从二氧化合物和多氧化合物产生芳香族化合物的制作方法
本申请是申请号为201480014871.7,申请日为2014年3月14日,发明名称为“从二氧化合物和多氧化合物产生芳香族化合物”的申请的分案申请。
相关申请的交叉引用
本申请要求美国临时申请号61,784,417的权益,该申请通过引用以其全部内容结合在此。
联邦资助的研究或开发
本发明是在美国能源部提供的基金(基金号de-ee0003044和de-ee0005006)下由政府支持完成的。政府对本发明具有某些权利。
本发明涉及但不限于从二氧化合物和多氧化合物产生芳香族化合物的方法。
发明背景
用于将生物质转化成液体燃料和化学品的先前的努力集中于使用缩合反应途径将各种氧合烃转换成所希望的产品。缩合反应可以例如在温和条件(即,80℃与600℃之间的温度以及等于或略大于大气压的压力)下使用一种沸石催化剂来催化。
用于将氧合烃转化成汽油范围烃的最常见的方法已知为甲醇制汽油(mtg)方法(莫尔比石油公司(mobiloilcorporation),加利福尼亚州,1980)。另外,莫尔比石油和康菲石油(conocophillips)已开发用于将生物质来源的碳水化合物(例如,葡萄糖、木糖、淀粉、蔗糖)和糖醇(山梨糖醇和木糖醇)转化成类似的汽油范围烃的方法并且获得了专利。然而,用沸石催化剂处理这些高度氧化的物种的一个主要缺点是产生高产率的不希望的焦炭,这会严重损害/限制催化剂性能以及最终产品的产率。
陈(chen)等人开发出有效氢碳比(h:ceff)作为一种工具来辅助确定使用沸石催化剂将氧合烃原料催化转化成烃的适合性(陈n·y·(n.y.chen)、j·t·f·德格南(j.t.f.degnan)以及l·r·凯尼格(l.r.koeing),化学技术(chem.tech.),1986,16,506)。h:ceff比是基于进料中的碳、氧和氢的量,并且计算如下:
h:
其中h表示氢原子数,o表示氧原子数,并且c表示碳原子数。水和分子氢(二原子氢,h2)排除在该计算之外。该h:ceff比既适用于单独的组分又适用于多种组分的混合物,但是对于含有除了碳、氢和氧之外的原子的组分是无效的。对于混合物,c、h、和o概括了除了水和分子氢之外的所有组分。术语“氢”指代任何氢原子,而术语“分子氢”限于二原子氢,h2。
张(zhang)等人使用一种zsm-5催化剂研究了h:ceff比对多种生物质来源的氧合烃转化为焦炭、烯烃和芳香族化合物的影响(张等人,用zsm-5将生物质来源的原料催化转化成烯烃和芳香族化合物:氢碳有效比(catalyticconversionofbiomass-derivedfeedstocksintoolefinsandaromaticswithzsm-5:thehydrogentocarboneffectiveratio),能源与环境科学(energyenviron.sci.),2011,4,2297)。张报道了具有在0与0.3之间的h:ceff比的生物质来源的原料产生高水平的焦炭,从而使得它对于将此类原料转化成芳香族化合物和化学品是不经济的。通过加氢处理原料以加入氢,张能够使用一种zsm-5催化剂以高于一种未使用氢化作用的方法的产率来产生芳香族化合物和烯烃。然而,烯烃与芳香族化合物之比还随着h:ceff比的增加而增加,其中对于所有原料,烯烃产率高于芳香族化合物的产率。据报道,在1.2的h:ceff比处还存在一个拐点,在此处该芳香族化合物和烯烃产率并未进一步增加。张指出,当根据所披露的方法使用沸石催化剂时,高价值芳香族化学品如苯、甲苯和二甲苯(btx)的产率至多可能限于24%。
来源于生物质的氧合烃如碳水化合物、糖和糖醇具有低h:ceff比。一种典型的碳水化合物或糖具有可以由化学式((ch2o)n)m表示的化学式,其中n典型地是等于3-6(即,丙糖、丁糖、戊糖、或己糖),并且m是1(即,单糖)与针对大型多糖的数万之间的任何数值。具有化学式((ch2o)n)m的分子将具有0的h:ceff比。糖醇类似地具有低h:ceff比。例如,c6和c5糖醇如山梨糖醇和木糖醇分别具有0.33和0.4的h:ceff,从而使得它们对于缩合反应而言是不希望的,因为过量焦炭会形成在缩合催化剂上。
为了克服将富氧的(可替代地,贫氢的)生物质来源的原料转化成烃的限制,已将生物质来源的原料转化成贫氧的(可替代地,富氢的)分子,如单氧合烃(醇、酮、环醚等),同时保持碳链完整。随后使用一种缩合催化剂将这些单氧化合物转化成汽油范围烃。参见,例如,美国专利号7,767,867、8,017,818、8,231,857以及美国专利申请号12/980,892和13/586,499,这些专利的内容以其全文结合在此。
在所述方法下,从生物质来源的氧合烃转化成单氧化合物产生具有接近2的总h:ceff比的含氧化合物混合物。该总h:ceff比是基于该含氧化合物混合物中的所有烃(氧合的和非氧和合的两者)的组合的h:ceff比。单羟基醇不管大小如何都具有2.0的h:ceff比,而环醚、酮、醛和烷烃的h:ceff比随着烃的长度而变化。例如,c6和c5环醚、酮和醛的h:ceff分别是1.67和1.6,而c6和c5烷烃的h:ceff分别是2.33和2.40。任何实质数量的烷烃都是特别不希望的,因为它们在缩合过程中进一步处理时大部分是不反应的并且会造成较高的h:ceff比。
虽然形成单氧化合物允许在催化剂上不产生过量焦炭的情况下进行含氧化合物的缩合,但该过程是有代价的。确切而言,单氧化合物的缩合经常导致以与芳香族分子的产生相当的产率产生大量烷烃。对于高度希望芳香族分子的应用,烷烃的大量产生会减少产生的总芳香族化合物,从而增加成品的总成本。
概述
因此,需要用于以高百分比产生芳香族化合物同时最小化烷烃产生的方法;用于产生对这些方法有用的含氧化合物混合物的方法;以及用于形成该含氧化合物混合物的方法中的催化剂。此外,对这些方法还具有低焦炭产率存在需要。
诸位发明人基于对总含氧化合物混合物进行的精制出人意料地发现了所有那些需求的解决方案。具体而言,诸位发明人已发现,具有在0.5至1.7范围内的h:ceff比以及一个或多个以下属性的含氧化合物混合物对改进芳香族化合物生产提供了意想不到的且有益的结果:(1)比单氧化合物更多的二氧化合物和多氧化合物,(2)比单氧化合物更多的二氧化合物,(3)比c5-6含氧化合物(尤其是单氧化合物)更多的c2-4含氧化合物(尤其是二氧化合物和多氧化合物),和/或(4)很少乃至不存在烷烃。
本发明提供了用于制备具有高产率的芳香族分子和低产率的烷烃的生物质来源的化学品和燃料的方法。本发明还提供了用于从生物质制备含氧化合物混合物的方法,该含氧化合物混合物在一种缩合催化剂存在下反应以产生具有高产率的芳香族分子和低产率的烷烃的化学品和燃料。此外,本发明还提供了在能够反应来产生高产率的芳香族分子和低产率的烷烃的含氧化合物混合物的生产中有用的催化剂。本发明的一个另外的方面在于这些方法在制备芳香族分子时产生低产率的焦炭。
本发明的第一实施例是一种用于产生具有高产率的芳基化合物和低产率的c4+烷烃的烃的方法:在存在一种缩合催化剂的情况下,使包含一种c2-6含氧化合物混合物的一种原料反应以产生一种烃混合物。该含氧化合物混合物包含选自由c2-6单氧化合物、c2-6二氧化合物、c2-6多氧化合物、以及它们的组合组成的组的一个成员,并且具有大于或等于0.5且小于或等于1.7的h:ceff比。该烃混合物包含大于或等于50%cf的芳基化合物以及小于或等于20%cf的c4+烷烃。
本发明的第二实施例是一种用于产生具有高产率的芳基化合物和低产率的烷烃的烃的方法:在包含第viii族金属和结晶氧化铝载体的一种催化剂存在下,使一种水性原料与氢气反应以产生一种c2-6含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物(hemiceullosicderivatives)、木质素衍生物、木质纤维素衍生物、以及它们的混合物;并且在存在一种缩合催化剂的情况下使该c2-6含氧化合物混合物反应以产生包含c4+烷烃和芳基化合物的一种烃混合物,其中该烃混合物包含大于或等于50%cf的芳基化合物以及小于或等于20%cf的c4+烷烃。该含氧化合物混合物包含选自由c2-6单氧化合物、c2-6二氧化合物、c2-6多氧化合物、以及它们的组合组成的组的一个成员。该含氧化合物混合物还可以具有大于或等于0.5且小于或等于1.7的h:ceff比。在某些实施例中,该催化剂可以进一步包含选自第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、以及第va族的一种第二金属。在某些实施例中,该催化剂可以包含大于或等于0.5wt%的第viii族金属以及大于或等于0.05wt%的第二金属。在某些实施例中,该结晶氧化铝催化剂可以是一种过渡氧化铝载体。在某些实施例中,该结晶氧化铝催化剂可以是一种θ-氧化铝载体。在某些实施例中,该结晶氧化铝载体可以是用选自由b、cr、ce、co、cu、fe、mg、mo、nb、w、zr、以及它们的混合物组成的组的一个成员来改性。
本发明的第三实施例是一种用于产生能够反应来产生具有高产率的芳基化合物和低产率的烷烃的烃的含氧化合物混合物的方法:在包含第viii族金属和结晶氧化铝载体的一种脱氧催化剂存在下,使一种水性原料与氢气反应以产生一种c2-6含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物。该含氧化合物混合物具有大于或等于0.5且小于或等于1.7的h:ceff比。在某些实施例中,该脱氧催化剂可以进一步包含选自由第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、以及第va族组成的组的一种第二金属。在某些实施例中,该催化剂可以包含大于或等于0.5wt%的第viii族金属以及大于或等于0.05wt%的第二金属。在某些实施例中,该结晶氧化铝载体可以是一种过渡氧化铝载体。在某些实施例中,该结晶氧化铝载体可以是一种θ-氧化铝载体。在某些实施例中,该载体可以是用选自由b、cr、ce、co、cu、fe、mg、mo、nb、w、zr、以及它们的混合物组成的组的一个成员来改性。
本发明的第四实施例是一种能够产生可以反应来产生具有高产率的芳基化合物和低产率的烷烃的烃的含氧化合物混合物的催化剂组合物,该催化剂组合物包含一种ninsnm合金和一种结晶氧化铝载体。在某些实施例中,n等于3并且m等于1或2。在某些实施例中,ni的wt%可以是大于或等于0.5wt%、大于或等于1.0wt%或大于或等于2.0wt%。在某些实施例中,ni的wt%可以是小于或等于20wt%、小于或等于15wt%、小于或等于12wt%、或小于或等于10wt%。在某些实施例中,该结晶氧化铝载体可以是一种过渡氧化铝载体。在某些实施例中,该结晶氧化铝载体可以是一种θ-氧化铝载体。在某些实施例中,该结晶氧化铝载体可以是用选自由b、cr、ce、co、cu、fe、mg、mo、nb、w、zr、以及它们的混合物组成的组的一个成员来改性。在某些实施例中,该催化剂对以下内容可能是有用的:在该催化剂存在下,使一种水性原料与氢气反应以产生一种含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物,其中该含氧化合物混合物的h:ceff比是大于或等于0.5且小于或等于1.7。在另一个实施例中,该催化剂对以下内容可能是有用的:在该催化剂存在下,使一种水性原料与氢气反应以产生一种含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物;并且在存在一种缩合催化剂的情况下使该含氧化合物混合物反应以产生包含c4+烷烃和芳基化合物的一种烃混合物,其中该烃混合物包含大于或等于50%cf的芳基化合物以及小于或等于20%cf的c4+烷烃。
本发明的第五实施例是一种可用于通过使一种水性原料反应来产生具有高产率的芳基化合物和低产率的烷烃的烃的物质组合物,该催化剂组合物包含一种ninsnm合金和一种结晶氧化铝载体、氧合烃、以及一种含氧化合物混合物。
在任何以上实施例中,该含氧化合物混合物可以具有选自下组的一个或多个属性,该组由以下各项组成:(i)二氧化合物和多氧化合物与单氧化合物的大于或等于0.5的%cf比,(ii)二氧化合物与单氧化合物的大于或等于0.5的%cf比,(iii)c2-4含氧化合物与c5-6含氧化合物的大于或等于1.0的%cf比,以及(iv)该含氧化合物混合物进一步包含小于或等于10%cf的烷烃。
在任何以上实施例中,该烃混合物包含大于或等于55%cf的芳基化合物、大于或等于60%cf的芳基化合物、或大于或等于65%cf的芳基化合物。在某些实施例中,该烃混合物包含小于或等于15%cf的c4+烷烃、小于或等于10%cf的c4+烷烃、或小于或等于5%cf的c4+烷烃。
在任何以上实施例中,该含氧化合物混合物的h:ceff比可以小于或等于1.6、小于或等于1.5、或小于或等于1.4。在某些实施例中,该含氧化合物混合物的h:ceff比可以大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、或大于或等于1.0。
在任何以上实施例中,该含氧化合物混合物可以包含大于或等于约30%cf的二氧化合物和多氧化合物、大于或等于40%cf的二氧化合物和多氧化合物、大于或等于50%cf的二氧化合物和多氧化合物、或大于或等于60%cf的二氧化合物和多氧化合物。在某些实施例中,该含氧化合物混合物可以包含大于或等于30%cf的二氧化合物、大于或等于40%cf的二氧化合物、大于或等于50%cf的二氧化合物、或大于或等于60%cf的二氧化合物。在某些实施例中,该含氧化合物混合物可以包含大于或等于20%cf的二醇、大于或等于30%cf的二醇、大于或等于40%cf的二醇、或大于或等于50%cf的二醇。
在任何以上实施例中,这些芳基化合物可以包括选自下组的一种或多种芳基化合物,该组由以下各项组成:苯、甲苯、二甲苯、对二甲苯、间二甲苯、邻二甲苯、以及乙苯。
本申请提供了以下内容:
项目1.一种用于产生烃的方法,该方法包括:在存在一种缩合催化剂的情况下使c2-6含氧化合物混合物反应以便产生包含c4+烷烃和芳基化合物的烃混合物;其中该c2-6含氧化合物混合物包含选自由c2-6单氧化合物、c2-6二氧化合物、c2-6多氧化合物、以及它们的组合组成的组的一个成员;其中该c2-6含氧化合物混合物具有大于或等于0.5且小于或等于1.7的h:ceff比;并且其中该烃混合物包含大于或等于50%cf的芳基化合物以及小于或等于20%cf的c4+烷烃。
项目2.如项目1所述的方法,其中该c2-6含氧化合物混合物具有选自下组的一个或多个属性,该组由以下各项组成:(i)二氧化合物和多氧化合物与单氧化合物的大于或等于0.5的%cf比,(ii)二氧化合物与单氧化合物的大于或等于0.5的%cf比,(iii)c2-4含氧化合物与c5-6含氧化合物的大于或等于1.0的%cf比,以及(iv)该c2-6含氧化合物混合物进一步包含小于或等于10%cf的烷烃。
项目3.如项目1所述的方法,其中该烃混合物包含大于或等于55%cf的芳基化合物、大于或等于60%cf的芳基化合物、或大于或等于65%cf的芳基化合物。
项目4.如项目1所述的方法,其中该烃混合物包含小于或等于15%cf的c4+烷烃、小于或等于10%cf的c4+烷烃、或小于或等于5%cf的c4+烷烃。
项目5.如项目1所述的方法,其中该c2-6含氧化合物混合物的h:ceff比是小于或等于1.6、小于或等于1.5、或小于或等于1.4。
项目6.如项目1所述的方法,其中该c2-6含氧化合物混合物的h:ceff比是大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、或大于或等于1.0。
项目7.如项目1所述的方法,其中该c2-6含氧化合物混合物包含大于或等于30%cf的二氧化合物和多氧化合物。
项目8.如项目7所述的方法,其中该c2-6含氧化合物混合物包含大于或等于40%cf的二氧化合物和多氧化合物、大于或等于50%cf的二氧化合物和多氧化合物、或大于或等于60%cf的二氧化合物和多氧化合物。
项目9.如项目1所述的方法,其中该c2-6含氧化合物混合物包含大于或等于30%cf的二氧化合物。
项目10.如项目9所述的方法,其中该c2-6含氧化合物混合物包含大于或等于40%cf的二氧化合物、大于或等于50%cf的二氧化合物、或大于或等于50%cf的二氧化合物。
项目11.如项目1所述的方法,其中这些芳基化合物包括选自下组的一种或多种芳基化合物,该组由以下各项组成:苯、甲苯、二甲苯、对二甲苯、间二甲苯、邻二甲苯、以及乙苯。
项目12.如项目1所述的方法,其中该c2-6含氧化合物混合物通过以下方式产生:在一种包含第viii族金属和结晶氧化铝载体的脱氧催化剂存在下,使一种水性原料与氢气反应,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物。
项目13.一种用于产生烃的方法,该方法包括:(a)在一种包含第viii族金属和结晶氧化铝载体的脱氧催化剂存在下,使一种水性原料与氢气反应以便产生c2-6含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物以及(b)在存在一种缩合催化剂的情况下使该c2-6含氧化合物混合物反应以便产生包含c4+烷烃和芳基化合物的烃混合物,其中该c2-6含氧化合物混合物包含选自下组的一个成员,该组由以下各项组成:c2-6单氧化合物、c2-6二氧化合物、c2-6多氧化合物、以及它们的组合并且其中该烃混合物包含大于或等于50%cf的芳基化合物以及小于或等于20%cf的c4+烷烃。
项目14.如项目13所述的方法,其中该c2-6含氧化合物混合物具有大于或等于0.5且小于或等于1.7的h:ceff比。
项目15.如项目13所述的方法,其中该c2-6含氧化合物混合物具有选自下组的一个或多个属性,该组由以下各项组成:(i)二氧化合物和多氧化合物与单氧化合物的大于或等于0.5的%cf比,(ii)二氧化合物与单氧化合物的大于或等于0.5的%cf比,(iii)c2-4含氧化合物与c5-6含氧化合物的大于或等于1.0的%cf比,以及(iv)该c2-6含氧化合物混合物进一步包含小于或等于10%cf的烷烃。
项目16.如项目13所述的方法,其中该烃混合物包含大于或等于55%cf的芳基化合物、大于或等于60%cf的芳基化合物、或大于或等于65%cf的芳基化合物。
项目17.如项目13所述的方法,其中该烃混合物包含小于或等于15%cf的c4+烷烃、小于或等于10%cf的c4+烷烃、或小于或等于5%cf的c4+烷烃。
项目18.如项目13所述的方法,其中该c2-6含氧化合物混合物的h:ceff比是小于或等于1.6、小于或等于1.5、或小于或等于1.4。
项目19.如项目13所述的方法,其中该c2-6含氧化合物混合物的h:ceff比是大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、或大于或等于1.0。
项目20.如项目13所述的方法,其中该脱氧催化剂进一步包含选自由第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、以及第va族组成的组的一种第二金属。
项目21.如项目13所述的方法,其中该第viii族金属是ni或pd。
项目22.如项目20所述的方法,其中该第二金属包括sn或mo。
项目23.如项目20所述的方法,其中该脱氧催化剂包含大于或等于0.5wt%的该第viii族金属以及大于或等于0.05wt%的该第二金属。
项目24.如项目13所述的方法,其中该结晶氧化铝载体是一种过渡氧化铝载体。
项目25.如项目13所述的方法,其中该结晶氧化铝载体是θ-氧化铝。
项目26.如项目13所述的方法,其中该结晶氧化铝载体是用选自由b、cr、ce、co、cu、fe、mg、mo、nb、w、zr、以及它们的混合物组成的组的一个成员来改性。
项目27.如项目13所述的方法,其中该c2-6含氧化合物混合物包含大于或等于30%cf的二氧化合物和多氧化合物、大于或等于40%cf的二氧化合物和多氧化合物、大于或等于50%cf的二氧化合物和多氧化合物、或大于或等于60%cf的二氧化合物和多氧化合物。
项目28.如项目13所述的方法,其中该c2-6含氧化合物混合物包含大于或等于30%cf的二氧化合物、大于或等于40%cf的二氧化合物、大于或等于50%cf的二氧化合物、或大于或等于60%cf的二氧化合物。
项目29.如项目13所述的方法,其中这些芳基化合物包括选自下组的一种或多种芳基化合物,该组由以下各项组成:苯、甲苯、二甲苯、对二甲苯、间二甲苯、邻二甲苯、以及乙苯。
项目30.如项目13所述的方法,其中该烃混合物进一步包含选自下组的一个成员,该组由以下各项组成:c4+烯烃、c5+环烷烃、c5+环烯烃、稠合芳基化合物、多环化合物、以及它们的混合物。
项目31.如项目13所述的方法,其中该氧合烃的h:ceff比是小于或等于0.4。
项目32.如项目13所述的方法,其中大于或等于50%cf的该氧合烃具有5或6个连续碳原子。
项目33.一种用于从氧合烃产生氧合化合物的方法,该方法包括:在一种包含第viii族金属和结晶氧化铝载体的催化剂存在下,使一种水性原料与氢气反应以便产生含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物,其中该含氧化合物混合物的h:ceff比是大于或等于0.5且小于或等于1.7。
项目34.如项目33所述的方法,其中该含氧化合物混合物具有选自下组的一个或多个属性,该组由以下各项组成:(i)二氧化合物和多氧化合物与单氧化合物的大于或等于0.5的%cf比,(ii)二氧化合物与单氧化合物的大于或等于0.5的%cf比,(iii)c2-4含氧化合物与c5-6含氧化合物的大于或等于1.0的%cf比,以及(iv)该含氧化合物混合物进一步包含烷烃且小于或等于10%cf的烷烃。
项目35.如项目33所述的方法,其中该含氧化合物混合物的h:ceff比是小于或等于1.6、小于或等于1.5、或小于或等于1.4。
项目36.如项目33所述的方法,其中该含氧化合物混合物的h:ceff比是大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、或大于或等于1.0。
项目37.如项目33所述的方法,其中该催化剂进一步包含选自第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、以及第va族的一种第二金属。
项目38.如项目33所述的方法,第viii族金属是ni或pd。
项目39.如项目37所述的方法,其中该第二金属包括sn或mo。
项目40.如项目37所述的方法,其中该催化剂包含大于或等于0.5wt%的该第viii族金属以及大于或等于0.05wt%的该第二金属。
项目41.如项目33所述的方法,其中该结晶氧化铝载体是一种过渡氧化铝载体。
项目42.如项目33所述的方法,其中该结晶氧化铝载体是θ-氧化铝。
项目43.如项目33所述的方法,其中该载体是用选自由b、cr、ce、co、cu、fe、mg、mo、nb、w、zr、以及它们的混合物组成的组的一个成员来改性。
项目44.如项目33所述的方法,其中该含氧化合物混合物包含大于或等于30%cf的二氧化合物和多氧化合物、大于或等于40%cf的二氧化合物和多氧化合物、大于或等于50%cf的二氧化合物和多氧化合物、或大于或等于60%cf的二氧化合物和多氧化合物。
项目45.如项目33所述的方法,其中该含氧化合物混合物包含大于或等于30%cf的二氧化合物、大于或等于40%cf的二氧化合物、大于或等于50%cf的二氧化合物、或大于或等于60%cf的二氧化合物。
项目46.如项目33所述的方法,该方法进一步包括在存在一种酸缩合催化剂的情况下、在缩合温度和缩合压力下使该反应产物的一部分反应以便产生选自下组的一种或多种c4+化合物,该组由以下各项组成:c4+烷烃、c4+烯烃、c5+环烷烃、c5+环烯烃、芳基化合物、稠合芳基化合物、多环化合物、以及它们的混合物。
项目47.如项目46所述的方法,其中这些c4+化合物包括选自下组的一种或多种芳基化合物,该组由以下各项组成:苯、甲苯、二甲苯、对二甲苯、间二甲苯、以及邻二甲苯。
项目48.如项目46所述的方法,其中该烃混合物包含大于或等于50%cf的芳基化合物以及小于或等于20%cf的c4+烷烃。
项目49.一种催化剂组合物,该催化剂包含ninsnm合金和结晶氧化铝载体。
项目50.如项目49所述的催化剂组合物,其中n等于3并且m等于1或2。
项目51.如项目49所述的催化剂组合物,其中ni的wt%是大于或等于0.5wt%。
项目52.如项目51所述的催化剂组合物,其中该ni的wt%是大于或等于1.0wt%或大于或等于2.0%。
项目53.如项目49所述的催化剂组合物,其中该ni的wt%是小于或等于20%。
项目54.如项目53所述的催化剂组合物,其中该ni的wt%是小于或等于15%、小于或等于12wt%、或小于或等于10wt%。
项目55.如项目49所述的催化剂组合物,其中该结晶载体是一种过渡氧化铝载体。
项目56.如项目49所述的催化剂组合物,其中该结晶载体是一种θ-氧化铝载体。
项目57.如项目49所述的催化剂组合物,其中该载体是用选自由b、cr、ce、co、cu、fe、mg、mo、nb、w、zr、以及它们的混合物组成的组的一个成员来改性。
项目58.一种物质组合物,该物质组合物包含如项目49所述的催化剂、氧合烃、以及一种含氧化合物混合物。
项目59.如项目58所述的物质组合物,其中该含氧化合物混合物的h:ceff比是大于或等于0.5且小于或等于1.7。
项目60.如项目59所述的物质组合物,其中该含氧化合物混合物的h:ceff比是小于或等于1.6、小于或等于1.5、或小于或等于1.4。
项目61.如项目59所述的物质组合物,其中该含氧化合物混合物的h:ceff比是大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、或大于或等于1.0。
项目62.如项目58所述的物质组合物,其中该含氧化合物混合物具有选自下组的一个或多个属性,该组由以下各项组成:(i)二氧化合物和多氧化合物与单氧化合物的大于或等于0.5的%cf比,(ii)二氧化合物与单氧化合物的大于或等于0.5的%cf比,(iii)c2-4含氧化合物与c5-6含氧化合物的大于或等于1.0的%cf比,以及(iv)该含氧化合物混合物进一步包含烷烃且小于或等于10%cf的烷烃。
项目63.如项目58所述的物质组合物,其中大于或等于50%cf的氧合烃具有5或6个连续碳原子。
项目64.一种用于从氧合烃产生氧合化合物的方法,该方法包括:在项目49所述的催化剂存在下,使一种水性原料与氢气反应以便产生含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物,其中该含氧化合物混合物的h:ceff比是大于或等于0.5且小于或等于1.7。
项目65.一种用于产生烃的方法,该方法包括:(a)在项目49所述的催化剂存在下,使一种水性原料与氢气反应以便产生含氧化合物混合物,该水性原料包含水和选自下组的一种或多种氧合烃,该组由以下各项组成:单糖、二糖、低聚糖、多糖、糖醇、糖降解产物、纤维素衍生物、半纤维素衍生物、木质素衍生物、木质纤维素衍生物、以及它们的混合物以及(b)在存在一种缩合催化剂的情况下使该含氧化合物混合物反应以便产生包含c4+烷烃和芳基化合物的烃混合物,其中该烃混合物包含大于或等于50%cf的芳基化合物以及小于或等于20%cf的c4+烷烃。
附图简述
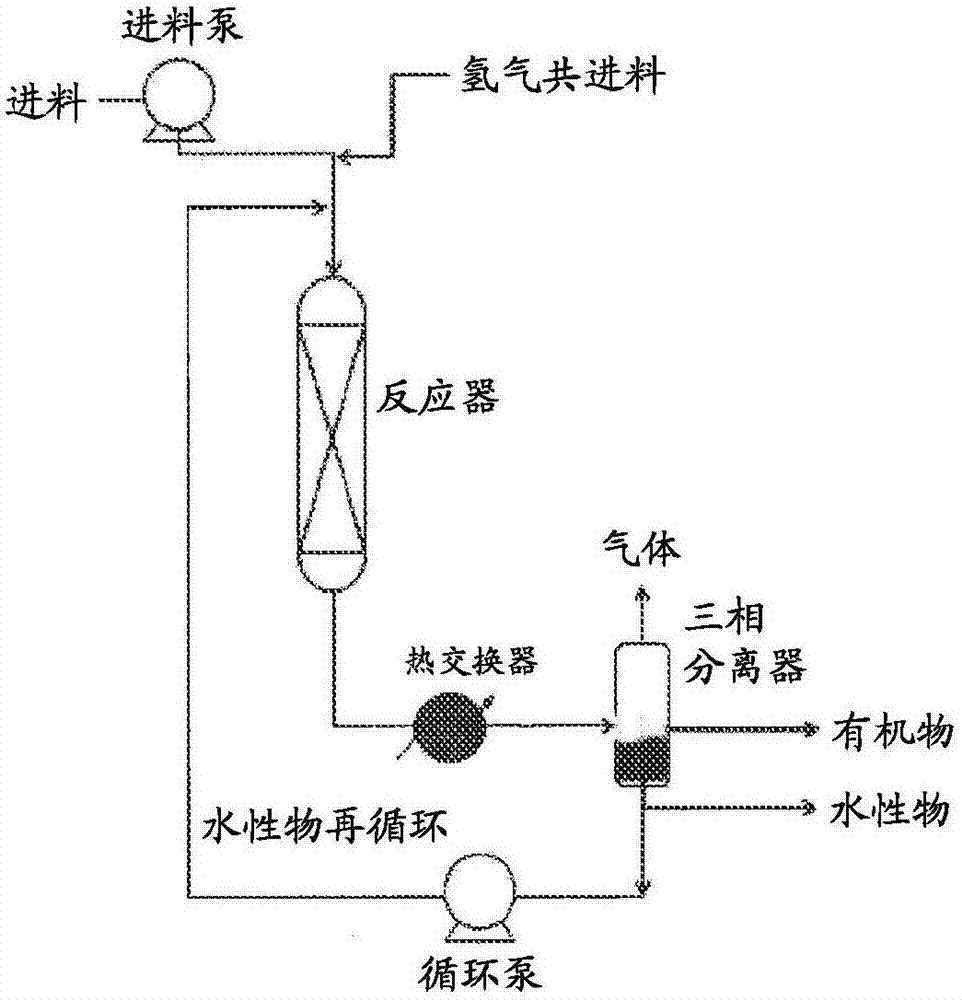
图1是用于将氧合烃转化成氧合化合物或用于将氧合化合物转化成烃的一个示例性流程图。
图2是用于将氧合烃转化成氧合化合物的包括一个任选的再循环液流的一个示例性流程图。
图3是用于将氧合烃转化成液体燃料和化学品的一个示例性工艺流程图,包括一个脱氧反应器、一个水性再循环液流、一个缩合反应器、以及一个蒸气相再循环液流。
图4是用于将氧合烃转化成液体燃料和化学品的一个示例性工艺流程图,包括一个脱氧反应器、一个水性再循环液流、以及一个缩合反应器。
图5是用于将氧合烃转化成液体燃料和化学品的一个示例性工艺流程图,包括一个脱氧反应器、一个水性再循环液流、一个缩合反应器、一个蒸气相再循环液流、以及一个液相(例如,蒸馏塔塔顶产物)再循环液流。
图6是说明脱氧催化剂组合物对产物分布的影响的一个示例性产物分布。该催化剂组合物是w-zro2上的2%pd2%mo0.5%sn(降温至300℃)以及w-zro2上的4%ni1%sn(降温至300℃)。
图7是说明脱氧催化剂载体对产物分布的影响的一个示例性产物分布。该催化剂组合物是mzro2上的2%pd2%mo0.5%sn13.5%w(降温至300℃)以及θ-氧化铝上的2%pd2%mo0.5%sn13.5%w(降温至300℃)。
图8是说明脱氧催化剂组合物和温度对产物分布的影响的一个示例性产物分布(例如,链烷烃、二氧化合物等)。
图9是说明脱氧催化剂组合物和温度对产物分布的影响的一个示例性产物分布(例如,酮、环醚等)。
图10是说明脱氧催化剂组合物和温度对产物分布的影响的识别的化合物的一个示例性碳分布。
图11是利用含有ni和sn的不同催化剂在一个分批反应器中生成的氧合化合物的一个比较。
图12是在0.5hr-1的whsv下利用含有ni和sn的不同催化剂在一个固定床反应器中生成的氧合化合物的一个比较。
图13是在1.0hr-1的whsv下利用含有ni和sn的不同催化剂在一个固定床反应器中生成的氧合化合物的一个比较。
图14是利用不同载体上含有ni3sn2合金催化剂的不同催化剂生成的氧合化合物的一个比较。
详细说明
本发明通常提供用于制备具有高产率的芳香族分子和低产率的烷烃和焦炭的生物质来源的化学品和燃料的方法。出人意料的是,本发明方法允许产生具有大于或等于50%芳香族分子,同时又具有小于或等于20%烷烃的一种烃混合物。
本发明还提供了用于从生物质制备含氧化合物混合物的方法,该含氧化合物混合物在一种缩合催化剂存在下反应以产生具有高产率的芳香族分子和低产率的烷烃的化学品和燃料。此外,本发明在产生芳香族化学品时还产生低产率的焦炭。该含氧化合物混合物通常会具有大于或等于0.5至小于或等于1.7的h:ceff比,这将实现出人意料地高的产率的芳香族分子,同时最小化烷烃的产率。该含氧化合物混合物还可以具有一个或多个以下属性:(1)比单氧化合物更多的二氧化合物和多氧化合物,(2)比单氧化合物更多的二氧化合物,(3)比c5-6含氧化合物(尤其是单氧化合物)更多的c2-4含氧化合物(尤其是二氧化合物和多氧化合物),和/或(4)很少乃至不存在烷烃。
该含氧化合物混合物可以源自任何来源,但也可以通过以下内容产生:在一种脱氧催化剂下,使含有具有三个或更多个碳原子的一种水溶性氧合烃的一种水性原料溶液与氢气反应来产生所希望的含氧化合物混合物。该含氧化合物混合物之后在一种缩合催化剂下,在有效引起一个缩合反应的温度和压力的条件下反应,该缩合反应产生高产率的芳香族分子和低产率的烷烃和焦炭。该氧合烃可以是单糖、二糖、多糖、纤维素、半纤维素、木质素、糖、糖醇或其他多羟基醇、糖降解产物,或可以来源于糖、糖醛、羧酸、酮、或呋喃的氢化,或糖、糖醇、多糖、单糖、二糖或多羟基醇的氢解。本发明还提供了对产生含氧化合物混合物有用的脱氧催化剂。
本发明的一个方面是产生具有高产率的芳香族分子和低产率的烷烃的一个烃液流。具体而言,本方法使得芳基化合物产率大于或等于50%cf,并且使得c4+烷烃产率小于或等于20%cf。在某些实施例中,该芳基化合物产率可以是大于或等于55%cf、大于或等于60%cf,或大于或等于65%cf。在某些实施例中,该c4+烷烃产率是小于或等于15%cf、小于或等于10%cf,或小于或等于5%cf。在某些其他实施例中,产物可以进一步包含总c1+烷烃产率小于或等于20%cf、小于或等于15%cf,小于或等于10%cf,或小于或等于5%cf的c1-3烷烃。%cf是通过将组分中的碳质量(例如,芳基化合物中的碳质量)除以进料中的碳质量再乘以100来计算。可替代地,%cf可以被记录为碳进料的百分比、碳百分比、或其他类似命名法。
在某些实施例中,该芳基化合物产率是大于或等于55%cf,并且该c4+烷烃产率是小于或等于15%cf。在另一个实施例中,该芳基化合物产率是大于或等于60%cf,并且该c4+烷烃产率是小于或等于10%cf。在另外的实施例中,该芳基化合物产率是大于或等于55%cf,并且该c1+烷烃产率是小于或等于15%cf。在又另一个实施例中,该芳基化合物产率是大于或等于60%cf,并且该c1+烷烃产率是小于或等于10%cf。
本发明中实现高芳香烃产率和低烷烃产率的出人意料的益处的一个方面是将含氧化合物进料混合物送入缩合反应器中。此外,本发明还实现了缩合催化剂上低焦炭产率的出人意料的益处。典型的生物质来源的来自糖、淀粉、半纤维素、纤维素等的氧合烃具有非常低的0.0左右的h:ceff比。由于这些生物质来源的化合物是如此富氧(相反地,贫氢),它们容易在缩合催化剂上堆积焦炭(coke-up)。另一方面,单氧化合物具有显著更高的h:ceff(对于醇,h:ceff等于2.0),并且容易导致大量烷烃产生,经常处于与所希望的芳香族分子相当的产率。用于产生高产率的芳香族分子,同时最小化产生的烷烃的量的一种理想的混合物将具有0.5与1.7之间的h:ceff比。通过比较,非常适于产生高产率的芳香族化合物和低产率的烷烃的氧合化合物具有2至4个碳原子以及2或3个氧原子。这些分子的h:ceff比通常是在0.5与1.5之间。实例包括c2-4二醇和三醇,如h:ceff为1的乙二醇、h:ceff为1.33的丙二醇、h:ceff为0.67的甘油、h:ceff为1.25的丁二醇、以及h:ceff为1的丁三醇。更小的二和/或多氧化合物如具有羧酸、羟基酮、或羟基醛部分的c2-4化合物和rcoor’酯(其中r是c1-3并且r’是c1-4)也可能落入所希望的h:ceff范围。
在不受任何特定理论限制的情况下,本发明人相信通过将相对贫氢的生物质来源的原料转化成c2-4o2-3含氧化合物变得可用的氢原子允许在缩合催化剂下利用在其他情况下并不可行的反应途径。这些反应途径包括可以直接产生烯烃中间体的反应。另外的烯烃中间体可以通过芳香族化合物形成时释放和转移氢来间接地生成,其中通过芳香族化合物的形成而释放的氢被转移至不饱和的含氧化合物,如酯、酮、醛、羧酸或其他氧合分子如二醇或多元醇。如在此所使用,将能够与氢以此方式反应的含氧化合物称为“氢受体”。据信,特征为具有小于2的h:ceff比的羰基化合物类、羧酸类、酯类、环醚类、二醇类、多元醇类、呋喃类以及其他含氧化合物能够直接地或者其他反应(如脱水)之后成为氢受体,这些其他反应已经将组分转化成氢受体。在接受氢之后,这些氢受体可以被转化成容易脱水形成烯烃或可能能够接受另外的氢的物质。
通常,该含氧化合物混合物将具有大于或等于0.5且小于或等于1.7的h:ceff比,以及一个或多个以下属性:(1)比单氧化合物更多的二氧化合物和多氧化合物,(2)比单氧化合物更多的二氧化合物,(3)比c5-6含氧化合物(尤其是单氧化合物)更多的c2-4含氧化合物(尤其是二氧化合物和多氧化合物),和/或(4)很少乃至不存在烷烃。
在大多数实施例中,该含氧化合物混合物将具有大于或等于0.5、大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、或大于或等于1.0的h:ceff比。该含氧化合物混合物还具有小于或等于1.7、小于或等于1.6、小于或等于1.5、或小于或等于1.4的h:ceff比。
该含氧化合物混合物通常还将具有大量二氧化合物和/或多氧化合物。在这类实施例中,该含氧化合物混合物可以具有大于或等于30%cf的二氧化合物和多氧化合物、大于或等于35%cf的二氧化合物和多氧化合物、大于或等于40%cf的二氧化合物和多氧化合物、大于或等于45%cf的二氧化合物和多氧化合物、大于或等于50%cf的二氧化合物和多氧化合物、大于或等于55%cf的二氧化合物和多氧化合物、大于或等于60%cf的二氧化合物和多氧化合物、大于或等于65%cf的二氧化合物和多氧化合物、大于或等于70%cf的二氧化合物和多氧化合物、大于或等于75%cf的二氧化合物和多氧化合物、大于或等于80%cf的二氧化合物和多氧化合物、大于或等于85%cf的二氧化合物和多氧化合物、大于或等于90%cf的二氧化合物和多氧化合物、或它们的任何间隔之间的任何%cf。在这种情况下,%cf是通过将组分中的碳质量(例如,二氧化合物和多氧化合物分子中的碳质量)除以该含氧化合物混合物中的碳质量再乘以100来计算。
在其他实施例中,该含氧化合物混合物可以具有大于或等于30%cf的二氧化合物、大于或等于35%cf的二氧化合物、大于或等于40%cf的二氧化合物、大于或等于45%cf的二氧化合物、大于或等于50%cf的二氧化合物、大于或等于55%cf的二氧化合物、大于或等于60%cf的二氧化合物、大于或等于65%cf的二氧化合物、大于或等于70%cf的二氧化合物、大于或等于75%cf的二氧化合物、大于或等于80%cf的二氧化合物、大于或等于85%cf的二氧化合物、大于或等于90%cf的二氧化合物、或它们的任何间隔之间的任何%cf。在这种情况下,%cf是通过将组分中的碳质量(例如,二氧化合物分子中的碳质量)除以该含氧化合物混合物中的碳质量再乘以100来计算。
在其他实施例中,该含氧化合物混合物可以具有大于或等于20%cf的二醇、大于或等于25%cf的二醇、大于或等于30%cf的二醇、大于或等于35%cf的二醇、大于或等于40%cf的二醇、大于或等于45%cf的二醇、大于或等于50%cf的二醇、大于或等于55%cf的二醇、大于或等于60%cf的二醇、大于或等于65%cf的二醇、大于或等于70%cf的二醇、大于或等于75%cf的二醇、大于或等于80%cf的二醇、或它们的任何间隔之间的任何%cf。在这种情况下,%cf是通过将组分中的碳质量(例如,二醇分子中的碳质量)除以该含氧化合物混合物中的碳质量再乘以100来计算。
在其他实施例中,该含氧化合物混合物可以具有小于或等于20%cf的单氧化合物、小于或等于15%cf的单氧化合物、小于或等于10%cf的单氧化合物、小于或等于9%cf的单氧化合物、小于或等于8%cf的单氧化合物、小于或等于7%cf的单氧化合物、小于或等于5%cf的单氧化合物、小于或等于5%cf的单氧化合物、小于或等于5%cf的单氧化合物、小于或等于4%cf的单氧化合物、小于或等于3%cf的单氧化合物、小于或等于2%cf的单氧化合物、小于或等于1%cf的单氧化合物、或它们的任何间隔之间的任何%cf。在这种情况下,%cf是通过将组分中的碳质量(例如,单氧化合物中的碳质量)除以该含氧化合物混合物中的碳质量再乘以100来计算。
在其他实施例中,该含氧化合物混合物可以具有小于或等于20%cf的醇、小于或等于15%cf的醇、小于或等于10%cf的醇、小于或等于9%cf的单氧化合物、小于或等于8%cf的醇、小于或等于7%cf的醇、小于或等于5%cf的醇、小于或等于5%cf的醇、小于或等于4%cf的醇、小于或等于3%cf的醇、小于或等于2%cf的醇、小于或等于1%cf的醇、或它们的任何间隔之间的任何%cf。在这种情况下,%cf是通过将组分中的碳质量(例如,醇中的碳质量)除以该含氧化合物混合物中的碳质量再乘以100来计算。
该含氧化合物混合物的第一可能的属性是二氧化合物和多氧化合物与单氧化合物的%cf比是大于或等于0.5,其中%cf比是通过将每个组分的%cf相除(即,%cf的二氧化合物和多氧化合物除以%cf的单氧化合物)来计算。在某些实施例中,二氧化合物和多氧化合物与单氧化合物的%cf比是大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、大于或等于1.0、大于或等于1.1、大于或等于1.2、大于或等于1.3、大于或等于1.4、大于或等于1.5、大于或等于1.6、大于或等于1.7、大于或等于1.8、大于或等于1.9、大于或等于2.0、大于或等于3.0、大于或等于4.0、大于或等于5.0、大于或等于6.0、大于或等于7.0、大于或等于8.0、大于或等于9.0、大于或等于10.0、大于或等于11.0、大于或等于12.0、大于或等于13.0、大于或等于14.0、大于或等于15.0、大于或等于20.0、大于或等于25.0、大于或等于35.0、大于或等于45.0、或它们的任何间隔之间的任何比例。二氧化合物和多氧化合物与单氧化合物的%cf比在某些实施例中还可以通过二醇和三醇与醇之比来更容易地测量。
该含氧化合物混合物的第二可能的属性是二氧化合物与单氧化合物的%cf比是大于或等于0.5,其中%cf比是通过将每个组分的%cf相除(即,%cf的二氧化合物除以%cf的单氧化合物)来计算。在某些实施例中,二氧化合物与单氧化合物的%cf比是大于或等于0.6、大于或等于0.7、大于或等于0.8、大于或等于0.9、大于或等于1.0、大于或等于1.1、大于或等于1.2、大于或等于1.3、大于或等于1.4、大于或等于1.5、大于或等于1.6、大于或等于1.7、大于或等于1.8、大于或等于1.9、大于或等于2.0、大于或等于3.0、大于或等于4.0、大于或等于5.0、大于或等于6.0、大于或等于7.0、大于或等于8.0、大于或等于9.0、大于或等于10.0、大于或等于11.0、大于或等于12.0、大于或等于13.0、大于或等于14.0、大于或等于15.0、大于或等于20.0、大于或等于25.0、大于或等于35.0、大于或等于45.0、或它们的任何间隔之间的任何比例。二氧化合物与单氧化合物的%cf比还可以通过二醇与醇之比来更容易地测量。如实例1中所示,这导致该含氧化合物混合物随着二醇与醇之比的增加而产生更大量的芳香族分子同时最小化不希望的烷烃的产生的出人意料的且意想不到的能力。在呈现最大比例时,缩合反应出人意料地产生大于或等于65cf%的芳香族化合物以及小于或等于10cf%的链烷烃。
该含氧化合物混合物的第三可能的属性是c2-4含氧化合物与c5-6含氧化合物的%cf比是大于或等于1.0,其中%cf比是通过将每个组分的%cf相除(即,%cf的c2-4含氧化合物除以%cf的c5-6含氧化合物)来计算。在某些实施例中,以水性碳原料的百分比计的c2-4含氧化合物与c5-6含氧化合物的%cf比是大于或等于1.0。在某些实施例中,c2-4含氧化合物与c5-6含氧化合物之比是大于或等于1.1、大于或等于1.2、大于或等于1.3、大于或等于1.4、大于或等于1.5、大于或等于1.6、大于或等于1.7、大于或等于1.8、大于或等于1.9、大于或等于2.0、大于或等于3.0、大于或等于4.0、大于或等于5.0、大于或等于6.0、大于或等于7.0、大于或等于8.0、大于或等于9.0、大于或等于10.0、大于或等于11.0、大于或等于12.0、大于或等于13.0、大于或等于14.0、大于或等于15.0、大于或等于20.0、大于或等于25.0、大于或等于35.0、大于或等于45.0、或它们的任何间隔之间的任何比例。在该含氧化合物混合物产生自具有大于或等于50%cf的c5-6氧合烃的生物质来源的氧合烃时,c2-4含氧化合物与c5-6含氧化合物的%cf比大于或等于1.0指示一些碳-碳键出现断裂。这反过来通过针对该含氧化合物混合物产生更多希望的分子来使h:ceff比增大。在不受任何特定理论限制的情况下,据信较短的c2-4含氧化合物能够更好地反应以形成所希望的芳香族分子,并且因此产生更少的不希望的烷烃。
该含氧化合物混合物的第四可能的属性是存在很少乃至不存在烷烃。在某些实施例中,该含氧化合物混合物可以包括烷烃,其中该含氧化合物混合物包括小于或等于10%cf的烷烃,其中%cf是通过将组分中的碳质量(例如,烷烃中的碳质量)除以该含氧化合物混合物中的碳质量再乘以100来计算。在其他实施例中,该含氧化合物混合物包括小于或等于9%cf的烷烃、小于或等于8%cf的烷烃、小于或等于7%cf的烷烃、小于或等于6%cf的烷烃、小于或等于5%cf的烷烃、小于或等于4%cf的烷烃、小于或等于3%cf的烷烃、小于或等于2%cf的烷烃、或小于或等于1%cf的烷烃。在某些实施例中,在该含氧化合物混合物产生自生物质来源的氧合烃时,烷烃构成小于或等于10%cf、小于或等于9%cf、小于或等于8%cf、小于或等于7%cf、小于或等于6%cf、小于或等于5%cf、小于或等于4%cf、小于或等于3%cf、小于或等于2%cf、或小于或等于1%cf,其中%cf是通过将组分中的碳质量(例如,烷烃中的碳质量)除以生物质来源的水性碳原料中的碳质量再乘以100来计算。
该含氧化合物混合物可以通过任何已知方法来产生。在一个实施例中,该含氧化合物混合物是使用催化重整技术生产的,如由维仁特公司(virent,inc.)(麦迪逊,威斯康星州(madison,wisconsin))开发的、并且在美国专利号7,767,867(科特赖特(cortright))、7,898,664(科特赖特)、8,053,615(科特赖特等人)、8,017,818(科特赖特等人)、以及7,977,517(科特赖特等人)中描述的生物重整技术,这些专利全部通过引用结合在此。替代的方法包括使用酶或微生物的发酵工艺、气化、热解、水热液化、溶剂解离、以及催化解构。该含氧化合物混合物还可以使用针对醇和其他混合含氧化合物的生产的一个或多个费托合成类型反应来源于天然气、合成气、或其他可再生或不可再生资源。用于产生含氧化合物混合物的其他已知方法对于本领域技术人员而言可能是已知的。该含氧化合物混合物还可以通过将源自多个过程和/或来源的含氧化合物组合来产生。
术语“生物重整”指代(非限制地)用于使用水相重整、氢化、氢解、氢化脱氧和/或涉及使用非均相催化剂的其他转化方法而将生物质和其他碳水化合物催化转化成分子量更低的烃类和氧合化合物的方法,如醇类、酮类、环醚类、酯类、羧酸类、醛类、二氧化合物类、以及其他聚氧合烃类。生物重整还包括将此类较低分子量的氧合化合物进一步催化转化成c4+化合物。
图1和图2提供了根据本发明能够产生一种含氧化合物混合物的生物重整系统的实例。图1、图3、图4、图5提供了根据本发明能够产生一种烃混合物的生物重整系统的实例。在这些示出的实施例中,一种水性原料在一种脱氧催化剂存在下与氢气反应,以产生h:ceff比大于或等于0.5且小于或等于1.7,且具有一种或多种上述属性的一种含氧化合物混合物,并且该含氧化合物混合物可以在一种缩合催化剂存在下反应以产生该烃混合物。
脱氧催化剂
脱氧催化剂通常是能够催化氢气与氧合烃之间的一个反应以产生所希望的含氧化合物混合物的一种异相催化剂。一般而言,该脱氧催化剂将包括结晶氧化铝载体和第viii族金属,如fe、co、ni、ru、rh、pd、os、ir、或pt。与第viii族金属单独作用相比,结晶氧化铝载体能够改变第viii族金属的活性,以有利地产生具有所希望的在0.5与1.7之间的h:ceff比并且在大多数情况下具有一个或多个上述属性的一种含氧化合物混合物。如实例33和图7中所示,在θ-氧化铝载体上制备脱氧催化剂产生基本上比在氧化锆载体上制备相同催化剂更多的二氧化合物和多氧化合物。使用结晶氧化铝载体相对于氧化锆载体基本上还降低单氧化合物的生产。最终,结晶氧化铝载体将烷烃的生产基本上降低至水性碳原料的10%以下。
该脱氧催化剂可以包括上述元素单独或结合第二金属,该第二金属来自第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、以及第va族,包括合金和它们的组合。该脱氧催化剂还可以包括另外的金属,它们来自第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、以及第va族,包括合金和它们的组合,这取决于具体原料和所希望的含氧化合物混合物。例如,该脱氧催化剂可以包括ni或pd,伴有第二金属sn或mo,或ni或pd,伴有第二金属sn和第三金属mo。在一个实施例中,该脱氧催化剂是pd或ni的一种异相催化剂以及一种结晶氧化铝载体。在另一个实施例中,该脱氧催化剂是ni和sn的一种异相催化剂以及一种结晶氧化铝载体,或一种ninsnm合金以及一种结晶氧化铝载体,如ni3sn1或ni3sn2。在又另一个实施例中,该脱氧催化剂是pd、mo和sn的一种异相催化剂以及一种结晶氧化铝载体,包括它们的合金。
第一第viii族金属的负载量是在0.25wt%至25wt%的范围内,其中具有0.10%与0.05%增量之间的重量百分比,诸如1.00%、1.10%、1.15%、2.00%、2.50%、5.00%、10.00%、12.50%、15.00%、以及20.00%。第二金属的优选的原子比是在0.25比1到10比1的范围内,包括之间的任何比率,如0.50、1.00、2.50、5.00以及7.50比1。该催化剂与该载体的组合是第viii族金属的0.25wt%至10wt%。
在一个实施例中,该催化剂载体是过渡氧化铝载体如θ-氧化铝载体。结晶氧化铝可以通过经由溶胶-凝胶加工或任何其他方法从铝盐进行沉淀而生产。该载体可以通过使用硝酸在有机粘合剂(如羟乙基纤维素)存在下使合适的氢氧化铝(优选勃姆石或假勃姆石)胶溶而制造。在形成之后,然后在900℃至1200℃之间,或大于或等于1000℃的最终温度下煅烧该载体。可以加入一种改性剂以改良氧化铝的构造或催化特性。这类改性剂包括而不限于:硫酸盐、二氧化硅、cr、nb、mg、zr、b、fe、ce、la、cu、co、mo、sn、或w。
也可以对载体进行处理或改性以增强其特性。例如,可以处理载体(如通过表面改性)以对表面部分(如氢和羟基)进行改性。表面的氢和羟基可能引起局部ph变化,这些变化影响了催化效率。也可以例如通过用硫酸盐、磷酸盐、钨、硅烷、镧系元素、碱性化合物或碱土化合物进行处理而对载体进行改性。
用于制备催化剂系统的常规方法在本领域中是众所周知的。常见方法包括初始润湿、蒸发浸渍、化学气相沉积、洗涤-涂布、磁控管溅射技术等。选择来制造脱氧催化剂的方法对该过程而言并不是至关重要的,前提条件是不同的催化剂和制备方法会产生不同的结果,这取决于考虑因素如总表面积、孔隙度等。在一个实施例中,该催化剂通过将第viii族金属与第二金属组合以产生一种混合金属氧化物来制备,该第二金属来自第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、以及第va族。混合金属氧化物之后沉积在结晶氧化铝载体上。在另一个实施例中,该催化剂通过以下各项制备:将来自第ib族、第iib族、第iiib族、第ivb族、第vb族、第vib族、第viib族、第viii族、第iiia族、第iva族、或第va族的一种金属与假勃姆石(氢氧化铝)混合组合,之后煅烧以制造一种混合氧化物载体,然后将第viii族金属沉积在载体上。在另一个实施例中,结晶氧化铝载体被煅烧以产生一个较小的表面积。在某些实施例中,氧化铝载体的煅烧可以在大于或等于800℃,或在800℃与1200℃之间的温度下执行。在一个实施例中,第viii族金属是ni,并且第二金属选自一种混合氧化铝载体上的第iva族或第viii族金属。在一个实施例中,第viii族金属是pd,并且第二金属选自第vib族或第iva族或者第vib族和第iva族两者。
在一个实施例中,在50与5000ml氢气/ml催化剂/hr之间的气体时空速度(ghsv)以及大气压与2000psig之间的压力下,该催化剂是利用氢气来还原。该催化剂是使用0.1℃/分钟与10℃/分钟之间的一个温度斜升达到20℃与600℃之间的一个温度来还原。一旦达到所希望的温度,接下来就是1小时到24小时的氢气浸透。在还原之后,使催化剂在一个惰性或还原环境中达到所希望的操作温度。在某些实施例中,在500至1000hr-1的ghsv、1.0℃/分钟至8.5℃/分钟的小时温度梯度、250℃与500℃之间的温度下用氢气还原该催化剂,之后是1至4小时的氢气浸透。
在一个替代的实施例中,该催化剂可以是预硫化的。在另一个替代的实施例中,该催化剂可以原位硫化。
原料
可用于本发明中的包含氧合烃的原料可以来源于任何来源,但是优选地是来源于生物质。原料可以是纯材料、纯化的混合物或原材料,如从对玉米、甘蔗、甜菜糖、稻谷、小麦、藻类或能源作物进行加工而衍生的糖和淀粉。原料也可以是作为大型工艺的一部分或在同一工艺中而形成的中间物,如在糖氢化的最初阶段产生的糖醇或从生物质的解构产生的糖降解产物。
如在此所使用,术语“木质纤维生物质”和“生物质”指代(而不限于)由植物(例如,木材、叶、根、种子、茎等)产生的有机材料、以及微生物和动物代谢废料。常见的生物质来源包括:(1)农作物残余物:如玉米秆、稻草、种子壳、甘蔗剩余物(sugarcaneleavings)、甘蔗渣、坚果壳、以及来自牛、家禽和猪的粪便;(2)木材材料:如木材、树皮、锯屑、木材碎片、以及工厂废品;(3)城市废物:如废纸和庭院修剪物;(4)能源作物:如杨树、柳树、松树、柳枝稷(switchgrass)、芒草、高粱、紫花苜蓿、草原蓝草(prairiebluestream)、玉米、大豆等;(5)来自工业过程的残余固体:如来自制浆工艺、酸水解、或酶水解的木质素;以及(6)藻类衍生的生物质,包括来自微藻类(例如,葡萄藻(botryococcusbraunii)、小球藻(chlorella)、杜氏盐藻(dunaliellatertiolecta)、江蓠属藻(gracilaria)、颗石藻(pleurochyrsiscarterae)、以及马尾藻(sargassum))和大型藻类(例如,海藻)的碳水化合物和脂类。该术语还指代以上的主要基础材料,即,木质素、纤维素、半纤维素、它们的衍生物、以及碳水化合物,如除了别的之外还有糖类(单糖、二糖、低聚糖、以及多糖)、糖(sugars)、以及淀粉。
术语“氧合烃”指代含有三个或更多个碳原子以及两个或更多个氧原子的水溶性烃,如碳水化合物(例如,单糖、二糖、低聚糖、多糖、以及淀粉),糖(例如,葡萄糖、蔗糖、木糖等),糖醇(例如,二醇、三醇、以及多元醇),以及糖降解产物(例如,羟甲基糖醛(hmf)、乙酰丙酸、甲酸、以及糠醛),它们各自在此表示为c3+o2+。如在此所使用,术语“氧合化合物”或“含氧化合物”指代具有两个或更多个碳原子以及一个或多个氧原子的分子(即,c2+o1+);术语“单氧化合物”指代含有两个或更多个碳原子以及一个氧原子的烃分子(即,c2+o1);术语“二氧化合物”指代含有两个或更多个碳原子以及两个氧原子的烃分子(即,c2+o2);并且术语“多氧化合物”指代含有两个或更多个碳原子以及三个或更多个氧原子的烃分子(即,c2+o3+)。
除了氧合烃之外,原料还可以包括木质素、一种或多种提取物、一种或多种灰分组分、或一种或多种有机物质(例如,木质素衍生物)。提取物包括萜类、芪类、类黄铜、酚酯(phenolics)、脂族化合物、木脂素类、烷烃、蛋白质材料、氨基酸、以及其他无机产品。灰分组分包括al、ba、ca、fe、k、mg、mn、p、s、si、zn等。其他无机物质包括4-乙基苯酚、4-乙基-2-甲氧基苯酚、2-甲氧基-4-丙基苯酚、香草醛、4-丙基丁香醇、维生素e、皮质类固醇、长链烃、长链脂肪酸、苗类化合物(stilbenoids)等。
一般而言,原料包括具有三个或更多个碳原子以及0.5:1至1:1.2之间的氧/碳比的任何氧合烃。在一个实施例中,该氧合烃具有3至12个碳原子或3至6个碳原子。在另一个实施例中,该氧合烃具有多于12个碳原子。对本发明而言优选的氧合烃是具有5或6个连续碳原子的氧合烃,包括总共具有多于5或6个碳原子的氧合烃。氧合烃的非限制性实例包括单糖、二糖、三糖、多糖、低聚糖、糖、糖醇、糖降解产物、多羟糖醇、半纤维素衍生物、纤维素衍生物、木质纤维素衍生物、木质素衍生物、淀粉、有机酸、多元醇等。在一个实施例中,该氧合烃包括多糖、低聚糖、三糖、二糖、单糖、糖、糖醇、糖降解产物、以及其他多羟基醇。在另一个实施例中,该氧合烃是三糖、二糖、糖,如葡萄糖、果糖、蔗糖、麦芽糖、乳糖、甘露糖或木糖;或是糖醇,如阿拉伯糖醇、赤藓糖醇、甘油、异麦芽酮糖醇、乳糖醇、麦芽糖醇、甘露糖醇、山梨糖醇、木糖醇、阿拉伯糖醇或乙二醇。这些氧合烃还可以包括通过对上述物进行氢化而衍生的醇。
在一个实施例中,原料可以包括通过一种溶剂来溶剂化的氧合烃。溶剂的非限制性实例包括:有机溶剂如离子性液体、丙酮、乙醇、4-甲基-2-戊酮、以及其他氧合烃;稀酸如乙酸、草酸、氢氟酸;生物重整溶剂;以及水。这些溶剂可以来自外部来源、进行再循环或原位生成,如原位生成的氧合化合物(例如,c2+o2+氧合烃)。
生产氧合化合物
生物重整的方法、工艺和技术在以下各项中进行了充分描述:美国专利号6,699,457;6,964,757;6,964,758;以及7,618,612(全部授予科特赖特等人,且名为“从氧合烃低温产生氢气(low-temperaturehydrogenproductionfromoxygenatedhydrocarbons)”);美国专利号6,953,873(授予科特赖特等人,且名为“从氧合烃低温产生烃(low-temperaturehydrocarbonproductionfromoxygenatedhydrocarbons)”);美国专利号7,767,867;7,989,664;8,198,486;8,492,595,以及美国专利申请公开号2013/0289302(全部授予科特赖特,且名为“用于产生多元醇的方法和系统(methodsandsystemsforgeneratingpolyols)”);美国专利号8,053,615;8,017,818;7,977,517;8,362,307;8,367,882;8,455,705以及美国专利申请公开号2011/0245542和2013/0185992(全部授予科特赖特和布鲁麦(blommel),且名为“从氧合烃合成液体燃料和化学品(synthesisofliquidfuelsandchemicalsfromoxygenatedhydrocarbons)”);美国专利号8,231,857(授予科特赖特,且名为“用于重整氧合化合物的催化剂和方法(catalystsandmethodsforreformingoxygenatedcompounds)”);美国专利号8,350,108(授予科特赖特等人,且名为“从生物质合成液体燃料(synthesisofliquidfuelsfrombiomass)”);美国专利申请序列号2011/0160482(授予永木(nagaki)等人,且名为“用于对多元醇氢化脱氧的改良的催化剂(improvedcatalystsforhydrodeoxygenationofpolyols)”);美国专利申请序列号2011/0009614(授予布鲁麦等人,且名为“用于将糖转化成糖醇的方法和反应器系统(processesandreactorsystemsforconvertingsugarstosugaralcohols)”);国际专利申请号pct/us2008/056330(授予科特赖特和布鲁麦,且名为“从氧合烃合成液体燃料和化学品”);共同拥有的美国专利号8,231,857(授予科特赖特等人,且名为“用于重整氧合化合物的催化剂和方法”);以及美国专利申请号13/586,499(授予布朗克(blank)等人,且名为“用于对氧合烃氢化脱氧的改良的催化剂(improvedcatalystsforhydrodeoxygenationofoxygenatedhydrocarbons)”),这些专利全部通过引用结合在此。本发明为当前的生物重整技术提供了一种改进,其在于上述催化剂能够产生一种含氧化合物混合物,以用于制备具有高产率的芳香族分子和低产率的烷烃的生物质来源的化学品和燃料。
为了产生含氧化合物混合物,将氧合烃与水组合以便提供一种水性原料溶液,该水性原料溶液具有的浓度有效地用于引起形成所希望的反应产物。水与碳之比按摩尔计优选地是从0.5:1至100:1,包括比率如1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1、15:1、25:1、50:175:1、100:1、以及它们之间的任何比率。该原料溶液的特征还可以是具有至少1.0重量百分比(wt%)的总溶液为氧合烃的一种溶液。例如,该溶液可以包括一种或多种氧合烃,其中该氧合烃在该溶液中的总浓度是按重量计是至少1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、或者更大,包括它们之间的任何百分比,并且这取决于所使用的氧合烃。在一个实施例中,该原料溶液包括按重量计至少10%、20%、30%、40%、50%、或者60%的一种糖,如葡萄糖、果糖、蔗糖、或者木糖,或者一种糖醇,如山梨糖醇、甘露糖醇、甘油或者木糖醇。在以上陈述范围之外的水与碳之比和百分比也被包括在内。
在一个实施例中,在脱氧催化剂的存在下,使该原料溶液与氢气在能有效产生所希望的含氧化合物混合物的温度、压力、以及重量时空速度下反应。所产生的特定的含氧化合物混合物将取决于不同的因素,包括原料溶液、反应温度、反应压力、水浓度、氢浓度、催化剂的反应性、以及原料溶液的流速(因为它影响了时空速度)(单位时间内单位催化剂中反应物的质量/体积)、气体时空速度(ghsv)、以及重量时空速度(whsv)。例如,流速的增加,以及因此随着时间的推移暴露于这些脱氧催化剂的原料的减少,将会限制可能发生的反应的程度,由此引起更高水平的二氧化合物和多氧化合物产率的增加,同时酮、醇以及环醚的产率降低。
优选地选择反应温度和压力以使反应器入口处的原料的至少一部分维持液相。然而,认识到也可以选择温度和压力条件以更有利地产生呈蒸气相的所希望的产物。一般而言,该反应应该在多种工艺条件下进行,其中建议反应的热力学是有利的。例如,使该原料的一部分维持处于液相所要求的最小压力将可能随着反应温度而变化。随着温度增加,一般将要求更高的压力来使该原料维持处于液相(如果希望的话)。以上用于使该原料维持处于液相(即,蒸气相)所要求的压力也是适合的操作条件。
一般而言,反应可以包括允许氧合烃原料在低于原料的焦糖化点的温度下部分脱氧的一个温度梯度。包括一个温度梯度有助于防止该原料中的氧合烃冷凝(例如,焦糖化)在该催化剂上,并且在整个反应器上产生可能导致反应器无法操作的一个大幅度压降。焦糖化点以及因此所要求的温度梯度将根据原料而变化。在一个实施例中,该温度梯度是在约300℃以下,或约80℃以上、或在约150℃至300℃之间、或在约200℃至290℃之间。在另一个实施例中,未采用一个温度梯度。
在冷凝相液体反应中,反应器内的压力必须足以使反应器入口处的反应物维持处于冷凝液相。对于液相反应,反应温度可以大于约80℃、或110℃、或120℃、或130℃、或140℃、或150℃、或160℃、或170℃、或180℃、或190℃、或200℃,并且小于约350℃、或325℃、或290℃、或280℃、或270℃、或260℃、或250℃、或240℃、或230℃、或220℃。反应压力可以大于约70psig、或85psig、或100psig、或115psig、或130psig、或145psig、或160psig、或175psig、或190psig、或205psig、或220psig、或235psig、或250psig、或265psig、或280psig、或295psig、或310psig、或325psig、或375psig、或425psig、或475psig、或550psig、或625psig、或775psig、或925psig、或1050psig,并且小于约3000psig、或2950psig、2900psig、2850psig、2800psig、2750psig、2700psig、2650psig、2600psig、2550psig、或2500psig、或2450psig、或2400psig、或2350psig、或2300psig、或2250psig、或2200psig、或2150psig、或2100psig、或2050psig、或2000psig、或1950psig、或1900psig、或1850psig、或1800psig。在某些实施例中,反应温度是在约120℃与300℃之间、或在约200℃与300℃之间、或在约270℃与290℃之间,并且反应压力是在约145与1950psig之间、或在约1000与1900psig之间、或在约1050与1800psig之间。
对于蒸气相反应,可以在氧合烃的蒸气压是至少约0.1atm,优选地更高(例如,350psig)并且反应的热力学保持有利时所处的一个温度下进行反应。这个温度将根据所使用的特定的氧合烃化合物和操作压力而变化,但是对于蒸气相反应通常大于约100℃、或120℃、或160℃、或200℃、或250℃,并且小于约600℃、或500℃、或400℃。在某些实施例中,反应温度是在约120℃与约500℃之间、或在约250℃与约400℃之间。
一般而言,该反应应该是在其中原料溶液在该催化剂上的停留时间是适当的以生成所希望的产物的条件下进行的。例如,用于反应的whsv可以是每克催化剂每小时至少约0.01克的氧合烃,并且更优选地该whsv是约0.01至40.0g/ghr,包括约0.01、0.025、0.05、0.075、0.1、0.25、0.5、0.75、1.0、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、3.0、3.1、3.2、3.3、3.4、3.5、3.6、3.7、3.8、3.9、4.0、4.1、4.2、4.3、4.4、4.5、4.6、4.7、4.8、4.9、5.0、6、7、8、9、10、11、12、13、14、15、20、25、30、35、40g/ghr,以及在它们之间的比率(包括0.77、0.78、0.79、2.61、2.62、2.63等)的whsv。
反应中使用的氢气优选地是外部氢气,但可以使用水相重整原位生成(原位生成的h2或aprh2),或aprh2、外部h2或再循环的h2的组合,或仅仅简单地是外部h2或再循环的h2。术语“外部h2”指代并非起源于原料溶液的,但是从一个外部来源添加到该反应器系统中的氢气。术语“再循环的h2”指代未消耗的氢气,它被收集并且然后再循环返回到该反应器系统中以用于进一步的使用。外部h2和再循环的h2还可以统称为或者单独地称为“补充的h2”。一般而言,可以添加补充h2以用于补充apr氢气,或者增加系统内反应压力,或者增加氢与碳和/或氧的摩尔比以便增强某些反应产物类型的生产产率的目的。
引入到该原料中的外部氢气或再循环的氢气的量(摩尔)可以是该原料中的一种或多种氧合烃的总摩尔数的约0%至2400%、5%至2400%、10%至2400%、15%至2400%、20%至2400%、25%至2400%、30%至2400%、35%至2400%、40%至2400%、45%至2400%、50%至2400%、55%至2400%、60%至2400%、65%至2400%、70%至2400%、75%至2400%、80%至2400%、85%至2400%、90%至2400%、95%至2400%、98%至2400%、100%至2400%、200%至2400%、300%至2400%、400%至2400%、500%至2400%、600%至2400%、700%至2400%、800%至2400%、900%至2400%、1000%至2400%、1100%至2400%、或1150%至2400%、或1200%至2400%、或1300%至2400%、或1400%至2400%、或1500%至2400%、或1600%至2400%、或1700%至2400%、或1800%至2400%、或1900%至2400%、或2000%至2400%、或2100%至2400%、或2200%至2400%、或2300%至2400%,包括它们之间的所有间隔。在原料溶液或它们的任何部分与原位生成的氢气和外部氢气或再循环的氢气反应时,原位生成的氢气与外部氢气(或再循环的氢气)的摩尔比是至少1:100、1:50、1:20、1:15、1:10、1:5、1:3、1:2、1:1、2:1、3:1、5:1、10:1、15:1、20:1、50:1、100:1以及它们之间的比率(包括4:1、6:1、7:1、8:1、9:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1以及19:1,并且反之亦然)。
含氧化合物再循环
再循环液流可以用于最大化产物产率并且降低催化剂失活。脱氧反应的产物包括所希望的c2+o2+氧合化合物和部分脱氧的烃(例如,二糖、单糖、糖、糖醇、多羟糖醇、重的有机酸、以及重的二醇、三醇和其他多元醇)。将这些部分脱氧的烃再循环返回到脱氧反应器系统中通过用部分脱氧的烃稀释富含碳水化合物的原料溶液来降低进入脱氧反应器系统中的碳水化合物的浓度。稀释高度反应性的碳水化合物进料流使脱氧反应器系统中的缩合反应最小化,以帮助避免原料冷凝在脱氧催化剂上,从而弄脏该催化剂,并且要求频繁的催化剂更换和/或再生。使用一个再循环液流还允许更高的进料流温度。在某些实施例中,优选的再循环与新鲜进料的重量比是在约0.25比1至10比1的范围内,包括它们之间的任何比率,如约0.50、1.00、2.50、4.00、5.00和7.50比1。
反应器系统
该脱氧反应可以在具有适合的设计的任何反应器中进行,包括连续流反应器、分批反应器、半分批反应器或多系统反应器,而不对设计、大小、几何形状、流速等进行限制。反应器系统还可以使用流化催化床系统、摇床系统、固定床系统、移动床系统、或前述的组合。在一个实施例中,该过程利用处于稳态平衡的一个连续流系统来进行。
图1(不具有一个水性再循环液流)和图2(具有一个水性再循环液流)是示出用于使用在一个载体上含有一种脱氧催化剂的单一反应器将一种生物质来源的氧合烃原料溶液转化成一种最终希望的产物的实施例的示意图。在一个实施例中,多个脱氧反应器用于控制反应放热。在某些实施例中,该原料溶液包括与一种或多种氧合烃组合的一种溶剂(例如,水、再循环的部分脱氧的烃等),该一种或多种氧合烃如碳水化合物(例如,单糖、二糖、低聚糖、多糖、以及淀粉),糖(例如,葡萄糖、蔗糖、木糖等),糖醇(例如,二醇、三醇、以及多元醇),和糖降解产物(例如,羟甲基糖醛(hmf)、乙酰丙酸、甲酸、以及糠醛)。如上所述,在某些实施例中,该原料还可以包括灰分组分、提取物、酚酯等。在一个实施例中,该原料经由一个泵馈送至一个载体上具有该脱氧催化剂的脱氧反应器系统,在其中该原料随后与氢气反应以生成一种含氧化合物混合物,该含氧化合物混合物具有大于或等于0.5且小于或等于1.7的h:ceff比,以及一个或多个上述属性。
在一个实施例中,使该混合物穿过一个三相分离器以将未冷凝的气体(如氢气、二氧化碳、甲烷、乙烷、以及丙烷)与一个有机产物流和一个水流分开来。未冷凝的气体是经由一个废气流去除。不可冷凝流可以被燃烧以产生工艺用热(即,用于驱动脱氧反应器中的反应的热),或被送到一个分离系统中,在其中氢气可以被回收用于再循环回到氢气流中。含有部分脱氧的烃的水流可以再循环回到反应器入口中。包括一些单氧化合物(例如,醇)的一个水性清洗流可以用于防止水积聚在反应器系统中。
缩合
通过上述方法产生的含氧化合物混合物可以收集并且用于工业应用中,或通过一种缩合催化剂催化的缩合反应转化成c4+化合物。具体而言,c4+化合物包括包含大于或等于水性碳原料的50%cf的芳基化合物以及包含小于或等于水性碳原料的20%cf的c4+烷烃。
不受任何特定理论限制,相信这些缩合反应总体上由一系列步骤组成,这些步骤涉及:(a)由含氧化合物脱水为烯烃;(b)烯烃的低聚;(c)裂解反应;(d)大量烯烃环化形成芳香族化合物;(e)烷烃异构化;(f)氢转移反应形成烷烃。这些反应也可以由一系列步骤组成,这些步骤涉及:(1)羟醛缩合形成β-羟基酮或β-羟基醛;(2)由β-羟基酮或β-羟基醛脱水形成共轭烯酮;(3)由共轭烯酮氢化形成酮或醛,它们可以参与后续的缩合反应或转化成醇或烃;以及(4)由羰基化合物氢化成醇,或反之亦然。其他缩合反应可以并行发生,包括羟醛缩合、普林斯反应(prinsreaction)、酸的酮化、以及狄尔斯-阿尔德缩合(diels-aldercondensation)。
缩合催化剂通常将是一种能够通过经由一个新的碳-碳键连接两种含氧物质,或其他官能化化合物(例如,烯烃)而形成更长链的化合物,并且将所得的化合物转化成烃、醇或酮的催化剂。该缩合催化剂可以包括而不限于:碳化物、氮化物、氧化锆、氧化铝、二氧化硅、硅铝酸盐、磷酸盐、沸石、氧化钛、氧化锌、氧化钒、氧化镧、氧化钇、氧化钪、氧化镁、氧化铈、氧化钡、氧化钙、氢氧化物、杂多酸、无机酸、酸改性的树脂、碱改性的树脂、以及它们的组合。该缩合催化剂可以包括以上物质,单独地或与一种改性剂相组合,该改性剂如ce、la、y、sc、p、b、bi、li、na、k、rb、cs、mg、ca、sr、ba、以及它们的组合。该缩合催化剂还可以包括一种金属,如cu、ag、au、pt、ni、fe、co、ru、zn、cd、ga、in、rh、pd、ir、re、mn、cr、mo、w、sn、os、它们的合金以及组合,以便提供一种金属官能性。
在某些实施例中,该缩合催化剂可以包括而不限于:碳化物、氮化物、氧化锆、氧化铝、二氧化硅、硅铝酸盐、磷酸盐、沸石(例如,zsm-5、zsm-11、zsm-12、zsm-22、zsm-23、zsm-35以及zsm-48)、氧化钛、氧化锌、氧化钒、氧化镧、氧化钇、氧化钪、氧化镁、氧化铈、氧化钡、氧化钙、氢氧化物、杂多酸、无机酸、酸改性的树脂、碱改性的树脂、以及它们的组合。该缩合催化剂还可以包括一种金属,如cu、ag、au、pt、ni、fe、co、ru、zn、cd、ga、in、rh、pd、ir、re、mn、cr、mo、w、sn、os、它们的合金以及组合,以便提供一种金属官能性。
该缩合催化剂可以是自负载的(即,该催化剂不需要另一种材料用作载体),或者可以要求一种单独的载体,该载体适合用于将该催化剂悬浮在该反应物流中。在某些实施例中,该载体是选自由氧化铝、二氧化硅、以及氧化锆组成的组。在其他实施例中,特别是在该缩合催化剂是一种粉末的情况下,该催化剂系统可以包括一种粘合剂以便辅助将该催化剂成形为一种所希望的催化剂形状。适用的成形方法包括挤出、粒化、油柱(oildropping)、或者其他已知的方法。还可以将氧化锌、氧化铝、以及一种造粒剂混合在一起并且挤出以便产生一种成形的材料。在干燥之后,将该材料在适合于成形催化活性相的温度下进行煅烧,这通常要求温度超过350℃。其他催化剂载体可以包括以下进一步详细描述的那些。
在一个实施例中,可以使用具有酸性官能性的一种催化剂进行该缩合反应。这些酸催化剂可以包括而不限于:硅铝酸盐(沸石)、氧化硅-氧化铝磷酸盐(sapo)、磷酸铝(alpo)、无定形氧化硅氧化铝、氧化锆、硫酸化氧化锆、钨酸化氧化锆、碳化钨、碳化钼、氧化钛、酸性氧化铝、磷酸化氧化铝、磷酸化氧化硅、硫酸化碳、磷酸化碳、酸性树脂、杂多酸、无机酸、以及它们的组合。在一个实施例中,该催化剂还可以包括一种改性剂,如ce、la、y、li、na、k、rb、cs、mg、ca、sr、ba、p、b、bi、以及它们的组合。该催化剂还可以通过添加一种金属如cu、ag、au、pt、ni、fe、co、ru、rh、zn、ga、in、pd、ir、re、mn、cr、mo、w、sn、os、它们的合金以及组合,和/或ti、zr、v、nb、ta、mo、cr、w、mn、re、al、ga、in、fe、co、ir、ni、si、cu、zn、sn、p、以及它们的组合的硫化物和氧化物进行改性以便提供金属官能性。钨酸化氧化锆(用于本方法的一种示例性催化剂)可以用cu、pd、ag、pt、ru、re、ni、sn、以及它们的组合进行改性。该酸催化剂可以是均相的、自负载的或者粘附到以下进一步描述的载体中的任一者上,这些载体包括含碳、二氧化硅、氧化铝、氧化锆、氧化钛、氧化钒、氧化铈、杂多酸、它们的合金和混合物的载体。
该缩合催化剂可以是一种沸石催化剂。如在此所使用的术语“沸石”不仅指代微孔结晶硅铝酸盐,而且还指代含微孔结晶金属的硅铝酸盐结构,如镓铝硅酸盐(galloaluminosilicate)和镓硅酸盐(gallosilicate)。在这类情况下,in、zn、fe、mo、ag、au、ni、p、y、ta、以及镧系元素也可以交换到沸石上以便提供所希望的活性。金属官能性可以通过金属,如cu、ag、au、pt、ni、fe、co、ru、zn、in、rh、pd、ir、re、mn、cr、mo、w、sn、os、它们的合金以及组合来提供。
该缩合催化剂可以包括一种或多种沸石结构,该一种或多种沸石结构包括氧化硅-氧化铝的笼样结构。沸石是具有明确限定的孔结构的、结晶的微孔材料。沸石含有可以在沸石的框架中产生的活性部位,通常是酸部位。这些活性部位的强度和浓度可以针对具体的应用而进行定制。用于缩合仲醇与烷烃的适合沸石的实例可以包括任选地用阳离子(如ga、in、zn、mo、以及此类阳离子的混合物)改性的硅铝酸盐,如在例如美国专利号3,702,886中所描述,该专利通过引用结合在此。如在本领域中认识到的,可以改变这种或这些特定沸石的结构以便在产物混合物中提供不同量的不同烃物质。取决于该沸石催化剂的结构,该产物混合物可以含有不同量的芳香烃和环烃。
适合的沸石催化剂的实例包括zsm-5、zsm-11、zsm-12、zsm-22、zsm-23、zsm-35以及zsm-48。沸石zsm-5和它的常规制备在美国专利号3,702,886;re.29,948(高硅质zsm-5);4,100,262以及4,139,600中进行了描述,这些专利通过引用结合在此。沸石zsm-11和它的常规制备在美国专利号3,709,979中进行了描述,该专利也通过引用结合在此。沸石zsm-12和其常规制备在美国专利号3,832,449中进行了描述,该专利通过引用结合在此。沸石zsm-23和其常规制备在美国专利号4,076,842中进行了描述,该专利通过引用结合在此。沸石zsm-35和其常规制备在美国专利号4,016,245中进行了描述,该专利通过引用结合在此。zsm-35的另一种制备在美国专利号4,107,195中进行了描述,该专利的披露内容通过引用结合在此。zsm-48和它的常规制备由美国专利号4,375,573来传授,该专利通过引用结合在此。沸石催化剂的其他实例在美国专利5,019,663和美国专利7,022,888中进行了描述,这些专利同样通过引用结合在此。一种示例性缩合催化剂是用cu、pd、ag、pt、ru、re、ni、sn、或它们的组合进行改性的zsm-5沸石。
如在美国专利7,022,888中所描述,该缩合催化剂可以是一种双官能五硅环(pentasil)沸石催化剂,该催化剂包括来自下组的至少一种金属元素:cu、ag、au、pt、ni、fe、co、ru、zn、cd、in、rh、pd、ir、re、mn、cr、mo、w、sn、os、它们的合金以及组合,或者一种来自下组的改性剂:in、zn、fe、mo、au、ag、y、sc、ni、p、ta、镧系元素、以及它们的组合。该沸石可以具有强酸部位,并且可以在低于580℃的温度下与含有一种氧合烃的反应物流一起使用。该双官能五硅环沸石可以具有由大量5元氧环(即,五硅环)组成的zsm-5、zsm-8或zsm-11类型的晶体结构。在一个实施例中,该沸石将具有一个zsm-5类型结构。
可替代地,固体酸催化剂,如用磷酸盐、氯化物、二氧化硅以及其他酸性氧化物改性的氧化铝可以用于该方法中。另外,硫酸化氧化锆、磷酸化氧化锆、二氧化钛氧化锆、或钨酸化氧化锆可以提供必要的酸性。re和pt/re催化剂对于促进将含氧化合物缩合成c5+烃和/或c5+单氧化合物也是有用的。re对于促进酸催化的缩合是足够酸性的。在某些实施例中,还可以通过添加硫酸盐或磷酸盐而将酸性添加至活性炭。
所产生的具体c4+化合物将取决于不同的因素,包括而不限于:反应物流中的氧合化合物的类型、缩合温度、缩合压力、催化剂的反应性、以及反应物流的流速(因为它影响了空间速度:ghsv、lhsv、以及whsv)。在某些实施例中,在对于产生所希望的烃产物适当的whsv下使该反应物流与该缩合催化剂相接触。在一个实施例中,whsv是反应物流中的至少0.1克的挥发性(c2+o1-3)含氧化合物每克催化剂每小时。在另一个实施例中,该whsv是0.1至10.0g/ghr之间,包括1、2、3、4、5、6、7、8、9、10g/ghr、以及它们之间的增量的whsv。
在某些实施例中,该缩合反应是在所提出的反应的热力学是有利的温度和压力下进行的。对于挥发性c2+o1-3含氧化合物,该反应可以在其中这些挥发性含氧化合物的蒸气压是至少0.1atm(并且优选显著更高)的温度下进行。该缩合温度将根据这些氧合化合物的特定组成而改变。该缩合温度通常将大于80℃、或100℃、或125℃、或150℃、或175℃、或200℃、或225℃、或250℃,并且小于500℃、或450℃、或425℃、或375℃、或325℃、或275℃。例如,该缩合温度可以是在约80℃至500℃之间、或在约125℃至450℃之间、或在约250℃至425℃之间。缩合压力通常将大于0psig、或10psig、或100psig、或200psig,并且小于2000psig、或1800psig、或1600psig、或1500psig、或1400psig、或1300psig、或1200psig、或1100psig、或1000psig、或900psig、或700psig。例如,该缩合压力可以是大于0.1atm、或在0与1500psig之间、或在0与1200psig之间。
缩合产物
本发明的缩合反应可以用于产生c4+烷烃、c4+烯烃、c5+环烷烃、c5+环烯烃、芳基化合物、稠合芳基化合物、多环化合物、c4+醇、c4+酮、c4+呋喃、以及它们的混合物,其中芳基化合物具有有利的高比例,而烷烃具有低比例。具体而言,使用上述含氧化合物混合物使得芳基化合物产率大于或等于水性碳原料的50%cf,并且使得c4+烷烃产率小于或等于水性碳原料的20%cf。在某些实施例中,该芳基化合物产率可以是大于或等于水性碳原料的55%cf、大于或等于60%cf,或大于或等于65%cf。在某些实施例中,该c4+烷烃产率是小于或等于水性碳原料的15%cf、小于或等于10%cf,或小于或等于5%cf。在某些其他实施例中,产物可以进一步包含总c1+烷烃产率小于或等于水性碳原料的20%cf、小于或等于15%cf、小于或等于10%cf、或小于或等于5%cf的c1-3烷烃。
在某些实施例中,该芳基化合物产率是大于或等于水性碳原料的55%cf,并且c4+烷烃产率是小于或等于水性碳原料的15%cf。在另一个实施例中,该芳基化合物产率是大于或等于水性碳原料的60%cf,并且该c4+烷烃产率是小于或等于水性碳原料的10%cf。在另外的实施例中,该芳基化合物产率是大于或等于水性碳原料的55%cf,并且c1+烷烃产率是小于或等于水性碳原料的15%cf。仍然在其他实施例中,该芳基化合物产率是大于或等于水性碳原料的60%cf,并且c1+烷烃产率是小于或等于水性碳原料的10%cf。
这些c4+烷烃和c4+烯烃具有从4个至30个碳原子(c4-30烷烃和c4-30烯烃)并且可以是支链的或直链的烷烃或烯烃。这些c4+烷烃和c4+烯烃还可以相应地包括c4-9、c7-14、c12-24烷烃和烯烃的部分,其中c4-9部分是针对汽油,c7-16部分是针对喷气燃料,并且c11-24部分是针对柴油燃料和其他工业应用如化学品。各种c4+烷烃和c4+烯烃的实例包括而不限于:丁烷、丁烯、戊烷、戊烯、2-甲基丁烷、己烷、己烯、2-甲基戊烷、3-甲基戊烷、2,2-二甲基丁烷、2,3-二甲基丁烷、庚烷、庚烯、辛烷、辛烯、2,2,4,-三甲基戊烷、2,3-二甲基己烷、2,3,4-三甲基戊烷、2,3-二甲基戊烷、壬烷、壬烯、癸烷、癸烯、十一烷、十一碳烯、十二烷、十二碳烯、十三烷、十三碳烯、十四烷、十四碳烯、十五烷、十五碳烯、十六烷、十六碳烯、十七烷、十七碳烯、十八烷、十八碳烯、十九烷、十九碳烯、二十烷、二十碳烯、二十一烷、二十一碳烯、二十二烷、二十二碳烯、二十三烷、二十三碳烯、二十四烷、二十四碳烯、以及它们的异构体。
c5+环烷烃和c5+环烯烃具有从5个至30个碳原子并且可以是未取代的、单取代的、或者多取代的。在单取代的和多取代的化合物的情况下,所取代的基团可以包括支链的c3+烷基、直链的c1+烷基、支链的c3+烯基、直链的c2+烯基、苯基或它们的组合。通过举例,所取代的基团中的至少一个包括支链的c3-12烷基、直链的c1-12烷基、支链的c3-12烯基、直链的c1-12烯基、直链的c2-12烯基、苯基或它们的组合。通过进一步举例,所取代的基团中的至少一个包括支链的c3-4烷基、直链的c1-4烷基、支链的c3-4烯基、直链的c1-4烯基、直链的c2-4烯基、苯基或它们的组合。所希望的c5+环烷烃和c5+环烯烃的实例包括而不限于:环戊烷、环戊烯、环己烷、环己烯、甲基-环戊烷、甲基-环戊烯、乙基-环戊烷、乙基-环戊烯、乙基-环己烷、乙基-环己烯、丙基-环己烷、丁基-环戊烷、丁基-环己烷、戊基-环戊烷、戊基-环己烷、己基-环戊烷、己基-环己烷、以及它们的异构体。
芳基化合物将总体上由处于未取代的(苯基)、单取代的、亦或多取代的形式的芳香烃组成。在单取代的和多取代的化合物的情况下,所取代的基团可以包括支链的c3+烷基、直链的c1+烷基、支链的c3+烯基、直链的c2+烯基、苯基或它们的组合。通过举例,所取代的基团中的至少一个包括支链的c3-12烷基、直链的c1-12烷基、支链的c3-12烯基、直链的c2-12烯基、苯基或它们的组合。通过进一步举例,所取代的基团中的至少一个包括支链的c3-4烷基、直链的c1-4烷基、支链的c3-4烯基、直链的c2-4烯基、苯基或它们的组合。各种芳基化合物的实例包括而不限于:苯、甲苯、二甲苯(xylene)(二甲基苯(dimethylbenzene))、乙基苯、对二甲苯、间二甲苯、邻二甲苯、c9+芳香族化合物、丁基苯、戊基苯、己基苯、庚基苯、辛基苯、壬基苯、癸基苯、十一烷基苯、以及它们的异构体。
稠合的芳基化合物将总体上由处于未取代的、单取代的、亦或多取代的形式的二环的和多环的芳香烃组成。在单取代的和多取代的化合物的情况下,所取代的基团可以包括支链的c3+烷基、直链的c1+烷基、支链的c3+烯基、直链的c2+烯基、苯基或它们的组合。通过举例,所取代的基团中的至少一个包括支链的c3-4烷基、直链的c1-4烷基、支链的c3-4烯基、直链的c2-4烯基、苯基或它们的组合。各种稠合的芳基化合物的实例包括而不限于:萘、蒽、以及它们的异构体。
多环化合物将总体上由处于未取代的、单取代的、亦或多取代的形式的二环的和多环的烃组成。虽然多环化合物通常包括如在此所使用的稠合芳基化合物,但多环化合物通常具有至少一个饱和的或部分饱和的环。在单取代的和多取代的化合物的情况下,所取代的基团可以包括支链的c3+烷基、直链的c1+烷基、支链的c3+烯基、直链的c2+烯基、苯基或它们的组合。通过举例,所取代的基团中的至少一个包括支链的c3-4烷基、直链的c1-4烷基、支链的c3-4烯基、直链的c2-4烯基、苯基或它们的组合。各种稠合的芳基化合物的实例包括而不限于:四氢化萘和十氢萘、以及它们的异构体。
c4+醇还可以是环状的、支链的或直链的,并且具有从4个至30个碳原子。一般而言,c4+醇可以是根据化学式r1-oh的化合物,其中r1是选自下组的一个成员,该组由以下各项组成:支链的c4+烷基、直链的c4+烷基、支链的c4+烯基、直链的c4+烯基、取代的c5+环烷烃、未取代的c5+环烷烃、取代的c5+环烯烃、未取代的c5+环烯烃、芳基、苯基、以及它们的组合。所希望的c4+醇的实例包括而不限于:丁醇、戊醇、己醇、庚醇、辛醇、壬醇、癸醇、十一烷醇、十二烷醇、十三烷醇、十四烷醇、十五烷醇、十六烷醇、十七烷醇、十八烷醇、十九烷醇、二十烷醇、二十一烷醇、二十二烷醇、二十三烷醇、二十四烷醇、以及它们的异构体。
c4+酮还可以是环状的、支链的或直链的,并且具有从4个至30个碳原子。一般而言,c4+酮可以是根据以下化学式的化合物
其中r3和r4独立地是选自下组的一个成员,该组由以下各项组成:支链的c3+烷基、直链的c1+烷基、支链的c3+烯基、直链的c2+烯基、取代的c5+环烷烃、未取代的c5+环烷烃、取代的c5+环烯烃、未取代的c5+环烯烃、芳基、苯基、以及它们的组合。所希望的c4+酮的实例包括而不限于:丁酮、戊酮、己酮、庚酮、辛酮、壬酮、癸酮、十一烷酮、十二烷酮、十三烷酮、十四烷酮、十五烷酮、十六烷酮、十七烷酮、十八烷酮、十九烷酮、二十烷酮、二十一烷酮、二十二烷酮、二十三烷酮、二十四烷酮、以及它们的异构体。
液体燃料与化学品
可以将源自上述缩合反应的c4+化合物分馏,并且用在富含芳香族化合物的液体燃料如汽油、喷气燃料(煤油)或柴油燃料之中。也可以将这些c4+化合物分馏,并且用在多个化学过程之中,如石油化学工业普遍已知的那些化学过程。例如,可以将来自该过程的产物流分馏,以便收集二甲苯,以用于在邻苯二甲酸、聚对苯二甲酸乙二酯(pet)、以及最终可再生的塑料或溶剂的生产中使用。还可以收集苯并进行处理以用于生产可再生的聚苯乙烯、聚碳酸酯、聚氨酯、环氧树脂、酚醛树脂、以及尼龙。可以收集甲苯并进行处理以用于生产尤其是二异氰酸甲苯酯、以及最终可再生的溶剂、聚氨酯泡沫或tnt。
在一个实施例中,源自该方法的c4+化合物通过针对液体燃料组合物已知的任何手段来分离成不同的馏分。在这类应用中,具有源自该方法的至少一种c4+化合物的产物流被分离成多于一个馏分,其中这些馏分中的至少一个是一个较轻的、中等的、或较重的部分。较轻的部分(主要是c4-9,即c4、c5、c6、c7、c8、以及c9)可以被分离用于汽油用途。中等的部分(主要是c7-14,即c7、c8、c9、c10、c11、c12、c13以及c14)可以被分离用作煤油例如喷气燃料用途。较重的部分(主要是c12-24,即c12、c13、c14、c15、c16、c17、c18、c19、c20、c21、c22、c23、以及c24)可以被分离用于柴油燃料用途。最重的部分(c25+和c30+,即,c25、c26、c27、c28、c29、c30、c31、c32、c33、c34、c35等)可以用作润滑剂、燃料油,或可以被裂解以便产生另外的多个部分,以用于在汽油、煤油和/或柴油部分中使用。
由于这些c4+化合物是来源于生物质,因此这些化合物、或含有这些化合物的部分的年龄是小于500岁、优选小于40岁、更优选小于20岁,如从组分的14c浓度所计算。
以下实例是说明性的,并且不应理解为限制本发明的保护范围,该保护范围由随附权利要求书来限定。
实例
实例1生产芳香族分子
由如表1中所定义的含氧化合物和水的混合物构成的进料在一种ni-改性的zsm-5催化剂的存在下反应,以便确定进料组成对产物选择性和产率的影响。图1中示出了反应器系统。催化剂作为8英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在350℃、100psig以及每小时每克催化剂2克氧合烃的重量时空速度(whsv)下。
表1进料组成。
表2示出了模拟进料(modelfeeds)至烃产物的转化。每种进料都展现出相当的进料转化率,并且至少98%的碳进料被转化成烃产物。虽然原料展现出类似的总转化率,但产物选择性会显著变化。由于进料中的丙二醇的含量增加,观察到了芳香烃的增加,以及烷烃生产的减少。正如表2所示,芳香烃生产的增加是以烷烃生产为代价的。
表2随着进料组成变化的产物组成。
实例2生产芳香族分子
由如表3中所定义的含氧化合物和水的混合物构成的进料在一种ni-改性的zsm-5催化剂的存在下反应,以便确定进料组成对产物选择性和产率的影响。图1中示出了反应器系统。催化剂作为8英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在350℃、100psig以及每小时每克催化剂2克氧合烃的重量时空速度(whsv)下。
表3进料组成
表4示出了模拟进料至烃产物的转化。每种进料都展现出相当的进料转化率,并且至少96%的碳进料被转化成烃产物。虽然原料展现出类似的总转化率,但产物选择性会显著变化。由于进料中的丙酸丙酯的含量增加,观察到了芳香烃的增加。相比之下,烷烃生产随着进料中丙二醇的增加而减少。由于h:ceff降低,芳香烃生产增加,并且烷烃生产减少。
表4随着进料组成变化的产物组成
实例3生产芳香族分子
由如表5中所定义的含氧化合物和水的混合物构成的进料在存在一种ni-改性的zsm-5催化剂的情况下反应,以便确定进料组成对产物选择性和产率的影响。图1中示出了反应器系统。催化剂作为8英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在350℃、100psig以及每小时每克催化剂2克氧合烃的重量时空速度(whsv)下。
表5进料组成
表6示出了模拟进料至烃产物的转化。每种进料都展现出相当的进料转化率,并且至少97%的碳进料被转化成烃产物。虽然原料展现出类似的总转化率,但产物选择性会显著变化。由于进料中的丙酸的含量增加,观察到了芳香烃的增加。相比之下,烷烃生产随着进料中丙二醇的增加而减少。由于h:ceff降低,芳香烃生产增加,并且烷烃生产减少。
表6随着进料组成变化的产物组成
实例4nisn催化剂合成
在一个烧杯中,用水将4.515g五水四氯化锡(tin(iv)chloridepentahydrate)(赖德尔·德·亨公司(riedeldehaen))稀释成45ml,并且经由初始润湿技术添加至75克的结晶氧化铝(诺普罗公司(norpro))。将材料转移到一个静态烘箱中,并且在120℃下干燥3小时。在干燥之后,将材料放置在配备有以30立方英尺/小时(scfh)流动的空气扫气的一个马弗炉中。通过按1.6℃/分钟斜升至400℃并保持6小时来煅烧该催化剂。在冷却之后,经由初始润湿将含有32.766g硝酸镍(ii)(阿法埃莎公司(alfaaesar)的45ml溶液添加至该催化剂。将材料转移到一个静态烘箱中,并且在120℃下干燥3小时。在干燥之后,将材料放置在配备有以30立方英尺/小时流动的空气扫气的一个马弗炉中。
实例5生产含氧化合物
来自实例4的脱氧催化剂在表7中概述的三种不同的温度分布下进行测试,以检查温度对进料转化和产物选择性的影响。图2中示出了反应器系统。催化剂作为一个10英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在表7中概述的所希望的温度分布、1050psig、每小时每克催化剂0.40克糖的重量时空速度(whsv)、每1摩尔碳进料约2摩尔h2的氢气共进料、以及1质量流速水再循环比1质量流速进料的水再循环比下。该进料是由45wt%葡萄糖、15wt%木糖、以及40wt%的具有约1000ppm丙酸的水构成以防止细菌生长。
表7温度分布。
如表8中所示,几乎所有糖在每一个温度分布下都进行转化。由于温度分布出现增加,观察到了产物选择性从糖醇至主要为二醇和未识别的水性化合物的转变。虽然产物组成的一部分未被识别,但未识别的化合物并不是单氧化合物并且最可能是二氧化合物和多氧化合物。
表8随着温度分布变化的产物组成。
实例6脱氧催化剂合成
在单斜晶氧化锆上使用初始润湿技术来制备含有1wt%pd、1wt%mo、0.25wt%sn、以及5wt%w的一种三金属催化剂。经由初始润湿将体积等于单斜晶氧化锆的初始润湿体积且含有12.18g氧化钨铵水合物(阿法埃莎公司)的一种水溶液添加至165g单斜晶氧化锆(诺普罗公司)。将催化剂在130℃下干燥2小时。之后在空气中以1.6℃/分钟将该催化剂从环境温度煅烧至400℃。一旦达到所希望的温度,就将催化剂浸透在空气中持续另外的6个小时。经由初始润湿将体积等于有待浸渍的催化剂的初始润湿体积且含有1.22g五水四氯化锡(赖德尔·德·亨公司)的2摩尔硝酸铵(西格玛·奥德里奇公司(sigmaaldrich))的一种水溶液添加至所煅烧的催化剂。将催化剂在130℃下干燥2小时。之后在空气中以2℃/分钟将该催化剂从环境温度煅烧至200℃。从200℃至220℃,温度斜升减缓至0.1℃/分钟。最终,从220℃至400℃,温度斜升率增加至2℃/分钟。一旦达到所希望的温度,就将催化剂浸透在空气中持续另外的6个小时。将体积等于有待浸渍的催化剂的初始润湿体积且含有3.064g四水合钼酸铵(西格玛·奥德里奇公司)的2摩尔硝酸铵(西格玛·奥德里奇公司)的一种水溶液添加至所煅烧的催化剂。将催化剂在130℃下干燥2小时。将体积等于有待浸渍的催化剂的初始润湿体积且含有4.193g硝酸钯(ii)水合物(阿法埃莎公司)的2摩尔硝酸铵(西格玛·奥德里奇公司)的一种水溶液添加至所煅烧的催化剂。将催化剂在130℃下干燥2小时。之后在空气中以2℃/分钟将该催化剂从环境温度煅烧至200℃。从200℃至220℃,温度斜升减缓至0.1℃/分钟。最终,从220℃至400℃,温度斜升率增加至2℃/分钟。一旦达到所希望的温度,就将催化剂浸透在空气中持续另外的6个小时。
实例7生产含氧化合物
实例8将通过来自实例4和实例6的两种不同的脱氧催化剂产生的含氧化合物产物组成进行比较。使用如图2中所示的一个反应器系统来对该催化剂进行操作以产生一种氧合烃混合物。
来自实例6的脱氧催化剂作为一个11.5英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在170℃至280℃的温度分布、1800psig、每小时每克催化剂0.40克糖的重量时空速度(whsv)、每1摩尔碳进料约1.5摩尔h2的氢气共进料、以及4质量流速水再循环比1质量流速进料的水再循环比下。该进料是由60wt%蔗糖和40wt%的具有约1000ppm丙酸的水构成以防止细菌生长。
来自实例4的脱氧催化剂作为一个10英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在175℃至270℃的温度分布、1050psig、每小时每克催化剂0.40克糖的重量时空速度(whsv)、每1摩尔碳进料约2摩尔h2的氢气共进料、以及1质量流速水再循环比1质量流速进料的水再循环比下。该进料是由45wt%葡萄糖、15wt%木糖、以及40wt%的具有约1000ppm丙酸的水构成以防止细菌生长。
表9示出了每种脱氧催化剂的产物分布是不同的。来自实例6的催化剂如由链烷烃和单氧化合物物质的增加的生产所显示导致更大程度的脱氧。相比之下,来自实例4的催化剂如由二氧化合物和未识别的水性物质的生产所突出显示产生烃与更多的氧。这些结果展示了来自实例4的催化剂从氧合烃进料去除一些氧以增加h:ceff比。这些结果还展示了实例4的催化剂比实例6的催化剂去除更少的氧,这导致实例4的催化剂产生具有比实例6的催化剂更低的h:ceff比的一种产物组成。
表9脱氧产物组成。
表10示出了该产物组成的更详细的分类(breakdown)。来自实例6的脱氧催化剂主要产生醇和环醚单氧化合物。相比之下,实例4的脱氧催化剂主要产生二醇和未识别的水性化合物,这些未识别的水性化合物被认为是二氧化合物和多氧化合物。
表10详细的产物组成。
表11示出了产物中识别的碳物质的碳数衰减值(breakdown)。来自实例6的催化剂维持糖进料的碳主链并且产生少量长链化合物。相比之下,实例4的催化剂产生主要是在c2-c4范围内的短链分子。
表11碳数衰减值。
总而言之,表9至表11示出了产物组成是不同的。实例4的脱氧催化剂的产物倾向于是具有2至4个碳原子以及2或3个氧原子的氧合化合物。这种类型的分子经常具有近似在0.5与1.5之间的h:ceff比。因此,实例4的脱氧催化剂的产物组成具有比实例6的催化剂更低的h:ceff。
实例8生产芳香族分子
在一种ni-改性的zsm-5缩合催化剂下,在下游处理来自实例7的产物,以评价烃产率和选择性、短期催化剂失活、以及方法可操作性。分别在图3和图4中所示的反应器系统中测试来自实例6的脱氧催化剂和来自实例4的脱氧催化剂。在作为两个13.5英寸的填充床加载到1英寸外径的inconel反应器中的一种ni-改性的zsm-5缩合催化剂下,处理来自实例6的脱氧催化剂的产物。条件设定在400℃、100psig、每小时每克催化剂0.4克糖的重量时空速度(whsv)、以及1.9质量流速蒸气再循环比1质量流速进料的蒸气再循环比下。
在作为一个12英寸的填充床加载到1英寸外径的inconel反应器中的一种ni-改性的zsm-5缩合催化剂下,处理来自实例4的脱氧催化剂的产物。条件设定在375℃、100psig、每小时每克催化剂0.4克糖的重量时空速度(whsv)、以及无蒸气再循环下。
表12示出了不同的含氧化合物进料对烃产物产率和选择性的影响。在与实例6的脱氧催化剂进行比较时,使用实例4的脱氧催化剂增加了芳香族化合物产率并且降低了链烷烃产率。最显著的是,通过使用实例4的脱氧催化剂极大地降低了链烷烃生产,并且几乎不到16%的碳进料最终转化成一种链烷烃产物。大部分这种降低归因于产生了相当少的不希望的c4-链烷烃。
还观察到了焦炭沉积的差异。通过处理使用来自实例6的脱氧催化剂和来自实例4的脱氧催化剂产生的产物,之后进行氧化再生来实现ni-改性的zsm-5缩合催化剂中的碳去除。来自实例6的脱氧催化剂产生了平均2%左右的作为焦炭馈送的碳,而来自实例4的脱氧催化剂产生了平均5%的作为焦炭馈送的碳。然而,焦炭产率的差异并未转换成短期催化剂失活的差异。由于通过来自实例4和实例6的脱氧催化剂产生的产物的性质,两个催化剂系统在要求再生之前都产生了一个48小时周期,以恢复催化剂活性,但这些催化剂系统之间的焦炭生产略有不同。因此,实例4的脱氧催化剂的含氧化合物产物产生了一个容许数量的焦炭。
表12进料对产物组成的影响。
根据来自实例1的进料研究,实例8展示了使用某些含氧化合物混合物来产生高产率的芳香烃,同时最小化链烷烃的产率的优点。
实例9脱氧催化剂合成
使用初始润湿技术来制备含有2wt%pd、2wt%mo、0.5wt%sn/w-zro2的一种第二四金属脱氧催化剂。将体积等于有待浸渍的钨酸化四方氧化锆(诺普罗公司)的初始润湿体积且含有1.186g五水四氯化锡(赖德尔·德·亨公司)的2摩尔硝酸铵(西格玛·奥德里奇公司)的一种水溶液添加至79.965g催化剂载体。将催化剂在130℃下干燥2小时。之后在空气中以2℃/分钟将该催化剂从环境温度煅烧至200℃。从200℃至220℃,温度斜升减缓至0.1℃/分钟。最终,从220℃至400℃,温度斜升率增加至2℃/分钟。一旦达到所希望的温度,就将催化剂浸透在空气中持续另外的6个小时。将体积等于有待浸渍的催化剂的初始润湿体积且含有3.001g四水合钼酸铵(西格玛·奥德里奇公司)的2摩尔硝酸铵(西格玛·奥德里奇公司)的一种水溶液添加至所煅烧的催化剂。将催化剂在130℃下干燥2小时。将体积等于有待浸渍的催化剂的初始润湿体积且含有4.105g硝酸钯(ii)水合物(阿法埃莎公司)的2摩尔硝酸铵(西格玛·奥德里奇公司)的一种水溶液添加至所煅烧的催化剂。将催化剂在130℃下干燥2小时。之后在空气中以2℃/分钟将该催化剂从环境温度煅烧至200℃。从200℃至220℃,温度斜升减缓至0.1℃/分钟。最终,从220℃至400℃,温度斜升率增加至2℃/分钟。一旦达到所希望的温度,就将催化剂浸透在空气中持续另外的6个小时。
实例10生产含氧化合物
来自实例9和实例4的脱氧催化剂用于产生一种氧合烃混合物,以用于在一种ni-改性的zsm-5缩合催化剂下进行下游处理。图2中示出了反应器系统。来自实例9的脱氧催化剂作为一个11.5英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在185℃至270℃的温度分布、1050psig、每小时每克催化剂0.5克糖的重量时空速度(whsv)、每1摩尔碳进料约2摩尔h2的氢气共进料、以及4质量流速水再循环比1质量流速进料的水再循环比下。该进料是由45wt%葡萄糖、15wt%木糖、以及40wt%的具有约1000ppm丙酸的水构成以防止细菌生长。
实例4中定义的催化剂作为一个10英寸的填充床加载到一个1英寸外径的inconel反应器中。在引入进料之前,将该催化剂原位还原。条件设定在175℃至270℃的温度分布、1050psig、每小时每克催化剂0.4克糖的重量时空速度(whsv)、每1摩尔碳进料约2摩尔h2的氢气共进料、以及1质量流速水再循环比1质量流速进料的水再循环比下。该进料是由45wt%葡萄糖、15wt%木糖、以及40wt%的具有约1000ppm丙酸的水构成以防止细菌生长。
表13示出了每种脱氧催化剂的不同的产物分布。来自实例9的催化剂如由链烷烃和单氧化合物物质的增加的生产所显示导致进一步程度的脱氧。在另一方面,来自实例4的催化剂如由二氧化合物和未识别的水性物质的生产所突出显示产生烃与更多的氧。
表13脱氧产物组成。
表14示出了该产物组成的更详细的分类(breakdown)。来自实例9的催化剂主要产生醇和环醚单氧化合物。相比之下,实例4的催化剂主要产生二醇和未识别的水性化合物,这些未识别的水性化合物被认为是二氧化合物和多氧化合物。
表14详细的产物组成。
表15示出了产物中识别的碳物质的碳数衰减值。来自实例9的催化剂维持糖进料的碳主链并且产生少量长链化合物。相比之下,实例4的催化剂产生主要是在c2-c4范围内的短链分子。
表15碳数衰减值。
总而言之,表13至表15示出了产物组成是不同的。实例4的脱氧催化剂的产物倾向于是具有2至4个碳原子以及2或3个氧原子的氧合化合物。这种类型的分子经常具有近似在0.5与1.5之间的h:ceff比。因此,实例4的催化剂的产物组成具有比实例9的催化剂更低的h:ceff。
实例11生产芳香族分子
在一种ni-改性的zsm-5缩合催化剂下,在下游处理来自实例10的产物,以评价烃产率和选择性、短期催化剂失活、以及方法可操作性。图4中示出了反应器系统,并且一种ni-改性的zsm-5缩合催化剂作为一个12英寸的填充床加载到1英寸外径的inconel反应器中。条件设定在350℃、100psig以及每小时每克催化剂0.5克糖的重量时空速度(whsv)下。
作为比较,在作为一个12英寸的填充床加载到1英寸外径的inconel反应器中的一种ni-改性的zsm-5缩合催化剂下,同样处理来自实例4的脱氧催化剂的产物。条件设定在375℃、100psig以及每小时每克催化剂0.4克糖的重量时空速度(whsv)下。
表16示出了不同的含氧化合物进料对烃产物产率和选择性的影响。在与使用实例9的脱氧催化剂产生含氧化合物混合物进行比较时,使用实例4的脱氧催化剂产生的含氧化合物混合物增加了芳香族化合物产率并且降低了链烷烃产率。最显著的是,通过使用来自实例4的催化剂的含氧化合物混合物极大地降低了链烷烃生产,其中几乎不到17%的碳进料最终转化成一种链烷烃产物。大部分这种降低归因于产生了相当少的不希望的c4-链烷烃。
还观察到了焦炭沉积的差异。通过处理来自实例9的脱氧催化剂和实例4的脱氧催化剂的含氧化合物混合物,之后进行氧化再生来实现ni-改性的zsm-5缩合催化剂中的碳去除。来自实例9的脱氧催化剂的含氧化合物混合物产生了平均2%左右的作为焦炭馈送的碳,而来自实例4的脱氧催化剂的含氧化合物混合物产生了平均5%的作为焦炭馈送的碳。然而,焦炭产率的差异并未转换成短期催化剂失活的差异。由于通过来自实例4的脱氧催化剂产生的产物的性质,两个催化剂系统在要求再生之前都产生了一个24小时周期,以恢复催化剂活性。因此,使用实例4的脱氧催化剂产生的含氧化合物混合物产生了一个容许数量的焦炭。
表16进料对产物组成的影响。
实例12生产含氧化合物
来自实例4的脱氧催化剂在两个不同的水再循环比下进行测试,以检查水再循环比对进料转化和产物选择性的影响。图2中示出了反应器系统。脱氧催化剂作为一个10英寸的填充床加载到一个1英寸外径的inconel反应器中。1质量流速水再循环比1质量流速进料的水再循环比的条件设定在175℃至270℃、1050psig、每小时每克催化剂0.4克糖的重量时空速度(whsv)、以及每1摩尔碳进料约2摩尔h2的氢气共进料下。该进料是由45wt%葡萄糖、15wt%木糖、以及40wt%的具有约1000ppm丙酸的水构成以防止细菌生长。4质量流速水再循环比1质量流速进料的水再循环比的条件设定在160℃至285℃、1050psig、每小时每克催化剂0.4克糖的重量时空速度(whsv)、以及每1摩尔碳进料约2摩尔h2的氢气共进料下。进料是相同的。
正如表17中所示,几乎所有糖在两种再循环比情况下都进行转化。在调节再循环比时,观察到了非常小的产物选择性的转变。在4:1的水再循环比的情况下,产生了略多的醇。然而,在主要产物二醇和未识别的水性化合物处于几乎相等的水平时,水再循环比的影响是最小的。
表17随着再循环比变化的产物组成。
实例13-25
实例13至实例25描述了根据本发明使用的催化剂载体的制备。这些材料(例如,中孔氧化铝和/或混合氧化物)具有小于200m2/克,更优选小于150m2/克的bet表面积。
实例13b
将测量的量的硼酸(阿法埃莎公司)添加至含有75g假勃姆石粉末的一个烧杯中,这样使得硼构成按假勃姆石计的重量的8%。在添加80ml2%的hno3溶液(处于约70℃)之后将盐混合约5分钟。在将上述溶液添加至混合固体之后,出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟(cfm)流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例14cr
将测量的量的硝酸铬(阿法埃莎公司)添加至含有75g假勃姆石粉末的一个烧杯中,这样使得铬构成按假勃姆石计的重量的8%。在添加80ml2%的hno3溶液(处于约70℃)之后将盐混合约5分钟。在将上述溶液添加至混合固体之后,出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例15ce
将七十五克假勃姆石(萨索尔公司(sasol))添加至一个烧杯。将含有20.25g硝酸铈(奥德里奇公司)的150ml溶液添加至这个烧杯。搅拌所得浆料直到出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例16co
将七十五克假勃姆石(萨索尔公司(sasol))添加至一个烧杯。将含有32.28g硝酸钴(ii)(阿法埃莎公司)的150ml溶液添加至这个烧杯。搅拌所得浆料直到出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例17cu
将七十五克假勃姆石(萨索尔公司)添加至一个烧杯。将含有24.91g硝酸铜(阿克洛斯公司(acros))的150ml溶液添加至这个烧杯。搅拌所得浆料直到出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例18fea
将七十五克假勃姆石(萨索尔公司)添加至一个烧杯。将含有47.27g硝酸铁(阿克洛斯公司)的150ml溶液添加至这个烧杯。搅拌所得浆料直到出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例19feb
将七十五克假勃姆石(萨索尔公司)添加至一个烧杯。将含有11.82g硝酸铁(阿克洛斯公司)的150ml溶液添加至这个烧杯。搅拌所得浆料直到出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例20mg
将七十五克假勃姆石(萨索尔公司)添加至一个烧杯。在这个烧杯中,添加含有38.648g硝酸镁(西格玛·奥德里奇公司)的150ml溶液,之后添加2ml10%的硝酸溶液以及0.5g羟乙基纤维素(佛鲁卡公司(fluka))。搅拌所得浆料直到出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例21mo
将测量的量的钼酸(西格玛·奥德里奇公司)添加至含有75g假勃姆石粉末的一个烧杯中,这样使得钼构成按假勃姆石计的重量的8%。在添加80ml2%的hno3溶液(处于约70℃)之后将盐混合约5分钟。在将上述溶液添加至混合固体之后,出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例22nb
将测量的量的五氯化铌(阿法埃莎公司)添加至含有75g假勃姆石粉末的一个烧杯中,这样使得铌构成按假勃姆石计的重量的8%。在添加80ml2%的hno3溶液(处于约70℃)之后将盐混合约5分钟。在将上述溶液添加至混合固体之后,出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例23θ-al2o3
将750g假勃姆石(萨索尔公司)添加至一个烧杯。用去离子水将假勃姆石稀释成1800ml,并且将浆料混合约5分钟。之后添加22.5g羟乙基纤维素以及20ml10%的hno3。在将上述溶液添加至混合固体之后,出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例24w
将测量的量的氧化钨(阿法埃莎公司)添加至含有75g假勃姆石粉末的一个烧杯中,这样使得氧化钨构成按假勃姆石计的重量的8%。在添加80ml2%的hno3溶液(处于约70℃)之后将盐混合约5分钟。在将上述溶液添加至混合固体之后,出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例25zr
将七十五克假勃姆石(萨索尔公司)添加至一个烧杯。将含有16.553g硝酸氧锆(西格玛·奥德里奇公司)的150ml溶液添加至这个烧杯。搅拌所得浆料直到出现胶凝。将材料转移至130℃静态烘箱中并且允许干燥过夜。接着在配备有以10立方英尺/分钟流动的空气扫气的一个马弗炉中进行煅烧,同时按2℃/分钟斜升至1000℃,并且保持4小时。
实例26载体特性
表18中总结了实例13至实例25中描述的催化剂载体的表面积、孔径、以及孔体积。标记noproal2o3的样品可商购自圣戈本诺普罗公司(saint-gobainnorpro)。
表18催化剂载体特性
实例27改性载体
在实例13、实例15、实例18、实例19、实例23和实例25中,ni被添加至载体,并且催化剂用于产生根据本发明的一种含氧化合物混合物。使用图1中所示的反应器系统,12克催化剂作为填充床加载到一个1/2英寸外径的inconel反应器中。在引入进料之前,使用处于700hr-1的空速的氢气、达到400℃的2小时温度梯度以及随后的1小时氢气浸透将该催化剂还原。条件设定在1050psig、每小时每克催化剂0.50克糖的重量时空速度(whsv)、以及每1摩尔碳进料约2摩尔h2的氢气共进料下。该进料是由20wt%葡萄糖、5wt%木糖以及75wt%的具有约1000ppm丙酸的水构成以防止细菌生长。初始反应温度设定在180℃至220℃的分布下,在一个4小时的时段内斜升至180℃至300℃,并且保持处于该分布,持续一个3小时的时段,在此时,对产物进行取样。
如表19中所示,不同金属氧化物的改性会影响催化剂选择性范围。比较来自实例18和实例19的催化剂,可以看出,不仅金属改性会影响性能,而且并入到θ氧化铝结构中的金属的量对于产物选择性而言也是很重要的。总之,金属改性以各种方式将θ氧化铝基载体(实例23)的选择性从来自实例18的催化剂的情况下的主要是氢化调节为来自实例25和实例15的催化剂的大体上更多的氢化脱氧和脱羰。
表19产物组成
实例28脱氧催化剂合成
将含有7.48克硝酸镍(ii)水合物(阿法埃莎公司)和0.71克氯化锡(iv)(赖德尔·德·亨公司)的50ml溶液添加至两个分开的等效的整分试样中的37克钨酸化氧化锆(圣戈本诺普罗公司)。在添加期间允许固体在静态烘箱中在120℃下干燥一个1小时的时段。在第二添加之后,允许润湿的固体在静态烘箱中在120℃下干燥一个3小时的时段。之后将固体转移至一个马弗炉,并且在流动空气(30立方英尺/小时)下进行煅烧。煅烧通过以下内容完成:使烘箱按1.6℃/分钟斜升直到达到400℃的一个最终温度,在此之后将该温度保持6小时。
实例29脱氧催化剂合成
经由初始润湿技术将含有0.53克五水四氯化锡(赖德尔·德·亨公司)和di水的13ml溶液添加至28.04克钨酸化氧化锆(圣戈本诺普罗公司)。将所得润湿的固体在静态烘箱中在120℃下干燥一个2小时的时段。在此之后,去除固体并且在配备有以30立方英尺/小时流动的空气扫气的一个马弗炉中进行煅烧。煅烧通过以下内容完成:使烘箱按1.6℃/分钟斜升直到达到400℃的一个最终温度,在此之后将该温度保持6小时。允许烘箱冷却,将固体去除并且经由初始润湿技术用含有1.03克四水合钼酸铵(西格玛·奥德里奇公司)的13ml溶液浸渍该固体。之后根据上述程序对润湿的固体进行干燥和煅烧。经由初始润湿技术通过添加含有1.41克硝酸钯(ii)水合物(阿法埃莎公司)的13ml溶液来完成钯浸渍。经由上述程序对所得固体进行干燥和煅烧。
实例30生产含氧化合物
0和实例29中提供的催化剂在按图1中的工艺流程图配置的一个反应器系统中进行测试。将反应器出口温度维持在270℃。以每小时每1克催化剂1克糖的速率供应糖原料。将反应器压力维持在1050psig。图6中提供了测试的结果。
实例31脱氧催化剂合成
经由初始润湿技术将含有5.691克氧化钨铵水合物(阿法埃莎公司)和di水的12ml溶液添加至26.021克θ氧化铝(圣戈本诺普罗公司)。将所得润湿的固体在静态烘箱中在120℃下干燥一个2小时的时段。在此之后,去除固体并且在配备有以30立方英尺/小时流动的空气扫气的一个马弗炉中进行煅烧。煅烧通过以下内容完成:使烘箱按1.6℃/分钟斜升直到达到400℃的一个最终温度,在此之后将该温度保持6小时。允许烘箱冷却,将固体去除并且经由初始润湿技术用含有0.385克五水四氯化锡(赖德尔·德·亨公司)的12ml溶液浸渍该固体。之后根据上述程序对润湿的固体进行干燥和煅烧。通过用含有0.975克四水合钼酸铵(西格玛·奥德里奇公司)的12ml溶液对所得固体进行初始润湿浸渍来完成钼添加。根据上述程序干燥所得固体,之后进行钯浸渍。经由初始润湿技术通过添加含有1.333克硝酸钯(ii)水合物(阿法埃莎公司)的12ml溶液来完成钯浸渍。经由上述程序对所得固体进行干燥和煅烧。
实例32脱氧催化剂合成
经由初始润湿技术将含有5.834克氧化钨铵水合物(阿法埃莎公司)和di水的12ml溶液添加至26.652克单斜晶氧化锆(圣戈本诺普罗公司)。将所得润湿的固体在静态烘箱中在120℃下干燥一个2小时的时段。在此之后,去除固体并且在配备有以30立方英尺/小时流动的空气扫气的一个马弗炉中进行煅烧。煅烧通过以下内容完成:使烘箱按1.6℃/分钟斜升直到达到400℃的一个最终温度,在此之后将该温度保持6小时。允许烘箱冷却,将固体去除并且经由初始润湿技术用含有0.395克五水四氯化锡(赖德尔·德·亨公司)的12ml溶液浸渍该固体。之后根据上述程序对润湿的固体进行干燥和煅烧。通过用含有1.000克四水合钼酸铵(西格玛·奥德里奇公司)的12ml溶液对所得固体进行初始润湿浸渍来完成钼添加。根据上述程序干燥所得固体,之后进行钯浸渍。经由初始润湿技术通过添加含有1.367克硝酸钯(ii)水合物(阿法埃莎公司)的12ml溶液来完成钯浸渍。经由上述程序对所得固体进行干燥和煅烧。
实例33生产含氧化合物
实例31和实例32中提供的催化剂在按图2中的工艺流程图配置的一个反应器系统中进行测试。将反应器出口温度维持在270℃。以每小时每1克催化剂1克原料的速率供应穿过离子交换柱且掺杂有1000ppm丙酸原料的50wt%43de玉米糖浆(食品配料公司(foodingredients,inc.))。将反应器压力维持在1050psig,并且还将再循环流速维持在按质量计的4:1的质量比。图7中提供了测试的结果。
实例34生产含氧化合物
在图2所示的反应器系统中测试来自实例4和实例9的两种脱氧催化剂。50wt%43de玉米糖浆(食品配料公司)在以每小时每克催化剂0.5克原料的流速供应之前穿过一个离子交换柱并且掺杂有1000ppm丙酸原料,并且再循环比是1:1质量比。对于两个催化剂系统,将反应器的入口保持在180℃。按图8至图9中所指有意图地修改出口温度以检查nisn催化剂的选择性。
实例35合成nisn合金催化剂
一种nisn合金脱氧催化剂制备如下。多步骤合成涉及(1)溶剂化,(2)载体浸渍,(3)沉淀,(4)结晶,以及(5)还原/活化。表20含有nisn合金合成中使用的所有试剂和它们的生厂商的列表。
表20试剂列表
无担载合金合成:将测量的量的试剂1和2添加至配备有一个冷凝器的一个圆底烧瓶。按表21中详细描述的量将含有去离子(di)水、乙醇和2-甲氧乙醇(2me)的一种溶液添加至该圆底烧瓶。之后在搅拌的同时,将反应混合物加热至50℃,持续一个30分钟的时段,以促进金属盐的溶解。接着,将3m的naoh溶液逐滴添加至冷凝器柱的顶部以使相应的金属氢氧化物沉淀,直到观察到>10的一个最终ph。对于结晶步骤,将反应混合物的全部内容物全部倒入到一个特氟龙(teflon)内衬中并且加载到一个密封高压釜中;帕尔仪器公司(parrinstrumentcompany)型号4566(300ml)。之后在自生压力下,在2小时的过程中将反应器加热至150℃。在保持温度持续另外一个18小时的时段之后,允许该反应器冷却。在结晶完成之后,经由真空过滤回收固体,并且用3等份(equivalents)(各自200ml)的di水进行洗涤,之后在静态烘箱中在120℃下进行干燥。在将干燥固体加载到放置到一个炉中的一个石英反应器中的过程中,将这些干燥固体准备用于还原。在流动氢气下,按2℃/分钟将该反应器加热至400℃的一个最终温度,并且保持一个4小时的时段。
表21合成试剂的详细量(无担载合金)
担载合金合成:表22中列出了用于担载合金制备的催化剂载体。
表22催化剂载体
将测量的量的试剂1和2添加至一个烧杯,之后是处于表23中详述的量的含有去离子(di)水、乙醇以及2-甲氧乙醇(2me)的一种溶液。之后在搅拌的同时,将反应混合物加热至100℃,持续一个10分钟的时段,以促进金属盐的溶解。然后将该溶液添加至测量的量的催化剂载体,这样使得载体的初始润湿是基于总溶液的体积来实现。接着,将3m的naoh溶液逐滴添加至加载有润湿的固体的一个特氟龙内衬。naoh的添加导致相应的金属氢氧化物的沉淀,该沉淀在观察到>10的一个最终ph之后停止。对于结晶步骤,将特氟龙内衬加载到一个密封高压釜中,该密封高压釜来自帕尔仪器公司型号4566(300ml)。之后在自生压力下,在2小时的过程中将反应器加热至150℃。在保持温度持续另外一个18小时的时段之后,允许该反应器冷却。在结晶完成之后,经由真空过滤回收固体,并且用3等份(各自200ml)的di水进行洗涤,之后在静态烘箱中在120℃下进行干燥。将干燥固体加载到放置到一个炉中的一个石英反应器中。在流动氢气下,按2℃/分钟将该反应器加热至400℃的一个最终温度,并且保持一个4小时的时段。
表23合成试剂的详细量(担载合金)
实例36生产含氧化合物
在分批服务中,连同一个基线担载的8%ni2%sn/θ-al2o3催化剂一起测试无担载nisn合金。将催化剂非原位还原,之后在水下将这些催化剂加载到一个搅拌槽反应器中。表24中示出了还原条件。所有实验都是在存在20%山梨糖醇原料的情况下进行。表25中的实验出于比较不同脱氧催化剂而进行。所列的压力是温度斜升结束时的目标压力,但这实际上会根据每种单独的催化剂的活性而略有不同。基于新鲜催化剂的icp结果,每个实验都被设计成使等量的ni加载到反应器中。
表24搅拌槽还原条件
表25搅拌槽反应条件
图10中示出了产物的一个比较。通过比较无担载催化剂可以看到,每种合金具有不同的活性和选择性。在转化与不具有催化剂的一个空白实验类似的情况下,仅ni材料转化仅约55%的山梨糖醇。大多数识别的产物是山梨聚糖和异山梨醇,它们可以通过热反应单独来产生。ni3sn合金更具活性,转化约75%的山梨糖醇,但并未显示出醇或二醇产物增加了多少。ni3sn2合金在无担载材料中表现最好,转化超过80%的山梨糖醇,并且产生约5%的二醇和醇两者。ni3sn4合金与其他合金催化剂相比较具有不同的活性,并且当它具有超过90%的山梨糖醇转化时,几乎所有识别的产物都是山梨聚糖和异山梨醇,它们简单地通过脱水反应来产生。
在比较无担载合金与担载催化剂的活性时,清楚的是,在θ-al2o3上担载金属极大地提高了催化剂的活性。通过将两种担载催化剂彼此比较,可以看到,ni3sn2合金催化剂尤其是在二醇生产方面远远胜过初始润湿催化剂。担载ni3sn2催化剂与超过20%的初始润湿催化剂相比较还几乎不产生山梨聚糖或异山梨醇。
实例37生产含氧化合物
在一个固定床反应器系统中进一步测试担载nisn合金脱氧催化剂。在等温条件和三个不同的whsv下,测试这些催化剂。在与先前在实验过程中检查失活相同的条件下进行最终重量检查(wc)。表26中示出了反应条件。
表26反应条件
图12示出了θ-al2o3上担载的nisn合金的脱氧催化剂与标准初始润湿催化剂在wshv=0.5hr-1下在一个流通型反应器系统中进行比较的结果。可以观察到与无担载催化剂类似的趋势。ni3sn和ni3sn2合金具有与初始润湿催化剂类似的产物分布,并且在产生二醇方面更具选择性。担载ni3sn4合金是测试到的最低活性催化剂。再次,这种催化剂看上去似乎仅催化脱水反应,因为大量识别的产物是异山梨醇和山梨聚糖。在这个whsv下,除ni3sn4合金之外的所有催化剂几乎实现了完全转化,并且因此所有产物分布是类似的。
图13比较了在whsv=1hr-1下担载nisn合金脱氧催化剂的性能。由于低转化和较差的二醇选择性,图12中省略了ni3sn4合金。在这个whsv下,ni3sn2担载合金明显是性能最好的催化剂。ni3sn2合金几乎维持了山梨糖醇的完全转化,而测试的其他催化剂各自下降至约85%的转化。此外,这种催化剂将超过30%的碳进料转化成二醇,这超过任何其他催化剂的1.5倍。事实上,whsv=1hr-1下的ni3sn2催化剂表现得与whsv=0.5hr-1下的8%ni2%sn/θ-al2o3催化剂几乎相同。
除θ-al2o3之外,α-al2o3、活性炭和单斜晶zro2也测试作为ni3sn2合金的载体,其中图14中示出了结果。就活性和选择性两者而言,α-al2o3和m-zro2两者的表现远远不如θ-al2o3担载材料。两种催化剂具有不到50%的转化,并且几乎所有识别的产物都是异山梨醇和山梨聚糖。活性炭担载ni3sn2合金确实具有约95%的转化,但是该催化剂对(逆羟醛(retro-aldol)缩合产物具有极低的选择性。在测试的材料当中,θ-al2o3载体对本发明而言是优选的。
- 还没有人留言评论。精彩留言会获得点赞!