高耐热透明可溶联苯型聚酰亚胺薄膜及其制备方法与应用与流程
本发明涉及聚酰亚胺薄膜技术领域,尤其涉及一种高耐热透明可溶联苯型聚酰亚胺薄膜及其制备方法与应用。
背景技术:
近年来,随着微电子和光电子领域的发展,有机电致发光显示器(oled)、太阳能电池、液晶显示等光电材料越来越轻量化、超薄化、柔性化。光电器件制造过程中的电极薄膜沉积和退火处理等工序的加工温度高达400℃,远超商品透明聚合物的玻璃化转变温度。用于太阳能电池的空间柔性薄膜,其耐高低温性能直接决定了太阳能电站的使用寿命。因此,制备玻璃化转变温度超过400℃的透明聚合物材料意义重大。
聚酰亚胺(pi)耐热性优异,是耐热透明膜的首选材料。但传统的芳香族pi,一方面由于刚性的分子链和强烈的分子间作用力,既不能在有机溶剂中溶解,也不能在高温下熔融,导致加工困难,另一方面由于分子内及分子间形成的电荷转移络合物(ctc),使得薄膜的紫外-可见光吸收波长红移,透光率下降。通常通过引入柔性链节、含氟基团、脂环结构、大侧基或非共面结构可以改善pi溶解性和透明性,但往往导致热性能下降。因此,通过调控pi结构刚性,开发高耐热透明可溶pi薄膜具有重要意义。
以3,3',4,4'-联苯四羧酸二酐(bpda)作为酸酐单体制备的pi可以达到很高的耐热性,但往往不能兼顾溶解性和透光率。中国发明专利cn1976912b公开的高纯度bpda与4,4'-二氨基二苯基醚缩聚得到的pi薄膜在400nm处的透光率仅为30%。中国发明专利申请cn109021234a利用bpda制备了含炔基的pi预聚物,交联后的树脂玻璃化转变温度达430℃以上,但不再具备可溶性,无法进行二次加工,且固化后的薄膜为棕色,透光率差。以4,4'-(六氟异丙烯)二酞酸酐(6fda)作为酸酐单体的氟酐型pi透明性好,中国发明专利申请cn104017214a公开了一种基于三联苯结构的氟酐型pi膜,在450nm处透光率可达82%,但玻璃化转变温度降低到300℃以下。中国发明专利cn105906808a公开了一种联苯型可溶pi,其制备方法是将含叔丁基的多元醚二胺、bpda、间甲酚和异喹啉混合,于85℃搅拌反应12小时生成聚酰胺酸溶液,然后依次升温至120℃、150℃、220℃反应5小时、5小时、15小时制得聚酰亚胺溶液,其加热缩聚时间长达30多小时,不利于工业化生产,且pi的玻璃化转变温度仅为283℃。微波辅助加热有着内部加热、高效、副反应少等优点,采用微波加热替代传统聚合,可大幅缩短聚合时间,减少生产过程中的能量消耗,同时制得的pi性能较好。
由上可知,现有技术制备的pi存在耐热性与溶解性和透光率不能兼顾的问题。
技术实现要素:
针对现有pi存在的耐热性与溶解性和透光率不能兼顾的问题,本发明以带极性吊环的的三芳基甲烷类二胺与bpda缩聚,提供了一种高耐热透明可溶联苯型聚酰亚胺薄膜及其制备方法。
本发明的另一目的在于提供上述高耐热透明可溶联苯型聚酰亚胺薄膜的应用。
本发明采用3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷和3,3′,5,5′-四甲基-4,4′-二胺基-4″-氰基三苯甲烷两种带极性吊环的三芳基甲烷类二胺与刚性酸酐bpda缩聚,得到的联苯型pi由于极性吊环的引入,分子链扭曲,自由体积较大,有利于溶剂分子进入分子链间,具有好的溶解性;甲基的引入使c-n键相连的酰亚胺环和苯环形成了较大的扭转角,增加了单键的旋转位垒,赋予了主链较强的刚性,从而保持了联苯型pi优异的耐热性;扭转角的存在还破坏了酰亚胺环和苯环的共轭作用,不利于形成分子内和分子间的电荷转移,使制备的pi薄膜透明度较高。此外,本发明采用微波辅助加热代替常规加热制备联苯型聚酰亚胺,聚合时间从常规加热缩聚所需的30多小时缩短到20~60分钟,且制备的聚酰亚胺性能优异。
本发明目的通过如下技术方案实现:
一种高耐热透明可溶联苯型聚酰亚胺薄膜,具有如下分子结构通式:
其中ar为:
所述的高耐热透明可溶联苯型聚酰亚胺的制备方法,包括以下步骤:
(1)在氮气气氛下,将带极性吊环的三芳基甲烷类二胺单体、3,3',4,4'-联苯四羧酸二酐单体、极性非质子溶剂和催化剂混合,在室温下搅拌均匀,得到澄清溶液;所述的催化剂为异喹啉、三乙胺或吡啶中的一种;
(2)将所述步骤(1)得到的澄清溶液,通过微波辐射加热,在50~100℃辐射反应10~30分钟,然后升温至170~210℃辐射反应10~30分钟,得到联苯型聚酰亚胺溶液;
(3)将所述步骤(2)得到的联苯型聚酰亚胺溶液滴加到质量为其15~30倍的乙醇中,有纤维状沉淀析出,过滤除去有机溶剂,将沉淀干燥,得到纤维状联苯型聚酰亚胺;
(4)将所述步骤(3)得到的纤维状联苯型聚酰亚胺溶解在极性非质子溶剂中,控制固含量为10~15wt%,充分搅拌溶解后涂敷于洁净的二氧化硅玻璃板上,真空条件静置0.5~2小时,干燥,冷却,得到高耐热透明可溶联苯型聚酰亚胺薄膜。
为进一步实现本发明目的,优选地,步骤(1)中所述的带极性吊环的三芳基甲烷类二胺单体为3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷或3,3′,5,5′-四甲基-4,4′-二胺基-4″-氰基三苯甲烷;3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷的结构式为
优选地,步骤(1)中所述的带极性吊环的三芳基甲烷类二胺单体与3,3',4,4'-联苯四羧酸二酐单体的摩尔比为1:1~1.1。
优选地,所述的极性非质子溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮或间甲酚中的一种。
优选地,步骤(1)中所述的极性非质子溶剂的用量为带极性吊环的三芳基甲烷类二胺单体和3,3',4,4'-联苯四羧酸二酐单体总质量的4~9倍。
优选地,步骤(1)中所述的催化剂用量为带极性吊环的三芳基甲烷类二胺单体摩尔数的0.4~0.8倍。
优选地,步骤(1)中所述在室温下搅拌的时间为30~60分钟
优选地,步骤(3)中所述的干燥为将沉淀置于90~110℃下真空干燥8~12小时;步骤(4)中所述的干燥为在50~80℃干燥2~5小时,再升温至100~150℃干燥2~5小时,继续升温至180~220℃干燥2~5小时。
优选地,步骤(2)中所述的微波的频率为2.45ghz;微波的功率为200~400w。
本发明所述高耐热透明可溶联苯型聚酰亚胺薄膜在柔性显示和柔性太阳能电池上的应用。
与现有技术相比,本发明的优点在于:
1)本发明以带极性吊环的三芳基甲烷类二胺与bpda缩聚,获得了高耐热透明可溶联苯型聚酰亚胺薄膜,其玻璃化转变温度超过400℃,450nm处的透光率超过82%,易溶于大部分常规有机溶剂。
2)本发明采用微波辅助加热代替常规加热制备联苯型聚酰亚胺,缩聚时间只需20~60分钟,远低于常规加热缩聚所需的30多小时,且制备的聚酰亚胺性能优异。
附图说明
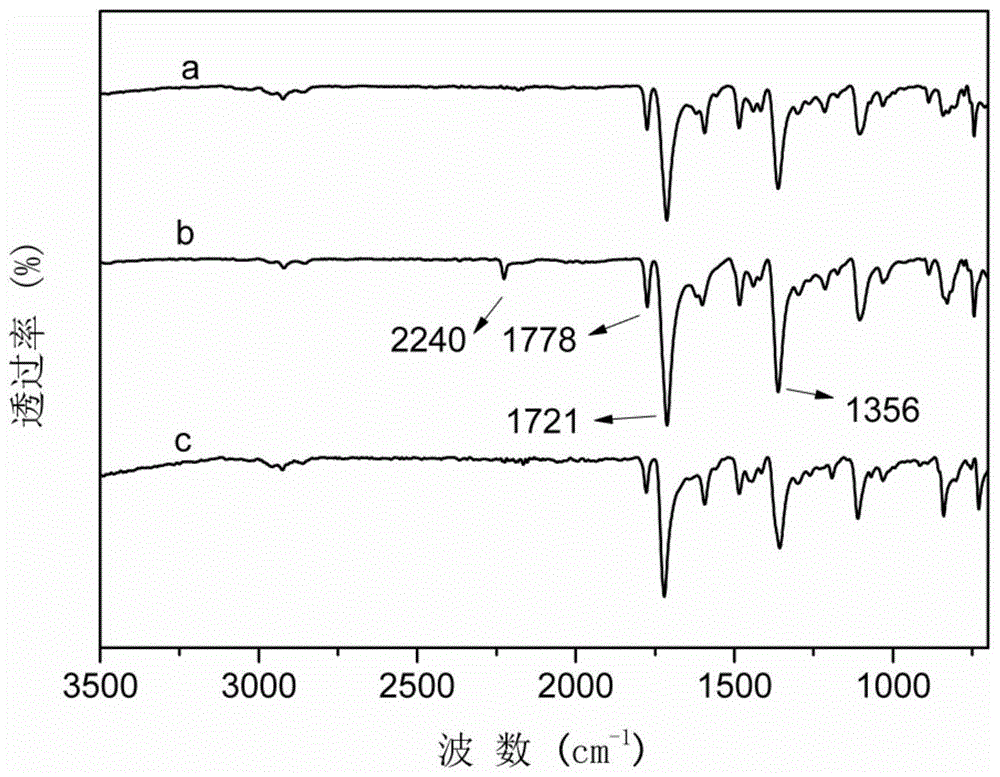
图1为实施例1、实施例5和对比例1所得到聚酰亚胺的红外光谱图,其中:a为实施例1所得聚酰亚胺薄膜产物,b为实例5所得聚酰亚胺薄膜产物,c为对比例1所得聚酰亚胺薄膜产物。
图2为实施例1所得聚酰亚胺(氘代二甲基亚砜作溶剂)核磁共振氢谱图。
图3为实施例5所得聚酰亚胺(氘代氯仿作溶剂)核磁共振氢谱图。
图4为对比例1所得聚酰亚胺(氘代氯仿作溶剂)核磁共振氢谱图。
具体实施方式
为更好地理解本发明,以下结合实施例对本发明的具体实施作进一步说明,但本发明的实施和保护范围不限于此。
实施例1
氮气保护下,在20ml反应管中加入0.6629g3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷(pydpm)、0.5884g3,3',4,4'-联苯四羧酸二酐(bpda)、7.51g间甲酚和0.13g异喹啉,然后封管,在室温下磁力搅拌60分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为400w,先在70℃辐射反应30分钟,再在200℃辐射反应30分钟,得到淡黄色粘稠状的聚酰亚胺溶液;
将该聚酰亚胺溶液滴加到177.8g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在90℃下真空干燥10小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为10%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置0.5小时,然后升温至50℃干燥5小时,再升温至100℃干燥5小时,继续升温至220℃干燥3小时,冷却后得到pydpm-bpda型聚酰亚胺薄膜。
该pydpm-bpda型聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1。将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得数均分子量为5.90×104g/mol,pdi=1.97,重复单元平均个数为100;采用动态热机械分析仪(dma)测得其玻璃化转变温度为400.3℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为82%。
该薄膜产物的红外(ir)光谱如附图1a所示,谱图上2960~2860cm-1处为甲基的c-h伸缩振动峰,1779cm-1和1720cm-1处分别对应酰亚胺环中c=o的不对称和对称伸缩振动的特征峰,1593cm-1(芳香环骨架振动),1337cm-1(c-n伸缩振动),1273cm-1(c-o伸缩振动);
该薄膜产物的核磁共振氢谱(400mhz,dmso-d6)如附图2.,各化学位移(ppm)归属为δ8.58(d,j=5.2hz,2h),8.48(s,2h),8.42(d,j=8.0hz,2h),8.14(d,j=8.0hz,2h),7.29(d,j=5.2hz,2h),7.15(s,4h),5.70(s,1h),2.10(s,12h);由上推知所得聚酰亚胺的分子结构式为:
实施例2
氮气保护下,在20ml反应管中加入0.6629g3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷(pydpm)、0.6061g3,3',4,4'-联苯四羧酸二酐(bpda)、11.42gn-甲基吡咯烷酮和0.08g三乙胺,然后封管,在室温下磁力搅拌40分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为300w,先在50℃辐射反应15分钟,再在210℃辐射反应20分钟,得到淡黄色粘稠状的聚酰亚胺溶液;
将该聚酰亚胺溶液滴加到383.1g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在90℃下真空干燥9小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为12%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置1小时,然后升温至70℃干燥2小时,再升温至100℃干燥4小时,继续升温至190℃干燥2小时,冷却后得到pydpm-bpda型聚酰亚胺薄膜。
该pydpm-bpda型聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1;将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为8.90×104g/mol,pdi=2.03,重复单元平均个数为151;采用动态热机械分析仪测得其玻璃化转变温度为401.6℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为82%;其红外谱图和核磁共振氢谱同实施例1。
实施例3
氮气保护下,在20ml反应管中加入0.6629g3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷(pydpm)、0.6355g3,3',4,4'-联苯四羧酸二酐(bpda)、5.19gn,n-二甲基甲酰胺和0.06g吡啶,然后封管,在室温下磁力搅拌50分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为200w,先在90℃辐射反应20分钟,再在190℃辐射反应15分钟,得到淡黄色粘稠状的聚酰亚胺溶液。
将该聚酰亚胺溶液滴加到131.1g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在100℃下真空干燥10小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为15%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置1.5小时,然后升温至80℃下干燥4小时,再升温至150℃干燥3小时,再升温至180℃干燥4小时,冷却后得到pydpm-bpda型聚酰亚胺薄膜。
该pydpm-bpda型聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1;将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为7.78×104g/mol,pdi=1.85,重复单元平均个数为132;采用动态热机械分析仪测得其玻璃化转变温度为400.9℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为82%;其红外谱图和核磁共振氢谱同实施例1。
实施例4
氮气保护下,在20ml反应管中加入0.6629g3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷(pydpm)、0.6473g3,3',4,4'-联苯四羧酸二酐(bpda)、7.86gn,n-二甲基乙酰胺和0.21g异喹啉,然后封管,在室温下磁力搅拌30分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为300w,先在100℃辐射反应10分钟,再在170℃辐射反应10分钟,得到淡黄色粘稠状的聚酰亚胺溶液。
将该聚酰亚胺溶液滴加到234.4g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在110℃下真空干燥12小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为15%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置2小时,然后升温至70℃干燥5小时,再升温至120℃干燥5小时,继续升温至200℃干燥2小时,冷却后得到pydpm-bpda型聚酰亚胺薄膜。
该pydpm-bpda型聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1;将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为6.25×104g/mol,pdi=2.13,重复单元平均个数为106;采用动态热机械分析仪测得其玻璃化转变温度为400.5℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为82%;其红外谱图和核磁共振氢谱同实施例1。
实施例5
氮气保护下,在20ml反应管中加入0.7110g3,3′,5,5′-四甲基-4,4′-二胺基-4″-氰基三苯甲烷(cytpm)、0.5885g3,3',4,4'-联苯四羧酸二酐(bpda)、5.20g间甲酚和0.15g异喹啉,然后封管,在室温下磁力搅拌30分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为300w,先在100℃辐射反应10分钟,再在200℃辐射反应15分钟,得到淡黄色粘稠状的聚酰亚胺溶液;
将该聚酰亚胺溶液滴加到166.3g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在100℃下真空干燥12小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为10%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置1小时,然后升温至60℃燥2小时,再升温至130℃干燥5小时,继续升温至210℃干燥2小时,冷却后得到cytpm-bpda型聚酰亚胺薄膜。
该cytpm-bpda聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1;将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为12.33×104g/mol,pdi=1.56,重复单元平均个数为201;采用动态热机械分析仪测得其玻璃化转变温度为411.3℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为85%;其红外光谱图如附图1b所示,谱图上2960~2860cm-1处为甲基的c-h伸缩振动峰,2225cm-1处为c≡n的伸缩振动峰,1775cm-1和1713cm-1处分别对应酰亚胺环中c=o的不对称和对称伸缩振动的特征峰,1602cm-1(芳香环骨架振动),1362cm-1(c-n伸缩振动),1296、1214cm-1(c-o伸缩振动);薄膜产物的核磁共振氢谱(600mhz,chloroform-d)如附图3,各化学位移(ppm)归属为δ8.20(s,2h),8.05(s,4h),7.59(d,j=8.1hz,2h),7.26(d,j=8.1hz,2h),6.87(s,4h),5.48(s,1h),2.09(s,12h)。由上推知所得聚酰亚胺的分子结构式为:
实施例6
氮气保护下,在20ml反应管中加入0.7110g3,3′,5,5′-四甲基-4,4′-二胺基-4″-氰基三苯甲烷、0.6179g3,3',4,4'-联苯四羧酸二酐(bpda)、9.30gn,n-二甲基甲酰胺和0.10g三乙胺,然后封管,在室温下磁力搅拌40分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为300w,先在80℃辐射反应30分钟,再在190℃辐射反应20分钟,得到淡黄色粘稠状的聚酰亚胺溶液;
将该聚酰亚胺溶液滴加到161.0g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在90℃下真空干燥8小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为12%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置0.5小时,然后升温至70℃干燥4小时,再升温至110℃干燥2小时,继续升温至200℃干燥3小时,冷却后得到cytpm-bpda型聚酰亚胺薄膜。
该cytpm-bpda型聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1;将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为10.74×104g/mol,pdi=1.56,重复单元平均个数为175;采用动态热机械分析仪测得其玻璃化转变温度为410.8℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为85%;其红外谱图和核磁共振氢谱同实施例5。
实施例7
氮气保护下,在20ml反应管中加入0.7110g3,3′,5,5′-四甲基-4,4′-二胺基-4″-氰基三苯甲烷、0.6355g3,3',4,4'-联苯四羧酸二酐(bpda)、8.08g间甲酚和0.11g吡啶,然后封管,在室温下磁力搅拌60分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为200w,先在50℃辐射反应20分钟,再在170℃辐射反应30分钟,得到淡黄色粘稠状的聚酰亚胺溶液;
将该聚酰亚胺溶液滴加到190.7g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在110℃下真空干燥9小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为12%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置1小时,然后升温至70℃干燥4小时,继续升温至120℃干燥3小时,再升温至200℃干燥5小时,冷却后得到cytpm-bpda型聚酰亚胺薄膜。
该cytpm-bpda型聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1;将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为10.12×104g/mol,pdi=1.93,重复单元平均个数为165;采用动态热机械分析仪测得其玻璃化转变温度为410.5℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为85%;其红外谱图和核磁共振氢谱同实施例5。
实施例8
氮气保护下,在20ml反应管中加入0.7110g3,3′,5,5′-四甲基-4,4′-二胺基-4″-氰基三苯甲烷、0.6473g3,3',4,4'-联苯四羧酸二酐(bpda)、12.22gn-甲基吡咯烷酮和0.21g异喹啉,然后封管,在室温下磁力搅拌60分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为300w,先在70℃辐射反应10分钟,再在210℃辐射反应10分钟,得到淡黄色粘稠状的聚酰亚胺溶液;
将该聚酰亚胺溶液滴加到413.7g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在110℃下真空干燥9小时得到纤维状聚酰亚胺;
取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为15%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置1小时,然后升温至50℃干燥5小时,再升温至100℃干燥3小时,继续升温至220℃干燥5小时,冷却后得到cytpm-bpda型聚酰亚胺薄膜。
该cytpm-bpda型聚酰亚胺薄膜可溶解在常见的极性溶剂中,具体如表1;将其溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为10.12×104g/mol,pdi=1.98,重复单元平均个数为143;采用动态热机械分析仪测得其玻璃化转变温度为410.3℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为85%;其红外谱图和核磁共振氢谱同实施例5。
对比例1
氮气保护下,在20ml反应管中加入0.6629g3,3′,5,5′-四甲基-4,4′-二胺基苯基-4″-吡啶甲烷(pydpm)、0.4362g均苯四甲酸二酐(pmda)、4.5g间甲酚和0.13g异喹啉,然后封管,在室温下磁力搅拌60分钟得到澄清溶液;将该反应管移至biotageinitiator+微波合成仪中,然后用频率为2.45ghz的微波,设置功率为400w,先在80℃辐射反应30分钟,再在200℃辐射反应10分钟,得到淡黄色粘稠状的聚酰亚胺溶液;
将该聚酰亚胺溶液滴加到171.8g乙醇中,有纤维状沉淀析出,静置过滤;将沉淀在90℃下真空干燥10小时得到纤维状聚酰亚胺;取部分干燥的纤维状聚酰亚胺溶解在n,n-二甲基乙酰胺中,控制固含量为10%,充分搅拌溶解后,均匀涂抹在干净的二氧化硅玻璃板上,抽真空静置1小时,然后升温至70℃干燥3小时,再升温至120℃干燥4小时,继续升温至200℃下干燥3小时,冷却后得到pydpm-pmda型聚酰亚胺薄膜。
该pydpm-pmda型聚酰亚胺薄膜可溶解于n,n-二甲基甲酰胺中,采用gpc法测得其数均分子量为5.23×104g/mol,pdi=1.92,重复单元的平均个数为102;采用动态热机械分析仪测得其玻璃化转变温度为473.3℃;采用紫外-可见分光光度计测得薄膜在450nm处的透光率为72%。薄膜产物的红外(ir)光谱如附图1c,谱图上2960~2860cm-1处为甲基的c-h伸缩振动峰,1780cm-1和1720cm-1处分别对应酰亚胺环中c=o的不对称和对称伸缩振动的特征峰,1593cm-1(芳香环骨架振动),1340cm-1(c-n伸缩振动),1272cm-1(c-o伸缩振动);该薄膜产物的核磁共振氢谱(400mhz,chloroform-d,ppm)如附图4,各化学位移归属为δ8.53(d,j=5.2hz,2h),8.47(s,2h),7.08(d,j=5.2hz,2h),6.89(s,4h),5.41(s,1h),2.07(s,12h).;由上推知所得聚酰亚胺的分子结构式为:
通过对比发现,本对比例制备的pydpm-pmda型聚酰亚胺薄膜的玻璃化转变温度为473.3℃;相对于实施例1~8得到的聚酰亚胺薄膜有一定的提高,但是在450nm处的透光率只有72%,明显低于实施例1~8得到的聚酰亚胺薄膜。
实施例1~8及对比例1制备的聚酰亚胺薄膜在不同溶剂中的溶解性能实验结果如表1。测试条件:将10mg的样品在室温或加热溶解在1ml溶剂中,静置24小时后观察其溶解状态。表1中“++”为室温可溶;“+”为加热至沸点可溶;“+-”为加热至沸点仍部分可溶。
从表1可知,实施例1~8的聚酰亚胺相比于对比例1有更好的溶解性能,不仅易溶于高沸点的n,n-二甲基乙酰胺、间甲酚、二甲基亚砜溶剂,而且在常见的低沸点溶剂二氯甲烷、三氯甲烷和四氢呋喃中也有优异的溶解性能。
表1实施例1~8及对比例1制备的聚酰亚胺薄膜的溶解性能
对比例2
发明专利cn104017214a公开了一种全芳香族浅色透明聚酰亚胺,其分子结构式如下:
本对比例2制备的聚酰亚胺树脂由动态热机械分析仪测得的玻璃化转变温度在280~310℃,薄膜在450nm处的透光率为82%。
通过对比发现,实施例1~8与对比例中的聚酰亚胺薄膜相比,在450nm处的透光率相近,但是在耐热性上,本发明制备的聚酰亚胺薄膜的玻璃化转变温度比对比例高约100℃。
此外,本发明采用微波辅助加热代替常规加热制备联苯型聚酰亚胺,缩聚时间只需20~60分钟,远低于常规加热缩聚的30多小时。
针对现有pi存在的耐热性与溶解性和透光率不能兼顾的问题,本发明以带极性吊环的的三芳基甲烷类二胺与3,3',4,4'-联苯四羧酸二酐单体在微波辐射下缩聚,只需20~60分钟,得到了一种高耐热透明可溶联苯型聚酰亚胺薄膜。其通过在三芳基甲烷类二胺上引入含吡啶基或苯氰基的极性吊环,该二胺单体与刚性二酐单体bpda缩聚形成的pi,分子链扭曲程度与自由体积较大,有利于溶剂分子进入分子链间,改善了溶解性;甲基的引入使c-n键相连的酰亚胺环和苯环形成了较大的扭转角,增加了单键的旋转位垒,赋予主链较大的刚性,从而保持了联苯型pi优异的耐热性;扭转角的存在还抑制了附近的酰亚胺环和苯环的共轭作用,不利于形成分子内和分子间的电荷转移,使制备的pi薄膜透明度较高。因此,本发明提供的联苯型聚酰亚胺兼具高耐热性、高透光率和优异的溶解性,其玻璃化转变温度在400℃以上,能适应较为苛刻的高温环境,薄膜在450nm处的透光率高于82%,且能溶于大部分有机溶剂,可以在较低的温度进行二次加工;在柔性显示和柔性太阳能电池领域潜力巨大。
本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!