑制剂的设计示意图;图6b:多肽抑制库的设计和对hela核提取物的酶活抑制效果的筛选;图6c:不同多肽抑制剂在hdac不同家族蛋白中酶活抑制效果。
图7为不同的多肽抑制剂在肿瘤细胞中的穿透能力图谱。
图8为不同的多肽抑制剂在各种肿瘤细胞以及正常细胞的毒性图谱。
图9为多肽抑制剂对肿瘤干细胞中hdac底物乙酰化水平的影响。
图10为多肽抑制剂对肿瘤干细胞内hdac家族的蛋白的结合能力的图谱。
图11为在肿瘤干细胞中多肽抑制剂穿膜能力随时间变化的图谱。
图12为多肽抑制剂对人红细胞溶血能力的鉴定图谱。
图13为多肽抑制剂对肿瘤干细胞细胞膜破损能力的图谱。
图14为多肽抑制剂对hdac底物乙酰化水平的时间梯度图谱。
图15为多肽抑制剂对肿瘤干细胞中hdac相关信号通路的影响。
图16为多肽抑制剂对肿瘤细胞诱导凋亡的能力的图谱。
图17为多肽抑制剂对肿瘤干细胞细胞周期阻滞中的影响。
图18为rna-seq技术分析多肽抑制剂对肿瘤干细胞整个信号通路的影响。
图19为多肽抑制剂对人类卵巢畸胎瘤异种移植裸鼠模型中肿瘤体积的变化。
图20为多肽抑制剂对人类睾丸胚胎癌异种移植裸鼠模型中肿瘤体积的变化。
图21多肽药物处理后通过免疫组化实验检测瘤h3k56ac,h4k16ac,hdac1,hdac6,caspase-3等的表达情况。
图22为多肽抑制剂处理后小鼠的器官组织部位h&e染色结果。
图23为多肽抑制剂对正常老鼠运动能力的影响。
具体实施方式
下面结合附图对本发明进一步说明。
实施例1:多肽抑制剂的设计
本发明采用多肽-小分子共价偶联的方式设计hdac多肽抑制剂,如图1所示,我们选择hdac1选择性底物h3k56(组蛋白h3的第56个氨基酸赖氨酸侧链氨基被乙酰化修饰)附近的一段多肽序列(tvalreirryqk(ac)),因为该多肽的k56在一段螺旋的c端,适合于我们用“n-末端天冬氨酸策略”方法对多肽进行稳定而不影响c端k56对靶点的识别。为了进一步提高底物多肽对hdac的结合,我们将k56的氨基酸突变为羟肟酸结构,如图1a所示。
该结构被报道可以与hdac酶催化区域的锌离子进行螯合而起到hdac酶活抑制的效果。hdac抑制剂基本结构有三个部分组成:锌离子螯合区,连接区,结构识别区。我们根据基本结构设计的多肽抑制剂也分为三个部分,羟肟酸为锌离子螯合区,连接锌离子的含5元碳的脂肪链为连接区,多肽底物作为结构识别区。如图1a所示。我们试图将设计的多肽抑制剂用于抗肿瘤干细胞的肿瘤,达到选择性抑制肿瘤干细胞增殖的目的,如图1b所示。
为了进一步验证多肽序列的重要性,我们筛选了一系列的多肽序列,如图6所示(图6a:稳定多肽抑制剂的设计示意图;图6b:多肽抑制库的设计和对hela核提取物的酶活抑制效果的筛选),有不同长度的多肽序列,相同长度不同氨基酸排布的序列,并研究这些多肽序列对hdac酶活的抑制效果,我们发现,原始的多肽序列酶活效果最佳,将其命名为16cyc-hxa。
为了进一步验证我们的设计,我们合成了一些负对照多肽,如图6b(多肽抑制库的设计和对hela核提取物的酶活抑制效果的筛选)所示包括含有羟肟酸结合的线性多肽,命名为16lin-hxa,目的是研究关环对酶活效果的影响,设计不含有羟肟酸结构的环肽,命名为16ka,目的是研究羟肟酸的存在对酶活效果的影响。如图6c所示(图6c:不同多肽抑制剂在hdac不同家族蛋白中酶活抑制效果)我们发现线性多肽的酶活效果不如环肽,不带羟肟酸的环肽酶活效果非常微弱,暗示多肽序列和羟肟酸的共价结合才能最大限度的抑制酶活。考虑到后续的细胞生物学功能,我们选择了长肽:16cyc-hxa,16lin-hxa和16ka为代表进行抗肿瘤的研究。
实施例2:多肽抑制剂的合成制备
我们采用国际上通用的固相多肽合成技术(spps)对多肽进行合成。其合成路线如图2所示。
具体操作步骤:
(1)带羟肟酸的非天然氨基酸(fmox-hx-oh)的合成:该合成路线完全按照文献报道的方法进行。
(2)多肽的固相合成:称取100mg2-chlorotity-n-fmoc-hydroxylamine树脂于10ml接肽管中,加入二氯甲烷(dcm),鼓氮气溶胀10min。加入50%(v/v)吗啡啉的n,n-二甲基甲酰胺(dmf)溶液,鼓氮气30min×2,脱去fmoc保护基团。用dmf和dcm交替洗涤6遍树脂之后,将配好的⑴fmoc-hx-oh(5eq,0.4m,dmf)溶液,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯(hctu)(5eq,0.38m,dmf)溶液,n,n-二异丙基乙胺(dipea)(10eq)混匀后加入树脂中鼓氮气3h。抽掉反应液,按上述方法洗树脂后进行下一步操作。
(3)在树脂氨基酸的碳端接ala:先将树脂上的氨基酸的ally基团脱保护,用二甲基巴比妥酸(4eq)与树脂在氮气保护下加入四三苯基膦钯的dcm溶液(1eq),避光搅拌2h。此时,树脂上的碳端cooh游离,先加入hctu(5eq,0.38m,dmf)溶液,n,n-二异丙基乙胺(dipea)(10eq)混匀将树脂的cooh活化5分钟,加入碳端用酰胺基封住、氮端氨基游离的ala,在n2鼓吹下反应3小时,重复两遍使反应充分。抽掉反应液,按上述方法洗树脂后进行下一步操作。
(4)后续的氨基酸操作步骤按照传统的spps方法进行反应,即抽掉反应液,按上述方法洗树脂后进行下一步操作,即在用吗啡啉脱掉树脂上fmoc保护基团之后,将配制好(3)fmoc-gln(trt)-oh或者(4)fmoc-tyr(tbu)-oh或(5)fmoc-arg-(pdf)-oh或者(6)fmoc-arg-(pdf)-oh或者(7)fmoc-ile-oh或者(8)fmoc-glu(tbu)-oh或者(9)fmoc-arg-(pdf)-oh或者(10)fmoc-dap(alloc)-oh或者(11)fmoc-ala-oh或者(12)fmoc-val-oh或者(13)fmoc-asp(oh)-oallyl,hctu和dipea溶液混匀后加入树脂中鼓氮气1h;抽掉反应液,按上述方法洗涤树脂后进行下一步操作。钯催化脱保护:二甲基巴比妥酸(4eq)与树脂在氮气保护下加入四三苯基膦钯的dcm溶液(1eq),避光搅拌2h。再重复一次。反应完后用二乙基二硫代氨基甲酸钠(0.5%,dmf)溶液洗5遍,再用dmf和dcm交替洗十遍。分子内酰胺键关环:加入六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(pybop,3.2eq),hobt(3.2eq),n-甲基吗啡啉(nmm,3.2eq)溶液,鼓氮气2h,可重复一次。
(5)接下来用吗啡啉脱去fmoc保护基团,将三个分别配好的fmoc-arg(pdf)-oh(5eq,0.4m,dmf)溶液,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯(hctu)(5eq,0.38m,dmf)溶液,n,n-二异丙基乙胺(dipea)(10eq)混匀后依次加入树脂中各鼓氮气2h。
(6)多肽纯化:用三氟乙酸(tfa),三异丙基硅烷(tips)和h2o(v:v:v=9.5:0.25:0.25)剪切液将多肽从树脂上切下来,除去剪切液。用高效液相色谱进行纯化分离。(图3和图4)
实施例3:多肽二级结构的鉴定及对hdac酶活抑制效果的研究
对于每种肽,用圆二色谱测定在水中的构象,证明通过末端侧链-尾端连接手性二酸修饰多肽的方法稳定的多肽在水中(如图5a)和在20%的tfe的溶液(如图5b)中具有稳定α螺旋构象,其中tfe(三氟乙醇)具有促进螺旋的作用。hdac酶活抑制效果的鉴定,用商业购买的hdac酶活检测试剂盒进行操作,每次操作至少重复三遍,该酶活结果如图6b(多肽抑制库的设计和对hela核提取物的酶活抑制效果的筛选)和图6c(图6c:不同多肽抑制剂在hdac不同家族蛋白中酶活抑制效果),分别是在不同的hdac家族蛋白中的测量结果。
实施例4:多肽在不同肿瘤细胞中穿膜能力的评价
采用流式细胞实验和共聚焦显微镜研究用fitc标记的多肽抑制剂的穿膜能力。在细胞中分别加入10μm的fitc荧光标记多肽化合物,在37度培养箱孵育4小时。对于流式细胞实验,用0.25%胰酶消化细胞10min后,收集细胞,将细胞重悬到pbs中,通过流式细胞仪来检测多肽的穿膜能力。(图7a)对于共聚焦成像实验,将接种在confocal载破片上的细胞用fitc的多肽处理4小时以后,用甲醛固定细胞制片,在激光共聚焦显微镜下鉴定多肽在肿瘤细胞内的分布情况(图7b)。随后我们检测了多肽在不同时间点的细胞穿膜能力,证明多肽随着时间的延长,穿膜能力提高,但是到24小时,由于多肽对细胞的毒性原因,其细胞的荧光下降,如图8所示。
实施例5:多肽抑制剂对不同肿瘤细胞选择性杀伤作用的评价
多肽抑制剂选择性抑制hdac1高敏感的肿瘤干细胞的增殖,而对普通的肿瘤细胞抑制效果比较微弱,如图9所示。细胞活力通过mtt测定法(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylt-etrazoliumbromide))。细胞在96孔板中以4×103接种,用溶于培养基(5%血清)的多肽处理24h或者48小时,将mtt加入培养基孵育4h。然后加入dmso溶解沉淀物,采用酶标仪在490nm测定吸光度。其中未处理的细胞存活率为100%。
结果表明,多肽抑制剂仅仅对肿瘤干细胞pa-1和ntera-2细胞具有明显的细胞增殖抑制以及浓度依赖性。对hela和hek293t细胞基本没有毒副作用。
实施例6:多肽抑制剂能显著提高hdac1和hdac6的底物乙酰化水平的变化
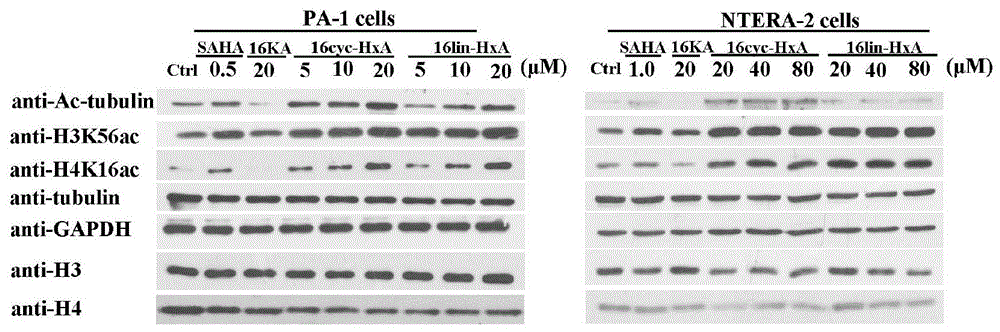
采用免疫印迹实验(wb)研究不同多肽在不同的给药浓度处理肿瘤干细胞后,对hdac底物乙酰化水平的影响。经过反复多次的wb实验发现,我们设计的多肽抑制剂可以明显引起和hdac1底物h3k56和h4k16,hdac6底物tubulin乙酰化水平的上调,如图10所示,揭示我们的多肽抑制剂可以明显抑制肿瘤干细胞中hdac1和hdac6的活性。随后我们检测了多肽处理pa-1细胞后,hdac底物乙酰化水平随时间的变化情况,发现在48小时内,底物乙酰化水平均很明显。如图14a和14b所示,图14b是用软件分析14a的结果的折线图,更直观的表现了底物乙酰化水平的变化情况。
实施例7:多肽抑制剂与内源性hdac家族的结合情况
在pa-1的细胞裂解液中,我们首先检测了hdac家族的不同蛋白的表达量,如图11a所示,证明hdac1的表达量很高。接着,我们采用链霉亲和素的磁珠进行pulldown实验,说明带biotin的多肽抑制剂可以与肿瘤干细胞内源的hdac家族结合,包括hdac1,hdac2,hdac3,hdac4,hdac5,hdac6,hdac8等,但是对nad+依赖的hdac家族结合较弱,说明了该多肽抑制剂仅能识别锌离子依赖的hdac家族。,如图11b。
实施例7:多肽抑制剂非特异性毒性的排除
采用ldh(乳酸脱氢酶)释放实验证明多肽抑制剂不会引起肿瘤干细胞的细胞膜损伤,说明该多肽不是通过裂解细胞膜来抑制肿瘤的(如图12所示,多肽抑制剂处理细胞4小时(图12a)和12小时(图12b)均不会明显引起细胞ldh的释放。溶血实验进一步说明,在高剂量的多肽处理下,小鼠不会出现溶血现象(如图13所示,a图是统计的溶血实验上清溶液的吸光值,该值越大说明溶血现象越严重,图b是溶血实验后得到的样品展示图)。这两个实验充分排除了该多肽抑制剂的非特异性毒性,暗示其高效低毒性。
实施例9:多肽抑制剂对肿瘤干细胞中hdac相关信号通路的影响。
采用免疫印迹和荧光定量pcr技术揭示,多肽抑制剂可以降低干性基因sox2的表达,提高分化基因的表达,上调p21和p53蛋白的表达等等。在wb水平,hdac的多肽抑制剂可以有效抑制lsd1的活性,导致其lsd1特异性底物h3k4甲基化水平的上调,暗示多肽抑制hdac1会影响lsd1的活性。如图15所示。为了进一步验证多肽抑制剂对肿瘤细胞的整个信号通路的影响,我们进行mrna芯片技术检测了多肽对肿瘤细胞整个基因表达的影响,发现该多肽对各个信号通路都有影响(图18a是rna微阵列分析加药组与不加药组的总基因表达的差异性,图18b是汇总的加药组和不加药组总上调基因和下调基因的个数,图18c是全基因组分析多肽抑制剂在肿瘤干细胞pa-1中影响的不同生物功能的信号通路。
实施例10:多肽16cyc-hxa对细胞凋亡和细胞周期阻滞的影响。
为了评价多肽药物是否引起caspase-3依赖的细胞凋亡,我们采用caspase-3凋亡试剂盒(如图16a)和fitcannexinv凋亡试剂盒检测(如图16b),发现多肽可以有效引起细胞caspase-3依赖的凋亡,通过wb实验进一步验证,加药组的细胞内激活的caspase-3的表达量会增加。(图16c)对于细胞周期实验,收集的细胞先用70%冰乙醇,在-20℃孵育过夜,pi染色,再用流式细胞仪分析。结果表明多肽16cyc-hxa可以明显引起肿瘤细胞发生g2期的细胞周期阻滞。(图17)
实施例11:多肽16cyc-hxa通过抑制hdac蛋白从而抑制体内肿瘤干细胞生长。
本项目采用裸小鼠为实验模型,通过在裸小鼠皮下注射培养pa-1肿瘤细胞悬浮液建立肿瘤模型,等肿瘤长到100-200mm3进行抑瘤实验。选取25只瘤体大小均一的裸小鼠分为5组,分别注射pbs、16cyc-hxa、16lin-hxa和16ka多肽抑制剂,小分子药物saha为正对照,每隔一天进行腹腔给药,给药剂量是50mg/kg,并且通过游标卡尺测量一次肿瘤的大小和小鼠的体重变化,统计数据。治疗21天后,通过对裸小鼠的组织切片以及免疫组化技术,检测多肽化合物的治疗效果和药物毒性。
结果如图19a、19b、19c所示,16cyc-hxa表现出高到80%的pa-1肿瘤抑制效果,而小鼠的体重没有显著变化(图19d)。为了进一步证明该多肽对小鼠没有神经毒性,我们采用了小鼠轮转实验,证明加药后不会影响小鼠的运转和记忆能力,如图23所示,图23a是轮转实验的小鼠体重不会出现明显变化,图23b是小鼠轮转实验的数据统计图。为了进一步验证多肽药物的抗肿瘤效果,我们采用另一种肿瘤干细胞ntera-2为小鼠模型,发现该多肽抑制剂对ntera-2细胞表现出非常显著的抑瘤效果(图20a、b、c所示)。而小鼠的体重没有显著变化(图20d)通过免疫组织化学也在体内证明该多肽能够引起hdac底物乙酰化的表达水平,再次证明该多肽是通过抑制hdac活性来发挥作用的(图21a是免疫组化检测小鼠肿瘤组织中h416乙酰化水平的变化,图21b是h3k56乙酰化水平的变化,图21c是caspase-3蛋白量的变化,图21d是hdac1蛋白量的变化,图21e是hdac6蛋白量的变化。另外,组织切片用苏木精和伊红(h&e)染色分析多肽的毒性,结果表明多肽对心,肝,脾,肺,肾和脑基本没有显著的毒性(图22不同药物处理后小鼠的器官组织的变化,图22a为pbs处理的组,图22b是saha处理组,图22c是16ka处理组,图22d是16cyc-hxa处理组,图22e是16lin-hxa处理组。小鼠轮转实验进一步证明该多肽对小鼠没有明显的神经毒性。
序列表
<110>北京大学深圳研究生院
<120>一种靶向hdac的稳定多肽类抑制剂及其用途
<130>jsp11901682
<160>1
<170>siposequencelisting1.0
<210>1
<211>12
<212>prt
<213>智人(homosapiens)
<400>1
thrvalalaleuarggluileargargtyrglnlys
1510
- 还没有人留言评论。精彩留言会获得点赞!
- 一种用于判断靶向单克隆抗体治疗肿瘤疗效的预测分子的制造方法与工艺
- 一种检测肺癌相关基因热点突变的试剂盒及其应用的制造方法与工艺
- 一种为肺癌提供离体个体化药物测试的方法及培养基与流程
- 一种谷胱甘肽/超声双重刺激响应纳米聚合物囊泡、其制备方法及应用与流程
- 一种多重刺激响应交联型聚合物纳米水凝胶、其制备方法及应用与流程
- 一种靶向CD47的免疫检查点抑制剂药物组合物及其制备方法与流程
- 具有淋巴靶向功能的生物自组装纳米晶注射剂及制备方法与流程
- 用于检测肝癌的基因标志物及其用途的制造方法与工艺
- 特异性shRNA筛选及其靶向Ang‑2基因抑制肺癌细胞的验证方法与流程
- 一种具有靶向功能仿病毒结构高分子囊泡及其制备与应用的制造方法与工艺
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺
- 一种线粒体靶向的粘度荧光探针及其制备方法和应用与制造工艺
- 一种兼具荧光可视、磁靶向和pH敏多功能的复合空心微球药物载体及其制备方法与制造工艺
- 一种用于修饰微泡的多肽以及靶向GBM的药物制剂的制造方法与工艺
- 白果内酯作为脑靶向增效剂在制备防治脑肿瘤药物的应用的制造方法与工艺
- 机器人辅助精确靶向穿刺软组织目标柔性针夹紧固定装置的制造方法