一种加氢裂化催化剂及其应用的制作方法
本发明涉及一种加氢裂化催化剂及其应用。
背景技术:
工业加氢裂化进料包括VGO等350-540℃馏分,反应温度一般在350-440℃之间,进料中高沸点组分在反应时以液体形式同催化剂进行接触。催化剂孔道结构就具有更重要的作用,这就需要增加载体的孔径,提高反应分子接触到催化剂内加氢活性金属的能力。由于分子筛和金属硫化物孔容较小,特别是满足石油加工所要求大分子烃类进料要求的中大孔较少,常规方法可通过在加氢裂化催化剂载体中引入大孔容耐热无机氧化物基质的方法来实现,例如引入氧化铝基质。
ZL97121663.0公开了一种特别适用于生产中间馏分油的加氢裂化催化剂,含有无定形氧化硅-氧化铝组分和小孔氧化铝粘合剂,无定形氧化硅-氧化铝的含量为30-60重量%,至少一种VIB族元素和至少一种Ⅷ族元素,加氢金属氧化物总含量为20-35重量%,余量为小孔氧化铝粘合剂,其特征在于催化剂比表面150-300米2/克,孔容0.25-0.50毫升/克,4-15纳米孔分布在60-90%,红外酸度0.30-0.50毫摩尔/克。
ZL01123765.1公开了一种柴油加氢处理催化剂,该催化剂含有一种载体和负载在该载体上的钼和/或钨及镍和/或钴,其特征在于,所述载体由氧化铝和沸石组成,氧化铝与沸石的重量比为90:10-50:50,所述氧化铝是由小孔氧化铝和大孔氧化铝按照75:25-50:50的重量比复合而成的氧化铝,其中,小孔氧化铝为直径小于80埃孔的孔体积占总孔体积95%以上的氧化铝,大孔氧化铝为直径60~600埃孔的孔体积占总孔体积70%以上的氧化铝。
技术实现要素:
本发明的目的是提供一种性能得到改性了的加氢裂化催化剂以及该催化剂应用。
本发明涉及的内容包括:
一种加氢裂化催化剂,含有载体和至少一种选自VIII族和至少一种选自VIB族的金属组分,所述载体含有氧化铝、Y型分子筛和低酸度硅铝,以载体为基准,Y型分子筛的含量为3重量%~70重量%,氧化铝的含量为20重量%~90重量%,低酸度硅铝含量为3重量%~60重量%,其特征在于,所述载体由Y 型分子筛、低酸度硅铝与拟薄水铝石混合、成型、干燥并焙烧得到,所述拟薄水铝石包括PB1,以X光衍射表征,所述PB1的κ1和κ2分别大于1至小于等于3,其中,κ1=h2/h1、κ2=h3/h2,h1、h2和h3分别为PB1的X光衍射谱图中在2θ角为24-30°、35-41°和46-52°的三个衍射峰的峰高。
根据本发明提供的催化剂,优选条件下,以载体为基准,Y型分子筛的含量为5重量%~65重量%,氧化铝的含量为30重量%~90重量%,低酸度硅铝的含量为5重量%~60重量%;以X光衍射表征,所述PB1的κ1和κ2分别为1.02~2.4;进一步优选,所述PB1的κ1为1.2~2.3,κ2为1.02~1.4;所述PB1的比表面积为100~350米2/克,孔容为0.7~1.2毫升/克,优选地,所述PB1的比表面积为150~280米2/克,孔容为0.85~1.12毫升/克。
根据本发明提供的催化剂,所述的低酸度硅铝的红外B酸酸度值0.01~0.1mmol/g,可以选自含氧化硅化合物和氧化铝化合物体通过化学反应所得到含硅氧化铝、无定型硅铝的一种或者数种,如采用德国Sasol公司生产的Siral系列硅铝粉(Siral-5,Siral-10,Siral-15,Siral-20,Siral-30,Siral-40等)为前身物制备得到的含硅氧化铝。以低酸度硅铝质量为基准,其硅含量以二氧化硅计为20重量%~60重量%,孔容0.5~1.0ml/g。;所述Y型分子筛为超稳Y型分子筛,所述Y型分子筛的晶胞常数为2.460~2.430,比表面积为550~700米2/克,孔容为0.30~0.45毫升/克。所述Y型分子筛可以含有磷,以所述分子筛为基准,磷的含量为0.1重量%~2.5重量%,优选为0.4重量%~2.0重量%。
本发明提供的催化剂中,所述VIII族的金属组分选自镍和/或钴,所述VIB族的金属组分选自钼/或钨,以氧化物计并以所述催化剂为基准,所述镍和/或钴的含量为1重量%~15重量%,优选为2重量%~10重量%,所述钼/或钨的含量为5重量%~40重量%,优选为10重量%~35重量%。
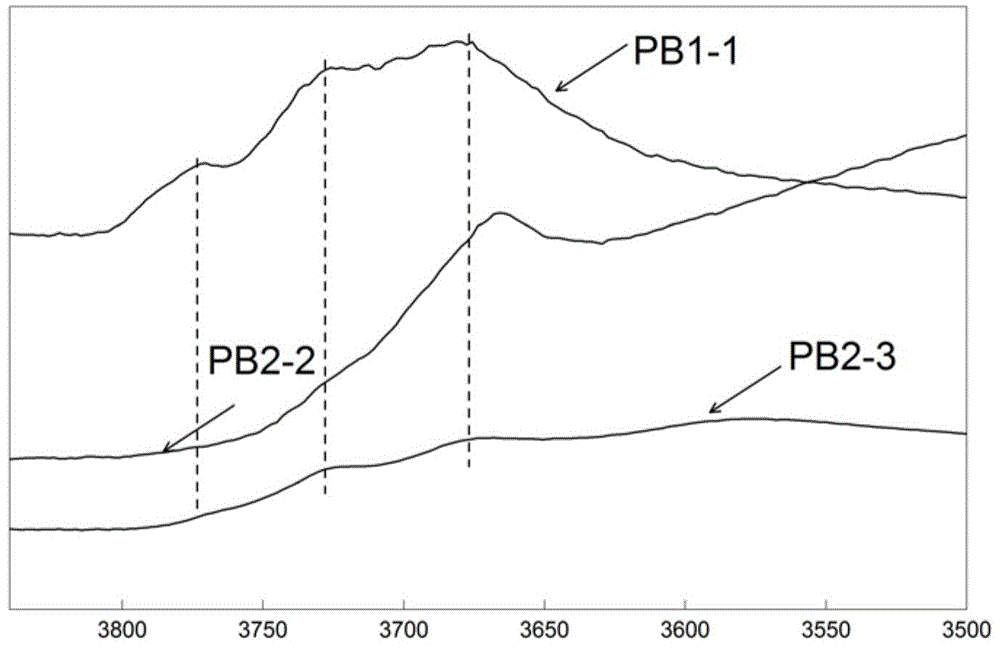
制备本发明所述催化剂的拟薄水铝石可以为PB1与PB2的混合物,以干基计并以载体为基准,PB1的含量为10~90重量%,PB2的含量为5~60重量%,以红外光谱表征,PB2的δ值为1.5~4.5,其中,δ=I1/(I2+I3),I1为所述拟薄水铝石的红外羟基谱中3665~3680cm-1处吸收峰的峰高,I2为3720~3730cm-1处吸收峰的峰高,I3为3760~3780cm-1处吸收峰的峰高;优选情况下,以干基计并以载体为基准,PB1的含量为20重量%~80重量%,PB2的含量为10重量%~50重量%,所述PB2的δ值为1.8~3.8。所述PB2为PB1的改性物,改性条件为PB1在70~400℃干燥0.5~14小时,优选地,改性条件为PB1在220~330℃干燥1~6小时。
制备本发明所述的催化剂,所述的干燥条件包括:温度为50~350℃,时间为1~24小时;所述的焙烧条件包括:温度为400~700℃,时间为0.5~6小时;优选条件下,所述的干燥条件包括:温度为80~200℃,时间为1~6小时;所述的焙烧条件包括:温度为550~650℃,时间为1~3小时。
本发明还提供了所述催化剂在烃油加工中的应用。
本发明中,所述的Y型分子筛选自HY(氢Y型分子筛)、REY(烯土Y型分子筛)、PY(含磷Y型分子筛)、USY(超稳Y型分子筛)、PUSY(含磷USY,包括PHY、REUSY(烯土超稳Y)),优选其中的USY、PUSY,进一步优选PUSY。所述分子筛可以是市售的商品,也可以采用任意的现有技术制备。例如,ZL00123139.1公开的制备USY的方法,Zl200410071122.6公开的制备PUSY的方法,这里均作为参考引用。
所述的低酸度硅铝的红外B酸酸度值0.01~0.1mmol/g,可以选自含氧化硅化合物和氧化铝化合物体通过化学反应所得到含硅氧化铝、无定型硅铝的一种或者数种,如采用德国Sasol公司生产的Siral系列硅铝粉(Siral-5,Siral-10,Siral-15,Siral-20,Siral-30,Siral-40等)为前身物制备得到的含硅氧化铝。以低酸度硅铝质量为基准,其硅含量以二氧化硅计为20重量%~60重量%,孔容0.5~1.0ml/g。
所述成型按常规方法进行,例如挤条成型。在挤出成型时可以加入适量助挤剂和/或胶粘剂,然后挤出成型。所述助挤剂、胶溶剂的种类及用量为本领域技术人员所公知,在此不赘述。
所述干燥和焙烧的方法为常规方法,例如,采用烘箱、网带、转炉加热方法进行干燥,干燥条件包括温度50~350℃,优选70~200℃,时间1~24小时,优选2~6小时;采用烘箱、网带、转炉加热方法进行焙烧,焙烧条件包括温度350~800℃,优选400~700℃,时间0.5~6小时,优选1~4小时。
本发明的发明人惊奇地发现,以X光衍射表征,采用包括一种满足κ1取值为1至小于等于3和κ2取值为1至小于等于3的拟薄水铝石PB1与Beta分子筛和Y型分子筛制备载体,由该载体制备加氢裂解催化剂时,催化剂具有更高的芳烃转化性能。
其中,κ1=h2/h1、κ2=h3/h2,h1、h2和h3分别为PB1的X光衍射谱图中在2θ角为24°~30°、35°~41°和46°~52°的三个衍射峰的峰高。
按照本发明,进一步优选所述PB1的κ1和κ2的值分别为1.02~2.3,更加优选所述PB1的κ1的值为1.2~2.3,κ2的值为1.02~1.4。
在足以满足本发明要求的前提下,本发明对PB1的来源没有特别限制,例如,可以是市售的商品或采用任意的现有方法制备。例如CN100999328B公开的一类采用硫酸铝和偏铝酸钠中和法制备的拟薄水铝石方法制备的拟薄水铝石可满足本发明的要求,这里作为参考引用。
本发明中,所述PB1在κ1和κ2的值满足要求的前提下,优选其中比表面积为100~350米2/克,孔容为0.7~1.2毫升/克,进一步优选比表面积为150~280米2/克,孔容为0.85~1.12毫升/克的拟薄水铝石。
本发明中,所述拟薄水铝石的孔容、比表面积等,是将所述拟薄水铝石于600℃焙烧4小时后,由BET氮吸附表征得到。
发明人进一步研究发现,在将满足前述要求的拟薄水铝石于70~400℃干燥0.5~14小时,优选于220~330℃干燥1~6小时后,得到PB1的改性物PB2,采用红外吸收光谱表征,PB2的δ值为1.5~4.5,优选为1.8~3.8。采用它与PB1以及Beta分子筛和Y型分子筛混合、成型制备的载体制备加氢裂解催化剂时,催化剂的性能进一步得到提高。其中,δ=I1/(I2+I3),I1为所述拟薄水铝石的红外羟基谱中3665~3680cm-1处吸收峰的峰高,I2为3720~3730cm-1处吸收峰的峰高,I3为3760~3780cm-1处吸收峰的峰高。
按照本发明提供的催化剂,所述选自VIII族和选自VIB族的金属组分的含量为加氢裂化催化剂惯常含量,例如,以催化剂为基准并以氧化物计,所述催化剂含有1重量%~15重量%的VIII族金属组分,5重量%~40重量%的VIB族金属组分;优选含有2重量%~10重量%的VIII族金属组分,10重量%~35重量%的VIB族金属组分。所述VIB族金属选自Cr、Mo或W中的一种或几种,优选Mo和/或W,所述VIII族金属组分选自Fe、Co或Ni中的一种或几种,优选Co和/或Ni。
在足以将所述至少一种选自VIII族和至少一种选自VIB族的金属组分负载于所述氧化铝载体上的前提下,对具体负载方法没有特别限制。优选的方法为浸渍法。包括分别或同时配制含选自VIII族和VIB族的金属组分化合物的浸渍溶液,所述的浸渍根据浸渍液用量不同可以是过量液浸渍、孔饱和浸渍,根据浸渍实现的方式不同可以是浸泡法浸、喷淋浸渍等。通过对浸渍溶液的浓度、用量或载体用量的调节和控制,可以制备出指定含量的所述催化剂,这是本领域技术人员所容易理解的,这里不赘述。
按照本发明提供的催化剂制备方法,所述的第VIB族金属化合物选自这些金属的可溶性化合物中的一种或几种,例如,可以是硅钨酸、硅钨酸盐、磷钼 酸、磷钼酸盐、钼酸盐、仲钼酸盐、钨酸盐、偏钨酸盐、乙基偏钨酸盐中的一种或几种。
所述的第VIII族金属化合物选自这些金属的盐,包括它们的无机酸盐或有机盐。例如,所述无机盐选自硝酸盐、碳酸盐、碱式碳酸盐、次磷酸盐、磷酸盐、硫酸盐、氯化物以及这些盐类的部分分解产物中的一种或几种,优选地,选自硝酸盐、碳酸盐或碱式碳酸盐中的一种或几种。所述有机盐为有机物与VIII族金属结合生成的盐类或可溶性络合物,所述有机物可以是有机碱、有机羧酸、胺类、酮类、醚类、烷基类,优选为有机羧酸盐。
按照本发明提供的催化剂,还可以含有有机添加物,以所述催化剂为基准,并以碳元素计,所述有机添加物的含量不超过10重量%,进一步优选不超过6重量%。
所述的有机添加物选自含氧和/或含氮的有机物中的一种或几种。所述含氧化合物选自有机醇、有机酸中的一种或几种,含氮有机化合物选自有机胺、有机铵盐中的一种或几种。具体地,选自含氧的有机物选自乙二醇、丙三醇、聚乙二醇(分子量为200~1500)、二乙二醇、丁二醇、乙酸、马来酸、草酸、氨基三乙酸、1,2-环己烷二胺四乙酸、柠檬酸、酒石酸、苹果酸中的一种或几种,其中优选乙二醇、丙三醇、聚乙二醇和柠檬酸;含氮的有机物选自乙二胺、二亚乙基三胺、环己二胺四乙酸、氨基乙酸、次氮基三乙酸、EDTA及其铵盐中的一种或几种,其中优选EDTA和次氮基三乙酸。所述的有机添加剂也可以同时包括含有机醇和有机酸。
按照本发明提供的制备方法,当本发明的催化剂进一步含有有机添加剂时,还包括引入有机化合物的步骤,所述有机化合物可以和第VIII族金属一起引入,也可以在引入第VIII族金属之后引入,并进行干燥,优选将有机物和第VIII族金属配制成混合溶液通过浸渍的方式同时引入,之后进行干燥。所述干燥温度和时间的选择除了不足以使VIII族金属盐或络合物分解外,还应允许在催化剂中保留大部分所引入的有机物,例如保留50%以上的有机物,更优选地,保留70%以上的有机物。其方法可以是现有技术中任意一种可实现上述目的的方法。例如,加热干燥,减压干燥以及加热结合减压干燥等方法。当所述干燥方法为加热干燥时,优选的干燥温度不超过250℃,进一步优选不超过200℃,更加优选不超过180℃。
按照本领域中的常规方法,所述加氢处理催化剂在使用之前,通常可在氢气存在下,于140~370℃的温度下用硫、硫化氢或含硫原料进行预硫化,这种 预硫化可在器外进行也可在器内原位硫化,将其所负载的活性金属组分转化为金属硫化物组分。
本发明提供的催化剂可用于各类烃油原料的加氢裂解或加氢改质。用于加氢裂解或加氢改质的反应条件为惯常的加氢裂解或加氢改质反应条件,例如反应温为度200~420℃、进一步优选为220~400℃,压力为2~18兆帕、进一步优选为2~15兆帕,液时空速为0.3~10小时-1、进一步优选为0.3~5小时-1,氢油体积比为50~5000、进一步优选为50~4000。
所述加氢裂解或加氢改质的反应的装置可以在任何足以使所述原料油在加氢处理反应条件下与所述催化剂接触反应的反应装器中进行,例如,在所述固定床反应器,移动床反应器或沸腾床反应器中进行。
与现有技术提供催化剂相比,本发明提供催化剂具有更好的加氢脱芳烃性能。例如,采用本发明提供的催化剂在6.4MPa中压条件下加工催化裂化尾油,尾油平衡芳烃含量相对降低可达10%~30%。
附图说明
附图用以对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于说明本发明,但并不构成对本发明的限制。在附图中:
图1(拟薄水铝石的XRD图)中的PB1-1是满足本发明要求的拟薄水铝石的XRD谱图;PB2-2为PB1-1的一种改性物;SB(Sasol公司生产的拟薄水铝石,商品名为SB粉)是一种不能满足本发明要求的拟薄水铝石的XRD谱图。
图2(拟薄水铝石的IR-OH图)中PB2-2为PB1-1的改性物的IR-OH谱图;PB2-3为PB1-1于600℃焙烧产物的IR-OH谱图。
具体实施方式
下面的实施例将对本发明做进一步的说明。
实验中,XRD在德国西门子公司的D5005型X射线衍射仪上进行,Cu靶,Ka辐射,固体探测器,40kV,40mA,步进扫描,步幅0.02°,扫描范围5°~70°。
IR-OH谱图在美国BIO-RAD公司FT3000型傅立叶变换红外光谱仪进行测定,15mm的自支撑片,并真空至10-3Pa,保持1h,冷至室温后扫描1000~4000cm-1波数范围。
重油中饱和烃和芳烃组成与含量采用SH/T0659-1998方法测定。
实施例及对比例中所使用的拟薄水铝石、制备方法或来源:
PB1-1
将澄清透明的500mL浓度为93g/L的硫酸铝和浓度为195g/L的偏铝酸钠 (苛性系数为1.6)同时缓慢加入2L带搅拌的反应釜中,控制反应温度为40℃,pH值7.3。反应完全后,搅拌加入浓度为0.5mol/L的碳酸钠至溶液pH值为9.7。恒温50℃反应1小时后,过滤,去离子水洗涤三次,烘箱中通风120℃温度干燥3hr,得到拟薄水铝石PB1-1。PB1-1的h1,h2,h3,k1,k2以及其孔容列于表1。
PB1-2
将澄清透明的600mL浓度为48g/L的硫酸铝和浓度8%的氨水同时缓慢加入2L带搅拌的反应釜中,反应过滤,再加入氨水控制反应温度为35℃,pH值6.0。反应完全后,过滤,将滤饼打浆,搅拌加入碳酸氢铵至溶液pH值为9.5。恒温35℃反应12小时后,过滤,去离子水洗涤三次,烘箱中通风120℃温度干燥3小时,得到拟薄水铝石PB1-2。PB1-2的h1,h2,h3,k1,k2以及其孔容列于表1。
PB1-3
将澄清透明的600mL浓度为93g/L的硫酸铝和浓度8%的氨水同时缓慢加入2L带搅拌的反应釜中,控制反应温度为50℃,pH值4.6。反应完全后,过滤,将滤饼打浆,搅拌加入碳酸钠至溶液pH值为8.4。恒温35℃反应0.5小时后,过滤,去离子水洗涤三次,烘箱中通风120℃温度干燥3小时,得到拟薄水铝石PB1-3。PB1-3的h1,h2,h3,k1,k2以及其孔容列于表1。
SB
Sasol公司生产的拟薄水铝石,商品名为SB粉。SB的h1,h2,h3,k1,k2以及其孔容列于表1。
表1拟薄水铝石XRD表征数据
其中,SB不能满足本发明要求。
实施例及对比例中所使用的拟薄水铝石改性物及其制备方法:
PB2-1
将300克PB1-1置于马弗炉中,加热至250℃并在该温度下恒温3小时,得到PB2-1。PB2-1的h1,h2,h3,k1,k2以及其孔容列于表1,PB2-1的I1, I2,I3和δ值列于表2。
PB2-2
将300克PB1-1置于马弗炉中,加热至300℃并在该温度下恒温3小时,得到PB2-2。PB2-2的h1,h2,h3,k1,k2以及其孔容列于表1,PB2-2的I1,I2,I3和δ值列于表2。
PB2-3
将300克PB1-1置于马弗炉中,加热至600℃并在该温度下恒温3小时,得到PB2-3。PB2-3的I1,I2,I3和δ值列于表2。
PB2-4
将300克PB1-2置于马弗炉中,加热至230℃并在该温度下恒温5小时,得到PB2-4。PB2-4的I1,I2,I3和δ值列于表2。
PB2-5
将300克PB1-3置于马弗炉中,加热至330℃并在该温度下恒温2小时,得到PB2-5。PB2-5的I1,I2,I3和δ值列于表2。
表2拟薄水铝石PB红外表征数据
对比例1
将233.3克拟薄水铝石SB(干基0.75,干基测定方法为将一定量的原粉置入带盖坩埚放入马弗炉中,升温至700℃恒温1hr后,计算剩余物同原粉的比值,下同)与333.3克超稳分子筛LAY(中石化催化剂分公司长岭催化剂厂生产,晶胞常数24.58,孔容0.39ml/g,干基0.75)、96g低酸度硅铝(Sasol德国生产,商品名Siral-40,二氧化硅含量40%,干基0.78,孔容0.9ml/g,红外B酸值0.04mmol/g,L酸值0.27mmol/g,)、田菁粉16克混合,挤成外接圆直径为1.6毫米的三叶形条,120℃烘干3小时,600℃温度下焙烧3小时,得载体,催化剂载体组成见表3。
降温至室温后,取100克载体用含偏钨酸铵(四川自贡硬质合金厂,氧化钨含量为82重量%)50克、碱式碳酸镍(江苏宜兴徐迟化工有限公司,氧化镍含量为51重量%)8.7克、10.5g柠檬酸68ml水溶液浸渍,180℃烘10小时, 得到催化剂R-1。以催化剂总量为基准,R1中金属组分的质量分数(为计算值,下同)列于表3。
实例1
将233.3克拟薄水铝石粉PB1-1(干基0.75)同333.3克超稳分子筛LAY、96gSiral-40、田菁粉16克混合,挤成外接圆直径为1.6毫米的三叶形条,120℃烘干3小时,600℃温度下焙烧3小时,得载体,催化剂载体组成见表3。
降温至室温后,取100克载体用含偏钨酸铵(四川自贡硬质合金厂,氧化钨含量为82重量%)51克、碱式碳酸镍(江苏宜兴徐迟化工有限公司,氧化镍含量为51重量%)8.7克、10.5g柠檬酸77ml水溶液浸渍,180℃烘10小时,得到催化剂C-1。以催化剂总量为基准,C-1中金属组分的质量分数列于表3。
实例2
将133克拟薄水铝石粉PB1-1同81.5克干基含量0.92的PB2-2,333.3g超稳分子筛LAY、96gSiral-40、田菁粉16克混合,挤成外接圆直径为1.6毫米的三叶形条,120℃烘干,600℃温度下焙烧3小时,得载体,催化剂载体组成见表3。
降温至室温后,取100g载体用含偏钨酸铵(四川自贡硬质合金厂,氧化钨含量为82重量%)51克、碱式碳酸镍(江苏宜兴徐迟化工有限公司,氧化镍含量为51重量%)8.7克、10.5g柠檬酸84ml水溶液浸渍,180℃烘10小时,得到催化剂C-2。以催化剂总量为基准,C-2中金属组分的质量分数列于表3。
实施例3
将197g克拟薄水铝石粉PB1-3(干基0.76)同28g干基含量0.89的PB2-1,308.6g含磷超稳分子筛PUSY(中石化催化剂分公司长岭催化剂厂生产,商品名USY-5,五氧化二磷含量1.2%,晶胞常数24.44,孔容0.40ml/g,干基0.81,)、96gSiral-40、田菁粉16克混合,挤成外接圆直径为1.6毫米的三叶形条,120℃烘干,600℃温度下焙烧3小时,得载体,催化剂载体组成见表3。
降温至室温后,取100g载体用含偏钨酸铵(四川自贡硬质合金厂,氧化钨含量为82重量%)51克、碱式碳酸镍(江苏宜兴徐迟化工有限公司,氧化镍含量为51重量%)8.7克、10.5g柠檬酸90ml水溶液浸渍,140℃烘3小时,得到催化剂C-3。以催化剂总量为基准,C-3中金属组分的质量分数列于表3。
实施例4
同实施例2,所不同的是用75gPB2-3代替PB-2-2,得到催化剂R-2。以催化剂总量为基准,C-4中金属组分的质量分数列于表3。
实施例5
将263.2g克拟薄水铝石粉PB1-3(干基0.76)同56g干基含量0.89的PB2-1,62g含磷超稳分子筛PUSY(中石化催化剂分公司长岭催化剂厂生产,商品名USY-5,五氧化二磷含量1.2%,晶胞常数24.44,孔容0.40ml/g,干基0.81,)、256gSiral-40、田菁粉16克混合,挤成外接圆直径为1.6毫米的三叶形条,120℃烘干,480℃温度下焙烧8小时,得载体,催化剂载体组成见表3。
降温至室温后,取100g载体用含偏钨酸铵(四川自贡硬质合金厂,氧化钨含量为82重量%)30克、碱式碳酸镍(江苏宜兴徐迟化工有限公司,氧化镍含量为51重量%)18克、25g柠檬酸90ml水溶液浸渍,110℃烘20小时,得到催化剂C-5。以催化剂总量为基准,C-5中金属组分的质量分数列于表3。
表3催化剂中载体和金属质量分数表征数据
实例6
本实例说明本发明提供催化剂的性能。
所用原料油为沙轻减二减压瓦斯油,其物化性质见表4。
本实例中,催化剂的评价方法是:将催化剂破碎成直径2-3毫米的颗粒,在30毫升固定床反应器中装入催化剂20毫升,反应前先在氢气氛下用含2重%二硫化碳的煤油按照如下程序进行硫化,然后切换反应原料反应。
硫化程序:升温至150℃,引入硫化油,恒温1hr待吸附温波通过两个反应器,以60℃/hr升温至230℃,稳定2hr,以60℃/hr升温至360℃,稳定6hr。置换反应油,调整反应温度至355℃,至少稳定20hr。反应氢分压为6.4兆帕,液时空速(LHSV)1小时-1,氢油比(体积)800,评价的催化剂及其结果列于表5中。
表4原料油性质
反应后将所得产品通过350℃进行减压蒸馏,所得产品进行质谱族组成分析,结果见表5所示:。
表5催化剂脱除尾油芳烃性能表征
由于催化剂评价反应压力较低,使得尾油芳烃接近所谓的热力学平衡区域,采用本方法后尾油芳烃含量相对降低10%~30%。
- 还没有人留言评论。精彩留言会获得点赞!