一类新型苯并桶烯五蝶烯配体、过渡金属催化剂、制备方法及在乙烯聚合中的应用与流程
本发明属于催化剂合成技术领域以及高分子合成领域,具体涉及一类新型苯并桶烯五蝶烯配体、过渡金属催化剂、制备方法及在乙烯聚合中的应用。
背景技术:
从1995年以来,由于独特的链行走机理,α-二亚胺后过渡金属(钯)催化剂(j.am.chem.soc.1995,117,6414)发展到现在已经成为一类非常有用的乙烯聚合催化剂。但是该体系仍然有一些问题尚未解决,例如催化剂的易热失活、聚合物微结构调控较难等问题。为了解决这些问题,目前常用手段是调节催化剂的轴向空间位阻,或调控配体的电子效应。
目前α-二亚胺钯催化剂在催化乙烯聚合方面,虽然通过调控配体的空间效应与电子效应,可以得到高分子量(达数百万)的聚合物,但是支化度却普遍不高(<130/1000c),支化度一般随反应条件(温度,压力)变化几乎保持不变,且很少表现出活性聚合的特征以及催化剂温度耐受性一般在110℃以下。从而在某种程度上限制了聚合物的应用范围。
技术实现要素:
本发明的目的是提供一类新型苯并桶烯五蝶烯配体、过渡金属催化剂、制备方法及在乙烯聚合中的应用。本发明的催化剂在保证催化剂热稳定性的基础上,通过催化乙烯聚合可以得到具备超高分子量(mw最高可达200万)、超高支化度(最高可达250/1000c)特征的聚合物。
本发明首先提供一类新型苯并桶烯五蝶烯配体,结构式如通式(i)所示:
通式(i)中,r1表示oh、c1~c20的烷氧基,r2表示h、ch3、tbu(叔丁基),x表示cl、br、i、h、tbu(叔丁基)、ph(苯基)、c1~c20的烷氧基,其中r2和x处于邻位-或间位-。
本发明还提供一类新型苯并桶烯五蝶烯配体的制备方法,包括:
将结构为(a)的二酮化合物、结构为(b)的苯胺化合物和催化剂在25-150℃下搅拌6小时-7天,得到通式(i)所述的一类新型苯并桶烯五蝶烯配体。
优选的是,所述的结构为(a)的二酮化合物和结构为(b)的苯胺化合物的摩尔比为1:n,其中n≥2。
优选的是,所述的催化剂为对甲苯磺酸一水合物、甲酸或者乙酸。
本发明还提供一类过渡金属催化剂,结构式如通式(ii)所示:
通式(ii)中,r1表示oh、c1~c20的烷氧基,r2表示h、ch3、tbu(叔丁基),x表示cl、br、i、h、tbu(叔丁基)、ph(苯基)、c1~c20的烷氧基,其中r2和x处于邻位-或间位-。
本发明还提供一类过渡金属催化剂的制备方法,包括:
将通式(i)所述的一类新型苯并桶烯五蝶烯配体和[cod]pdmecl(cod=1,5-环辛二烯)溶解于溶剂中,得到的混合物在20~50℃下搅拌3-30天,得到结构式如通式(ii)的一类过渡金属催化剂。
优选的是,所述的通式(i)所述的一类新型苯并桶烯五蝶烯配体和[cod]pdmecl的摩尔比为1:1。
优选的是,所述的溶剂为二氯甲烷或氯仿。
本发明还提供上述一类过渡金属催化剂在乙烯聚合中的应用。
优选的是,所述的应用方法包括:
将与高压气体管线连接的玻璃压力反应器干燥,然后将玻璃压力反应器调节至0-130℃,在惰性气氛下将溶剂和nabarf加入到反应器中,然后将上述一类过渡金属催化剂溶解在溶剂中通过注射器注入到聚合体系中,在快速搅拌下,通入乙烯并保持在8-20atm,30-480分钟后,排空压力反应器,加入酸性甲醇或酸性乙醇溶液淬灭聚合反应,得到聚合物。
本发明的有益效果
本发明提供一类新型苯并桶烯五蝶烯配体、过渡金属催化剂、制备方法及在乙烯聚合中的应用,本发明采用苯并桶烯骨架衍生物及五蝶烯胺类衍生物合成了一类新型的苯并桶烯五蝶烯配体,该新型配体可在一定范围内改变自身结构从而调节其性质,然后使用该类新型配体合成了一类新型α-二亚胺后过渡金属催化剂,使用苯并桶烯五蝶烯配体与金属络合,调控电子效应及金属轴向位阻。
本发明的催化剂的优点:a.该催化剂可在反应温度为30℃时实现乙烯的活性聚合,即可以在很大范围内调节聚合物的分子量,保证200支化度的情况下,mw可达200万。b.该催化剂可在非常高温度(130℃)下仍有较高活性,催化剂耐高温性强。c.该催化剂催化乙烯聚合可以得到超高支化度(250/1000c)的聚乙烯。d.该催化剂催化乙烯聚合在聚合温度升高时,所得聚合物的支化度降低。
另外,本发明高分子量高支化度的聚合物在某种程度上可以作为聚烯烃弹性体使用。聚烯烃弹性体不但具有高弹性、耐老化、耐油性等各项优异性能,同时又具备普通塑料加工方便、加工方式广泛的特点,既简化加工过程,又降低加工成本,是一种更具人性化的新型合成聚烯烃材料。
附图说明
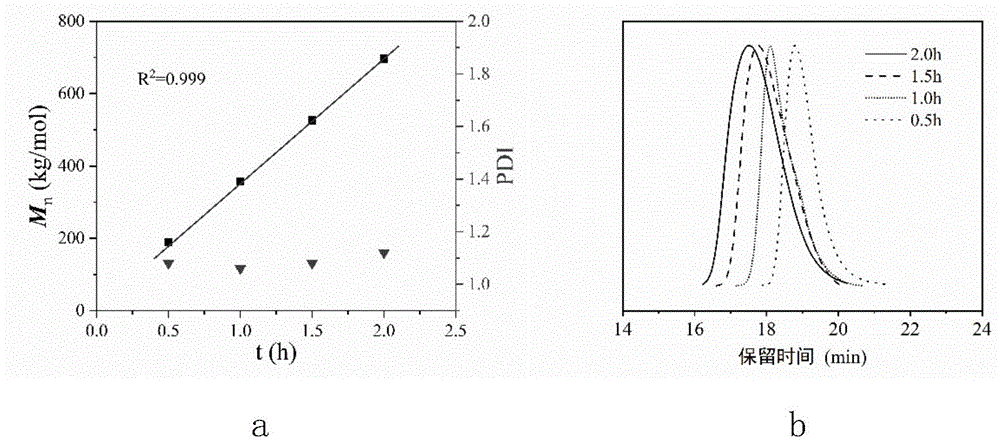
图1为本发明表3中条目11-14所得聚合物的数均分子量及分子量分布关于时间图(a图)和高温凝胶色谱图(b图)。
图2为本发明实施例4制备得到的催化剂的单晶衍射图。
图3为本发明实施例4制备得到的催化剂的核磁共振氢谱图。
图4为表3中条目1的聚合物的核磁共振氢谱图。
具体实施方式
本发明首先提供一类新型苯并桶烯五蝶烯配体,结构式如通式(i)所示:
通式(i)中,r1表示oh、c1~c20的烷氧基,优选为oh或och3,r2表示h、ch3、tbu(叔丁基),x表示cl、br、i、h、tbu(叔丁基)、ph(苯基)、c1~c20的烷氧基,优选为cl、br、i、h、tbu(叔丁基)或och3,其中r2和x处于邻位-或间位-。
本发明还提供一类新型苯并桶烯五蝶烯配体的制备方法,包括:
将结构为(a)的二酮化合物、结构为(b)的苯胺化合物溶于有机溶剂中,所述的有机溶剂优选为甲苯、二甲苯、氯苯、二氯甲烷、氯仿或者乙腈,然后加入催化剂在25-150℃下搅拌6h-7天,优选为2-5天,冷却至室温,旋蒸蒸发溶剂直至出现黄色固体,加入过量的甲醇或乙醇析出产品,过滤分离黄色固体,用甲醇或乙醇洗涤三次并在真空下干燥得到通式(i)所述的一类新型苯并桶烯五蝶烯配体。所述结构为(a)的二酮化合物和结构为(b)的苯胺化合物的摩尔比为1:n,其中n≥2,更优选为1:1-100;最优选为1:1-10。其中n越大,反应时间越短,且可提高产物转化;所述的催化剂优选为对甲苯磺酸一水合物、甲酸或者乙酸。
反应过程如下:
本发明所述的结构为(a)的二酮化合物的制备方法可以参考文献mondal,r.;shah,b.k.;neckers,d.c.,photogenerationofheptaceneinapolymermatrix.j.am.chem.soc.2006,128,9612-9613.以及zhong,l.;du,c.;liao,g.;liao,h.;zheng,h.;wu,q.;gao,h.,effectsofbackbonesubstituentandintra-ligandhydrogenbondinginteractiononethylenepolymerizationswithα-diiminenickelcatalysts.j.catal.2019,375,113-123.
本发明所述的结构为(b)的苯胺化合物的制备方法可以参考文献liao,y.;zhang,y.;cui,l.;mu,h.;jian,z.,pentiptycenylsubstituentsininsertionpolymerizationwithα-diiminenickelandpalladiumspecies.organometallics2019,38,2075-2083。
本发明还提供一类过渡金属催化剂,结构式如通式(ii)所示:
通式(ii)中,r1表示oh、c1~c20的烷氧基,r2表示h、ch3、tbu,x表示cl、br、i、h、tbu(叔丁基)、ph(苯基)、c1~c20的烷氧基,其中r2和x处于邻位-或间位-。
本发明还提供一类过渡金属催化剂的制备方法,包括:
将通式(i)所述的一类新型苯并桶烯五蝶烯配体和[cod]pdmecl(cod=1,5-环辛二烯)溶解于溶剂中,所述的溶剂优选为二氯甲烷或氯仿,得到的混合物在20~50℃下搅拌3-30天,优选3-10天,更优选3-5天,优选旋蒸蒸发溶剂,用正己烷或乙醚和二氯甲烷或氯仿重结晶,过滤分离固体,用己烷或乙醚洗涤三次并在真空下干燥,得到结构式如通式(ii)的一类过渡金属催化剂。所述的通式(i)所述的一类新型苯并桶烯五蝶烯配体和[cod]pdmecl的摩尔比为1:1。
本发明还提供上述一类过渡金属催化剂在乙烯聚合中的应用。
按照本发明,所述的应用包括:
将与高压气体管线连接的玻璃压力反应器干燥,然后将玻璃压力反应器调节至0-130℃,在惰性气氛下将溶剂和nabarf加入到反应器中,然后将上述一类过渡金属催化剂溶解在溶剂中通过注射器注入到聚合体系中,在快速搅拌下(750转以上),通入乙烯并保持在8-20atm,30-480分钟后,排空压力反应器,加入酸性甲醇或酸性乙醇溶液淬灭聚合反应,得到聚合物。
下面结合具体实施例对本发明做进一步详细的说明。
实施例1苯并桶烯五蝶烯配体的合成
1、4-甲氧基五蝶烯苯胺的合成
将4-羟基五蝶烯苯胺(3g,6.50mmol)溶解于100ml的二甲基甲酰胺中,在氮气气氛下加入氢化钠(468mg,19.5mmol),搅拌直至没有气泡生成,再加入碘甲烷(0.6ml,9.75mmol)。氮气气氛搅拌1-10天。倒入200ml水中,使用100ml二氯甲烷萃取3-20次,分液取有机层,无水硫酸镁干燥10min-3h,过滤取液体,旋蒸蒸发溶剂直至出现黄色固体,过滤分离黄色固体并在真空下干燥得到黄色固体产物(2.60g,84.10%收率)。
1hnmr(500mhz,298k,cdcl3,7.26ppm):δ=7.27-7.36(m,8h,aryl-h),6.98-6.88(m,8h,aryl-h),5.67(s,2h,chph2),5.39(s,2h,chph2),3.85(s,3h,och3)ppm.
2、苯并桶烯五蝶烯配体的合成
将上述4-甲氧基五蝶烯苯胺(6.5g,13.67mmol),苯并桶烯(1.3g,5.50mmol)和对甲苯磺酸(10mg)的甲苯(250ml)溶液在145℃下搅拌回流并保持72小时,冷却至室温,旋蒸蒸发溶剂直至出现黄色固体,加入过量的乙醇析出产品,过滤分离黄色固体,用乙醇洗涤三次并在真空下干燥得到黄色固体产物(8.00g,50.92%收率)。
1hnmr(500mhz,298k,dmso-d6,2.50ppm):δ=7.61-7.57(d,4h,aryl-h),7.49-7.43(d,8h,aryl-h),7.31-7.12(m,16h,aryl-h),7.07-7.03(m,4h,aryl-h),6.97-6.90(m,8h,aryl-h),5.94(s,4h,chph2),5.11(s,4h,chph2),5.09(s,2h,chph2),4.01(s,3h,och3)ppm。
实施例2叔丁基苯并桶烯五蝶烯配体的合成
将4-甲氧五蝶烯苯胺(6.5g,13.67mmol),叔丁基苯并桶烯(1.9g,5.50mmol)和对甲苯磺酸(10mg)的甲苯(250ml)溶液在145℃下搅拌回流并保持72小时,冷却至室温,旋蒸蒸发溶剂直至出现黄色固体,加入过量的乙醇析出产品,过滤分离黄色固体,用乙醇洗涤三次并在真空下干燥得到黄色固体产物(4.31g,62.15%收率)。
1hnmr(500mhz,298k,dmso-d6,2.50ppm):δ=7.59-7.53(d,2h,aryl-h),7.47-7.41(d,4h,aryl-h),7.38-7.31(m,10h,aryl-h),7.16-7.11(m,4h,aryl-h),7.07-6.98(m,4h,aryl-h)6.91-6.80(m,10h,aryl-h),6.77-6.70(d,2h,aryl-h),5.88(d,4h,chph2),5.34(s,2h,chph2),5.06(d,2h,chph2),4.07(s,3h,och3),1.41(d,2h,c(ch3)3)ppm。
实施例3桶烯邻位a式甲基五蝶烯配体及桶烯邻位a式甲基五蝶烯羟基配体的合成
1、将1,8-二甲基蒽(7.51g,36.4mmol),苯醌(1.94g,18mmol),四氯苯醌(8.85g,36mmol)的thf(500ml)溶液加热回流1-30天,冷却后过滤,乙醚(100ml)洗3-5次,滤渣真空干燥,得黄色固体产物顺-邻四甲基五蝶烯对苯二醌(8.18g,87.92%收率)。
2、将顺-邻四甲基五蝶烯对苯二醌(8.18g,15.8mmol)同盐酸羟胺(2.2g,31.7mmol)水(50ml)浓盐酸(37%2.3ml)溶于thf(500ml),溶液加热回流1-30天,冷却后旋蒸去除溶剂。二氯甲烷(200ml)溶解固体,水洗(100ml)萃取三次,其中萃取分液取有机层,无水硫酸镁干燥10min–5h,旋蒸去除溶剂,真空干燥,以乙酸乙酯:石油醚为1:1-50的比例过硅胶柱得黄色固体产物顺-邻四甲基3、五蝶烯对苯醌肟(3.36g,40%收率)。
3、将上步黄色固体产物顺-邻位a式四甲基五蝶烯对苯醌肟(8.41g,15.8mmol)同钯/碳(1g)分散于thf(500ml)中,缓慢滴加水合肼(4.2ml,86.9mmol),加热回流3h-5天,冷却后过滤取固体,将固体分散到二氯甲烷(200ml)中,搅拌1h-3天,过滤取液体,旋蒸去除溶剂真空干燥得黄色固体产物顺-邻位a式四甲基五蝶烯对羟基苯胺(5.73g,70.05%收率)。
4、将上述顺-邻位a式四甲基五蝶烯对羟基苯胺(5.2g,10.00mmol),苯并桶烯(0.9g,4.00mmol)和对甲苯磺酸(10mg)的甲苯(250ml)溶液在145℃下搅拌回流并保持72小时,冷却至室温,旋蒸蒸发溶剂直至出现黄色固体,加入过量的乙醇析出产品,过滤分离黄色固体,用乙醇洗涤三次并在真空下干燥得到黄色固体产物桶烯邻位a式甲基五蝶烯羟基配体(3.70g,75.00%收率)。
5、将上述顺-邻位a式四甲基五蝶烯对羟基苯胺(5.73g,11.07mmol)溶解于100ml的二甲基甲酰胺中,在氮气气氛下加入氢化钠(797mg,33.2mmol),搅拌直至没有气泡生成,再加入碘甲烷(1.0ml,16.7mmol)。氮气气氛搅拌1-10天。倒入200ml水中,使用100ml二氯甲烷萃取3-20次,分液取有机层,无水硫酸镁干燥10min-3h,过滤取液体,旋蒸蒸发溶剂直至出现黄色固体,过滤分离黄色固体并在真空下干燥得到黄色固体产物顺-邻位a式四甲基五蝶烯对甲氧基苯胺(4.95g,84.10%收率)。
6、将上述顺-邻位a式四甲基五蝶烯对甲氧基苯胺(7.09g,13.67mmol),苯并桶烯(1.3g,5.50mmol)和对甲苯磺酸(10mg)的甲苯(250ml)溶液在145℃下搅拌回流并保持72小时,冷却至室温,旋蒸蒸发溶剂直至出现黄色固体,加入过量的乙醇析出产品,过滤分离黄色固体,用乙醇洗涤三次并在真空下干燥得到黄色固体产物桶烯邻位a式甲基五蝶烯配体(8.00g,50.92%收率)。
实施例4苯并桶烯五蝶烯钯甲基氯(对应表格1中的14和表格2中的1)
将实施例1制备的苯并桶烯五蝶烯配体(2.00g,1.74mmol)和[cod]pdmecl(462mg,1.74mmol)(cod=1,5-环辛二烯)的混合物在20ml二氯甲烷中25℃下搅拌72小时。反应完成后减压蒸发溶剂得到红棕色固体,然后过滤并用二氯甲烷/己烷重结晶得到纯化合物,为红棕色固体(2.00g,82.10%收率)单晶衍射图如图2所示,核磁氢谱图如图3所示。
实施例5苯并桶烯五蝶烯钯甲基氯(对应表格2中的6)
将实施例2制备的叔丁基苯并桶烯五蝶烯配体(2.00g,1.59mmol)和[cod]pdmecl(422mg,1.59mmol)(cod=1,5-环辛二烯)的混合物在20ml二氯甲烷中25℃下搅拌72小时。反应完成后减压蒸发溶剂得到红棕色固体,然后过滤并用二氯甲烷/己烷重结晶得到纯化合物,为红棕色固体(1.70g,75.47%收率)。
实施例6桶烯邻位a式甲基五蝶烯羟基钯甲基氯(对应表格1中的6)
将实施例3制备的桶烯邻位a式甲基五蝶烯羟基配体(2.15g,1.74mmol)和[cod]pdmecl(462mg,1.74mmol)(cod=1,5-环辛二烯)的混合物在20ml二氯甲烷中25℃下搅拌72小时。反应完成后减压蒸发溶剂得到红棕色固体,然后过滤并用二氯甲烷/己烷重结晶得到纯化合物,为红棕色固体(1.97g,81.23%收率)。
实施例7桶烯邻位a式五蝶烯钯甲基氯(对应表格1中的19)
将实施例3制备的桶烯邻位a式五蝶烯配体(2.20g,1.74mmol)和[cod]pdmecl(462mg,1.74mmol)(cod=1,5-环辛二烯)的混合物在20ml二氯甲烷中25℃下搅拌72小时。反应完成后减压蒸发溶剂得到红棕色固体,然后过滤并用二氯甲烷/己烷重结晶得到纯化合物,为红棕色固体(1.97g,80.21%收率)。
实施例8表格1中1和表格2中7的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,制备桶烯五蝶烯羟基配体所用的原料为对羟基苯胺和苯并桶烯,制备催化剂所用原料为桶烯五蝶烯羟基配体,最终得到产物收率为79.36%。
实施例9表格1中2的催化剂的制备
制备条件和步骤同实施例3和6,不同之处在于制备配体所用的最初始原料为2,7-二甲基蒽,最终得到产物收率为77.28%。
实施例10表格1中3的催化剂的制备
制备条件和步骤同实施例9,不同之处在于最终得到产物收率为77.28%。
实施例11表格1中4的催化剂的制备
制备条件和步骤同实施例3和6,不同之处在于制备配体所用的最初始原料为2,6-二甲基蒽,最终得到产物收率为80.18%。
实施例12表格1中5的催化剂的制备
制备条件和步骤同实施例11,不同之处在于,最终得到产物收率为75.14%。
实施例13表格1中7的催化剂的制备
制备条件和步骤同实施例3和6,不同之处在于最终得到产物收率为79.78%。
实施例14表格1中8的催化剂的制备
制备条件和步骤同实施例3和6,不同之处在于制备配体所用的最初始原料为2,7-二叔丁基蒽,最终得到产物收率为71.45%。
实施例15表格1中9的催化剂的制备
制备条件和步骤同实施例14,不同之处在于,最终得到产物收率为79.63%。
实施例16表格1中10的催化剂的制备
制备条件和步骤同实施例3和6,不同之处在于制备配体所用的最初始原料为2,6-二叔丁基蒽,最终得到产物收率为83.45%。
实施例17表格1中11的催化剂的制备
制备条件和步骤同实施例16,不同之处在于最终得到产物收率为77.13%。
实施例18表格1中12的催化剂的制备
制备条件和步骤同实施例3和6,不同之处在于制备配体所用的最初始原料为1,8-二叔丁基蒽,最终得到产物收率为81.75%。
实施例19表格1中13的催化剂的制备
制备条件和步骤同实施例3和6,不同之处在于制备配体所用的最初始原料为1,8-二叔丁基蒽,最终得到产物收率为75.35%。
实施例20表格1中15的催化剂的制备
制备条件和步骤同实施例9,不同之处在于苯胺对位为甲氧基,最终得到产物收率为81.33%。
实施例21表格1中16的催化剂的制备
制备条件和步骤同实施例10,不同之处在于苯胺对位为甲氧基,最终得到产物收率为80.73%。
实施例22表格1中17的催化剂的制备
制备条件和步骤同实施例11,不同之处在于苯胺对位为甲氧基,最终得到产物收率为81.37%。
实施例23表格1中18的催化剂的制备
制备条件和步骤同实施例12,不同之处在于苯胺对位为甲氧基,最终得到产物收率为80.15%。
实施例24表格1中20的催化剂的制备
制备条件和步骤同实施例13,不同之处在于苯胺对位为甲氧基,最终得到产物收率为83.35%。
实施例25表格1中21的催化剂的制备
制备条件和步骤同实施例14,不同之处在于苯胺对位为甲氧基,最终得到产物收率为82.17%。
实施例26表格1中22的催化剂的制备
制备条件和步骤同实施例15,不同之处在于苯胺对位为甲氧基,最终得到产物收率为81.71%。
实施例27表格1中23的催化剂的制备
制备条件和步骤同实施例16,不同之处在于苯胺对位为甲氧基,最终得到产物收率为80.73%。
实施例28表格1中24的催化剂的制备
制备条件和步骤同实施例17,不同之处在于苯胺对位为甲氧基,最终得到产物收率为73.36%。
实施例29表格1中25的催化剂的制备
制备条件和步骤同实施例18,不同之处在于苯胺对位为甲氧基,最终得到产物收率为82.32%。
实施例28表格1中26的催化剂的制备
制备条件和步骤同实施例19,不同之处在于苯胺对位为甲氧基,最终得到产物收率为86.93%。
实施例29表格2中2的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为邻-二碘代苯并桶烯最终得到产物收率为81.82%。
实施例30表格2中3的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为邻-二溴代苯并桶烯最终得到产物收率为87.34%。
实施例31表格2中4的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为邻-二氯代苯并桶烯最终得到产物收率为86.31%。
实施例32表格2中5的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为邻-二甲氧基苯并桶烯最终得到产物收率为89.11%。
实施例33表格2中8的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为间-二碘代苯并桶烯和对羟基五蝶烯苯胺,最终得到产物收率为81.03%。
实施例34表格2中9的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为间-二溴代苯并桶烯和对羟基五蝶烯苯胺,最终得到产物收率为80.13%。
实施例35表格2中10的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为间-二氯代苯并桶烯和对羟基五蝶烯苯胺,最终得到产物收率为81.41%。
实施例36表格2中11的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为间-二甲氧基苯并桶烯和对羟基五蝶烯苯胺,最终得到产物收率为81.48%
实施例37表格2中12的催化剂的制备
制备条件和步骤同实施例1和实施例4,不同之处在于,所用的原料为间-二叔丁基苯并桶烯和对羟基五蝶烯苯胺,最终得到产物收率为87.72%。
应用实施例1
首先将与高压气体管线连接的350ml玻璃压力反应器在90℃下真空干燥至少1小时。然后将反应器调节至30℃,在惰性气氛下将98ml甲苯和7.5μmolnabarf加入到反应器中,然后将5μmol的钯催化剂溶解在2ml二氯甲烷或氯仿中通过注射器注入到聚合体系中。在快速搅拌下(750转以上),通入乙烯并保持在8atm。30分钟后,排空压力反应器,加入大量酸性甲醇或酸性乙醇(5%以上的盐酸醇溶液)溶液淬灭聚合反应,过滤聚合物,并在真空烘箱中干燥至恒重。如表1所示:
表1.不同钯催化剂(改变取代基r1、r2)对乙烯聚合的影响
表1所有数据至少是基于两条平行试验得出的结果(除非另有说明)。活性:以106gmol-1h-1为单位。mw、mw/mn:分别为重均分子量、聚合物分散性指数,150℃下,在1,2,4-三氯苯中通过gpc测定,相对于聚苯乙烯标准物。支化度=每1000个碳支化的个数,由核磁共振氢谱测定。
注:r2处于间位a:
r2处于间位c:
r2处于邻位a:
表1说明:当控制催化剂取代基x不变,改变取代基r1及r2时,在相同的聚合条件下(时间、温度、压力以及助催化剂浓度一致),r1如果是甲氧基、相比于羟基拥有更高的活性、分子量以及支化度。r2在相同位置的时候,在同等聚合条件下(时间、温度、压力以及助催化剂浓度一致),其空间位阻越大(叔丁基>甲基>氢),那么聚合活性、聚合物分子量和支化度越高。其中当r1=och3,r2=tbu处于邻位a(条目25)时,实现了超高的支化度(达250/1000c)。
应用实施例2
首先将与高压气体管线连接的350ml玻璃压力反应器在90℃下真空干燥至少1小时。然后将反应器调节至30℃,在惰性气氛下将98ml甲苯和7.5μmolnabarf加入到反应器中,然后将5μmol的钯催化剂溶解在2ml二氯甲烷(或氯仿)中通过注射器注入到聚合体系中。在快速搅拌下(750转以上),通入乙烯并保持在8atm。30分钟后,排空压力反应器,加入大量酸性甲醇或酸性乙醇(5%以上的盐酸醇溶液)溶液淬灭聚合反应,过滤聚合物,并在真空烘箱中干燥至恒重。如表2所示:
表2.不同钯催化剂(改变取代基r1、x)对乙烯聚合的影响
表2所有数据至少是基于两条平行试验得出的结果(除非另有说明)。活性:以106gmol-1h-1为单位。mw、mw/mn:分别为重均分子量、聚合物分散性指数,150℃下,在1,2,4-三氯苯中通过gpc测定,相对于聚苯乙烯标准物。支化度=每1000个碳支化的个数,由核磁共振氢谱测定。
注:条目1:钯催化剂(r1=och3,r2=h,x=i
表2说明:当控制催化剂r2不变,改变取代基r1和x时,在相同的聚合条件下(时间、温度、压力以及助催化剂浓度一致),r1如果是甲氧基、相比于羟基拥有更高的活性、分子量以及支化度。除此之外,在相同的聚合条件下(时间、温度、压力、助催化剂浓度一致),x如果为吸电子基(i,br,cl)相比其为供电子基团(och3,tbu)有着更高的活性、分子量及支化度。其中,当r1=och3,r2=h,x=i(邻位)时,实现了超高支化度(达250/1000c)。
应用实施例3
首先将与高压气体管线连接的350ml玻璃压力反应器在90℃下真空干燥至少1小时。然后将反应器调节至0-130℃,在惰性气氛下将98ml甲苯和7.5μmolnabarf加入到反应器中,然后将5μmol的钯催化剂溶解在2ml二氯甲烷(或氯仿)中通过注射器注入到聚合体系中。在快速搅拌下(750转以上),通入乙烯并保持在8-20atm。30-480分钟后,排空压力反应器,加入大量酸性甲醇或酸性乙醇(5%以上的盐酸醇溶液)溶液淬灭聚合反应,过滤聚合物,并在真空烘箱中干燥至恒重。如表3所示:
表3.不同反应条件对α-二亚胺钯催化剂催化乙烯聚合的影响
表3所有数据至少是基于两条平行试验得出的结果(除非另有说明)。活性:以106gmol-1h-1为单位。mw、mw/mn:分别为重均分子量、聚合物分散性指数,150℃下,在1,2,4-三氯苯中通过gpc测定,相对于聚苯乙烯标准物。支化度=每1000个碳支化的个数,由核磁共振氢谱测定。
注:条目5:钯催化剂(0.5μmol,r1=och3,r2=h,x=h),甲苯/二氯甲烷或氯仿(148ml/2ml);条目6:钯催化剂(0.5μmol,r1=och3,r2=h,x=h),甲苯/二氯甲烷或氯仿(148ml/2ml)。
表3说明:控制钯催化剂不变(5μmol,r1=och3,r2=h,x=h),nabarf(7.5μmol);当保持压力不变(8atm),时间不变(30min),随着反应温度的升高,聚合活性和分子量有先升高后下降的趋势,在70℃达到最高活性(2.19×106gmol-1h-1)、50℃分子量最高(62万)。聚合产物分子量分布也随着反应温度的升高而变大(最低1.07),然而其支化度却随着反应温度的升高而降低(之前报道规律均为温度升高支化度几乎保持不变),在0℃得到了最高支化度(220/1000c);在130℃得到了最低支化度(125)。在反应时间延长之后得到了更高分子量的聚合物,其中条目6:聚合温度(50℃)、聚合时间(8h)时,得到了较高的分子量(mw达200万)。当保持压力不变(8atm),温度不变(30℃),随着聚合时间的延长,聚合产物的产量、分子量均呈线性增长,分子量分布在1.1左右,聚合活性稳定不降,符合活性聚合特征(如下图1),另外其支化度也稳定在200左右。当保持温度不变(30℃),时间不变(30min),随着聚合压力的增加,聚合产物的产量和活性有较小幅度的提高,分子量也会有轻微微的变大,但支化度却基本没有变化。
图1为表3条目11-14所得聚合物的数均分子量及分子量分布关于时间图(a图)和高温凝胶色谱轨迹图(b图)。a图显示随着时间增加,所得聚合物的分子量呈线性增长,且分子量分布保持在1.1左右,b图显示随着聚合时间的延长,聚合物的高温凝胶色谱轨迹向左平移,且轨迹形状没有变化,呈现活性聚合的特征。说明在该反应温度下,催化聚合反应是活性聚合。
表3中条目1催化剂催化得到的聚合物的核磁共振氢谱图如图4所示。
nmegroups/1000c=聚合物每1000个碳中甲基的数量;
nbranches/1000c=聚合物每1000个碳中支化的数目;
ch2backbone=聚合物核磁谱图中的ch2骨架峰;
应用实施例4
首先将与高压气体管线连接的350ml玻璃压力反应器在90℃下真空干燥至少1小时。然后将反应器调节至指定温度,在惰性气氛下将98ml甲苯或98ml二甲苯或98ml苯甲醚或98ml氯苯或98ml己烷或98ml环己烷或98ml甲基环己烷或98ml二氯甲烷或98ml四氯乙烷或98ml氯仿,以及7.5μmolnabarf加入到反应器中,然后将5μmol的钯催化剂溶解在2ml二氯甲烷或氯仿中通过注射器注入到聚合体系中。在快速搅拌下(750转以上),通入乙烯并保持在指定压力。在指定时间后,排空压力反应器,加入大量酸性甲醇或酸性乙醇(5%以上的盐酸醇溶液)溶液淬灭聚合反应,过滤聚合物,并在真空烘箱中干燥至恒重。如表4所示:
表4.不同溶剂对α-二亚胺钯催化剂催化乙烯聚合的影响
表4所有数据至少是基于两条平行试验得出的结果(除非另有说明)。活性:以106gmol-1h-1为单位。mw、mw/mn:分别为重均分子量、聚合物分散性指数,150℃下,在1,2,4-三氯苯中通过gpc测定,相对于聚苯乙烯标准物。支化度=每1000个碳支化的个数,由核磁共振氢谱测定。
表4说明:控制钯催化剂不变(5μmol,r1=och3,r2=h,x=h),nabarf(7.5μmol);压力不变(8atm),时间不变(30min),温度不变(30℃),在不同溶剂下,数据显示,使用甲苯的活性、分子量以及支化度均占优势。
- 还没有人留言评论。精彩留言会获得点赞!
- 6,7,8,9‑四氢‑5H‑苯并[7]轮烯‑2‑烷基胺类化合物及其用途的制造方法与工艺
- 用于PHOLED的具有氮杂‑二苯并噻吩或氮杂‑二苯并呋喃核的新材料的制造方法与工艺
- 多孔ZSM‑5沸石与γ‑Al2O3复合材料的合成及制备加氢脱硫催化剂的制造方法与工艺
- 一种含有氟噻虫砜和五氟虫腙的杀虫组合物的制造方法与工艺
- 苯并双(噻二唑)衍生物、包含其的油墨、以及使用了其的有机电子装置的制造方法
- 苯并噻二唑胺的制造方法与工艺
- 用于有机电致发光器件的材料的制造方法与工艺
- 用于有机电致发光器件的材料的制造方法与工艺
- 一种苯并噻咯类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并噻吩并咔唑类衍生物及其制备方法和有机发光器件与制造工艺