细菌多环芳烃代谢产物高效提取及降解途径判定方法与流程
本发明属于生物化学分析领域,具体涉及一种简单高效提取培养基中多环芳烃降解产物的方法及一种判定多环芳烃降解途径比例的方法。
背景技术:
多环芳烃(polycyclicaromatichydrocarbons,pahs)是指两个及两个以上苯环以稠环形式相连的化合物,属国际公约规制的持久性有机污染物(persistentorganicpollutants,pops),主要来源于化石燃料、煤气和煤焦油的生产、木料加工、石油泄漏以及废物焚烧等,具有致癌性、致畸性、致突变性,可长久存在于大气、水体和土壤中,给人类健康和生态环境带来危害。因此,如何治理环境中的pahs,降低环境风险,日益受到人们的重视。
治理环境中多环芳烃的技术可分为物理方法、化学方法、生物方法。其中,生物方法中的微生物修复,因其成本低、高效、二次污染少,已逐渐成为有效解决多环芳烃污染的主要手段。研究微生物对多环芳烃的代谢转化,阐明其降解机理,对进一步利用微生物修复多环芳烃环境污染,并评估原位修复中中间代谢产物可能带来的风险具有重要意义。
提取微生物多环芳烃代谢产物作为较为关键的步骤,直接影响了下游分析中对代谢途径的揭示。目前,较为普遍的多环芳烃降解产物提取方法有:一、高速离心去除菌体后对培养基上清进行萃取提取。二、直接向培养基中添加等体积的有机溶剂(甲醇、正己烷、乙酸乙酯等)萃取或溶解提取。上述两种方法均未考虑菌体体内的代谢产物,且部分提取方法还需按酸碱性分步提取,操作繁琐耗时长,大大降低了对代谢产物提取效率。如何优化代谢产物提取方法尽可能完整地获取代谢产物,是探索多环芳烃代谢产物方法中较为关键的环节,也是进一步对降解产物进行鉴定的前提。高效提取降解产物后,如何进一步对降解产物进行分析,是评估降解菌株价值的基础。目前,较为高效的一种检测分析手段为高效液相色谱-质谱连用技术(highperformanceliquidchromatography-mass,hplc-ms),该技术分离效率高、选择性好、检测灵敏度高,在揭示降解途径中具有一定优势。通过优化高效液相色谱测定条件,提高多种降解产物分离度,为进一步解析降解产物、探究新降解途径具有重要意义;与此同时,如何与已报道降解途径进行比对,判定菌株代谢途径中已报道降解途径所占比例,是为进一步判定新降解途径存在可能的依据。两者皆是解析多环芳烃降解菌降解途径中重要的步骤,对于高效利用微生物修复多环芳烃环境污染具有重要意义。
技术实现要素:
本发明要解决的技术问题是提供一种高效提取细菌多环芳烃代谢产物并判定降解途径比例的方法。
为了解决上述技术问题,本发明提供一种测定多环芳烃降解产物的方法,包括以下步骤:
1)、以菲/芘为唯一碳源对降解菌进行培养:
制备浓度为(1~10)×108cfuml-1的菲或芘的降解菌菌悬液;
将1ml的上述降解菌菌悬液与9ml的含菲/芘的矿质培养基在容器内混合后放入恒温振荡器中于28±1℃、130±30rpm条件下培养至污染物被降解完全(视菌株降解情况而定,约为12~72小时);
含菲的矿质培养基中,菲浓度为100±10mgl-1;
含芘的矿质培养基中,芘浓度为50±5mgl-1;
备注说明:
菲的降解菌菌悬液配用菲的矿质培养基,芘的降解菌菌悬液配用芘的矿质培养基;
判断污染物是否被降解完全,可依据现有的hplc测定法;
2)、提取降解产物:
将步骤1)所得的装有以菲/芘为唯一碳源菌液的容器从恒温振荡器中取出;
向容器(含10ml培养体系)中添加1ml6moll-1naoh溶液,超声10±2min;添加2ml6moll-1hcl溶液混匀;添加10ml乙酸乙酯,超声10±2min;
将容器内的所有液体转移至棕色玻璃离心管ⅰ(50ml的规格)中,并用5ml乙酸乙酯润洗原容器内壁后将其合并至棕色离心管ⅰ中;室温条件下以3500±500rpm转速离心;
将离心所得的上层有机相通过含有无水na2so4的针筒后,收集至棕色玻璃离心管ⅱ(50ml的规格)中,氮吹至乙酸乙酯完全挥发;再加入2ml乙腈从而溶解洗涤并溶解棕色玻璃离心管ⅱ内的物质;过0.22μm有机相膜后上机测定;
备注说明:10ml培养体系包含菌体、未降解的菲或芘、中间降解产物、矿质培养基中的盐分和水;
3)、hplc上机检测:
分别配置流动相a(有机流动相)和流动相b(无机流动相),a为乙腈+1‰甲酸(v/v);b为水+1‰甲酸(v/v),脱气30±5min后使用;
按照下表设置hplc流动相梯度洗脱:
柱温为30℃,紫外检测器分别检测254nm和280nm波段吸光度变化;
通过标样确定物质出峰时间及检测下限,并通过标线换算降解产物含量,计算污染物降解途径比例。
备注说明:上述流动相的含量,以“0~12min流动相a”为例,是指从“10%”均匀递增至“34%”。
作为本发明的测定多环芳烃降解产物的方法的改进:
所述步骤1)中的矿质培养基为:
称取0.50gnano3,1.00gkh2po4,0.02gcacl2,0.20gmgso4,0.50g(nh4)2so4,1.00gnah2po4·h2o溶解于1l去离子水中,搅拌混匀后,调节ph至6.5(采用浓度为6moll-1的氢氧化钠溶液进行调节)。
作为本发明的测定多环芳烃降解产物的方法的进一步改进:
所述步骤1)中浓度为(1~10)×108cfuml-1的菲或芘的降解菌菌悬液的制备方法为:
吸取200μl25%甘油保存菌种于20mllb培养基中,在28℃130rpm条件下活化培养12h;
吸取200μl活化菌液转入新的20mllb培养基中,在28℃130rpm条件下培养14~16h获得菌液;
在4℃5000rpm条件下将菌液离心10min,获得降解菌;
将获得菌体采用无菌水涡旋,将菌体重悬于无菌水中;
再次离心,重复该步骤两次,最后将菌体重悬于无菌水中获得菌悬液;
调节菌悬液在紫外分光光度计od600nm的值,从而获得所需浓度的菌悬液(较佳为od600nm=1.0,此时菌体浓度约为5×108cfuml-1)。
作为本发明的测定多环芳烃降解产物的方法的进一步改进:所述步骤3)中的各物质出峰时间、检测下限、标线如下表所示:
本发明还同时提供了利用上述方法测定所得的多环芳烃降解产物含量来判定降解途径比例的方法:获得降解产物含量后,按以下公式进行降解途径比例换算(以10ml培养体系为例):
1-羟基-2-萘甲酸途径比例=[1-羟基-2-萘甲酸浓度(mgl-1)/1000×10ml/1000/188.18]÷[降解芘/菲浓度(mgl-1))/1000×10ml/1000/202.25或178.23];
邻苯二甲酸途径比例=[邻苯二甲酸浓度(mgl-1)/1000×10ml/1000/166.13]÷[降解芘/菲浓度(mgl-1))/1000×10ml/1000/202.25或178.23];
水杨酸途径比例=[邻苯二甲酸浓度(mgl-1)/1000×10ml/1000/138.12]÷[降解芘/菲浓度(mgl-1))/1000×10ml/1000/202.25或178.23];
2,2’-联苯二羧酸途径比例=[2,2’-联苯二羧酸浓度(mgl-1)/1000×10ml/1000/242.23]÷[降解芘/菲浓度(mgl-1)/1000×10ml/1000/202.25或178.23]。
本发明以多环芳烃菲/芘降解途径为例,采用菲/芘的多种中间代谢产物为标记,以指示菲/芘的不同降解途径;在解析新降解产物的同时,通过将降解产物与已报道降解途径进行比对,集定性定量分析于一体,综合判定常规降解途径比例,以此预测新降解途径存在可能,属于生物化学分析领域。
本发明主要包括如下内容:降解产物的提取方法;对降解产物物质进行鉴定;通过几种物质分别代表不同的降解途径(已报道降解途径),通过测定已知降解产物判定当菌株降解完100mg/l的菲或50mg/l的芘时,已报道降解途径所占总消耗菲/芘的比例,从而判断存在新降解途径的可能。新途径如果更为高效,在利用菌体直接治理多环芳烃污染或基因工程对某高效基因进行改造利用从而治理多环芳烃等方面具有重要意义。
本发明解决了现有技术的不足,以利用多环芳烃污染物菲或芘为唯一碳源的菌体培养物为例,通过本发明提供的碱性裂解法破碎细菌菌体后萃取提取菲或芘的中间降解产物,浓缩后进行上机测定。该提取方法更为简单高效,可兼得细胞内外的中间降解产物,获取降解产物更为完整;与此同时,本发明还通过优化hplc梯度洗脱方法,并以菲三种主要降解途径(也同为芘的下游降解途径)中的四种物质作为标记,指示其降解途径;该方法实现了分离污染物母体(菲和芘)及其他五种中间产物[1-羟基-2-萘甲酸(同分异构体:3-羟基-2-萘甲酸),邻苯二甲酸、水杨酸、2,2’联苯二羧酸]及这些物质的定性定量检测,七种物质均具有较好的分离度,峰形完整无拖尾,为后续质谱检测物质结构奠定了良好的基础。通过与已有报道途径进行定性定量比对,评估菌株存在新降解途径的可能,为进一步探索降解途径、采用微生物治理多环芳烃污染提供理论基础。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细说明。
图1为芘和菲的几条主要降解途径,框中为选取的表征不同降解途径的标志性代谢产物;
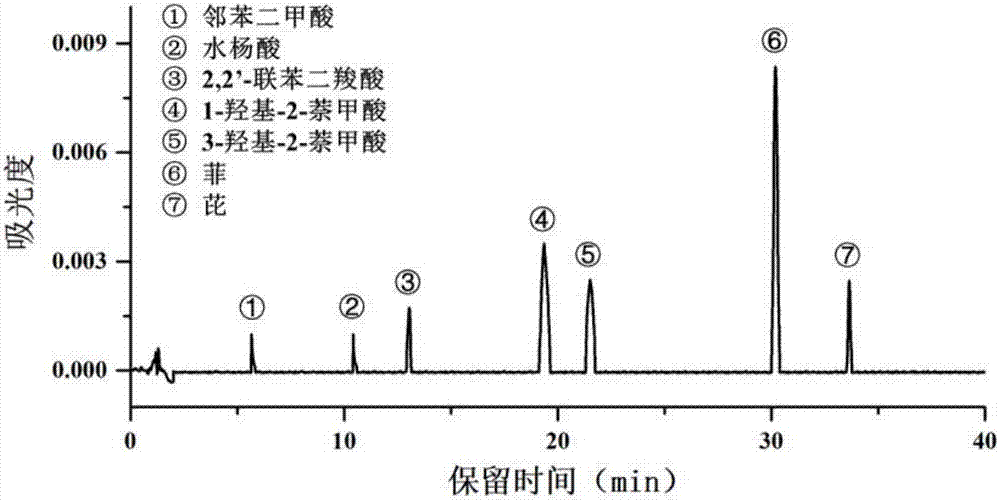
图2为菲和芘及五种降解产物hplc色谱图;
图3为菲和芘及五种降解产物hplc标准曲线;
图4为mycobacteriumsp.wy6以芘为唯一碳源的代谢产物hplc-ms质谱图;
图5为mycobacteriumsp.wy6以芘为唯一碳源降解途径示意图;
图6为mycobacteriumsp.wy6降解芘的代谢产物gc-ms质谱图;
图7为massiliasp.wg5以菲为唯一碳源的降解产物hplc-ms质谱图;
图8为massiliasp.wg5以菲为唯一碳源降解途径比例示意图;
图9为massiliasp.wg5以菲为唯一碳源的降解产物gc-ms质谱图。
具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此。
本发明中所采用仪器设备及化学试剂如下:
仪器:agilent1290液相色谱-6460质谱联用仪;
试剂:盐酸(hcl)、氢氧化钠(naoh)、乙酸乙酯(c4h8o2)、甲酸(ch2o2)、乙腈(c2h3n)、硫酸钠(na2so4)
色谱柱:
标准品的化学名、结构式及来源如下表1:
表1
所涉及的菌株如下:
多环芳烃高效降解菌wy6,为分枝杆菌wy6(mycobacteriumsp.wy6),其保藏号为:cctccno:m2017373。
多环芳烃高效降解菌wy6,保藏名称为:分枝杆菌wy6mycobacteriumsp.wy6,保藏单位:中国典型培养物保藏中心,保藏地址:中国武汉武汉大学,保藏编号:cctccno:m2017373,保藏时间2017年6月26日。
massiliasp.wg5,cctccab2015362。
一、以菲/芘为唯一碳源对降解菌进行培养:
1、培养基配置:
lb培养基:称取5glb粉末溶解于200ml去离子水中,搅拌混匀后分装20ml于50ml锥形瓶中,于121℃条件下高温高压灭菌15min后待用;此为常规技术。
以菲/芘为唯一碳源的矿质培养基:称取无机盐(0.50gnano3,1.00gkh2po4,0.02gcacl2,0.20gmgso4,0.50g(nh4)2so4,1.00gnah2po4·h2o)溶解于1l去离子水中;搅拌混匀后,采用浓度为6moll-1的氢氧化钠溶液调节溶液ph至6.5;121℃高温高压灭菌20min待用,即得灭菌矿质培养基;吸取200μl浓度为5000ppm的菲母液(称取0.5g菲粉末溶解于100ml丙酮中,即得5000ppm菲母液)或添加100μl浓度为5000ppm的芘母液(称取0.5g芘粉末溶解于100ml丙酮中,即得5000ppm芘母液)至50ml灭菌锥形瓶底部;待丙酮完全挥发在锥形瓶内底部形成一层菲/芘污染物薄膜后,加入9ml上述灭菌矿质培养基,即得以菲或芘为唯一碳源的矿质培养基(培养时菲和芘在矿质培养基中的终浓度为100mgl-1和50mgl-1)。
2、菌种活化:
吸取200μl25%甘油保存菌种于20mllb培养基中,在28℃130rpm条件下活化培养12h;吸取200μl活化菌液转入新的20mllb培养基中,在28℃130rpm条件下培养14~16h获得菌液。
3、菌种转接:
在4℃5000rpm条件下将菌液离心10min,获得降解菌;将获得菌体采用无菌水涡旋,将菌体重悬于无菌水中;再次离心,重复该步骤两次,最后将菌体重悬于无菌水中获得菌悬液;
调节菌悬液在紫外分光光度计od600nm=1.0(此时菌体浓度约为5×108cfuml-1);
吸取1mlod600nm=1.0菌液加入9ml含100mgl-1菲或50mgl-1芘矿质培养基中加入50ml三角瓶中,放入恒温振荡器中在28℃130rpm条件下培养。
培养直至污染物菲/芘被降解完全,视菌株降解情况而定,培养约为12~72小时;判断污染物是否被降解完全,可依据现有的hplc法;当满足培养体系中污染物浓度低于1mgl-1条件时,判定菲/芘被降解完全。
二、降解产物提取:
步骤一的培养结束后,将50ml三角瓶从恒温振荡器取出,向三角瓶内(包含10ml培养体系)添加1ml6moll-1的naoh,超声10min(超声)后添加2ml6moll-1的hcl,混匀。
向50ml三角瓶中添加10ml乙酸乙酯,超声10min。
将上述50ml三角瓶中所有液体转移至50ml棕色玻璃离心管ⅰ中,并用5ml乙酸乙酯润洗三角瓶内壁后将其合并至50ml棕色玻璃离心管ⅰ中,室温条件下以3500rpm转速离心10分钟,使有机相与水相分离;
将有机相(位于上层)吸出,并使有机相通过含有无水na2so4的针筒后(除水),将其收集至新的50ml棕色玻璃离心管ⅱ中,氮吹至乙酸乙酯完全挥发;
加入2ml乙腈溶解洗涤并溶解上述棕色离心管ⅱ内的物质,该步骤可超声助溶;
过0.22μm有机相膜后上机测定。
三、hplc-ms分离鉴定:
分别配置流动相a和流动相b,a为乙腈+1‰甲酸(v/v);b为水+1‰甲酸(v/v),脱气30min后使用;
各称取20mg七种标准物质(邻苯二甲酸、水杨酸、2,2’-联苯二羧酸、1-羟基-2-萘甲酸、3-羟基-2-萘甲酸、菲、芘)溶解于乙腈中,再用乙腈定容至100ml,配置成200mgl-1的标准物质混合母液。吸取1ml浓度为200mgl-1的标准物质混合母液,添加9ml乙腈混匀后获得浓度为20mgl-1的标准物质混合母液,将20mgl-1的混合母液按下表2稀释,配制标线:
表2
按照下表3设置hplc流动相梯度洗脱:
表3
柱温为30℃,紫外检测器分别检测254nm和280nm波段吸光度变化。
通过标样确定物质出峰时间及检测下限,并通过标线换算降解产物含量。
根据本发明中条件设置,各物质出峰时间及检测下限如表4所示:
表4
四、降解途径比例判定
获得降解产物含量后,按以下公式进行降解途径比例换算(以10ml培养体系为例):
1-羟基-2-萘甲酸途径比例=[1-羟基-2-萘甲酸浓度(mgl-1)/1000×10ml/1000/188.18]÷[降解芘/菲浓度(mgl-1))/1000×10ml/1000/202.25或178.23];
邻苯二甲酸途径比例=[邻苯二甲酸浓度(mgl-1)/1000×10ml/1000/166.13]÷[降解芘/菲浓度(mgl-1))/1000×10ml/1000/202.25或178.23];
水杨酸途径比例=[邻苯二甲酸浓度(mgl-1)/1000×10ml/1000/138.12]÷[降解芘/菲浓度(mgl-1))/1000×10ml/1000/202.25或178.23];
2,2’-联苯二羧酸途径比例=[2,2’-联苯二羧酸浓度(mgl-1)/1000×10ml/1000/242.23]÷[降解芘/菲浓度(mgl-1)/1000×10ml/1000/202.25或178.23]。
实施例1、提取mycobacteriumsp.wy6(cctccno:m2017373)以芘为唯一碳源的降解产物,依次进行如下步骤:
1)、吸取200μl25%(v/v)甘油保存菌种于20mllb培养基中,在28℃130rpm条件下活化培养12h;吸取200μl活化菌液转入新的20mllb培养基中,在28℃130rpm条件下培养14~16h获得菌液。
2)、在4℃5000rpm条件下将菌液离心10min,获得降解菌;将获得菌体采用无菌水涡旋,将菌体重悬于无菌水中;再次离心,重复该步骤两次,最后将菌体重悬于无菌水中获得菌悬液;
3)、调节菌悬液在紫外分光光度计od600nm=1.0(此时菌体浓度约为5×108cfuml-1);
4)、吸取1mlod600nm=1.0菌液加入9ml含50mgl-1芘的矿质培养基中,于28℃130rpm条件下培养。
5)、根据菌株对污染物的降解情况,在污染物降解完全前,每12h采样一次;按上述“二、降解产物提取”中方法对降解产物进行提取降解产物;即,每次所得采样物分别进行后续步骤。
6)、按照“三、hplc-ms分离鉴定”中条件设置仪器参数,对所提取降解产物进行定性定量分析。
通过上述实施例1,采用本发明中的碱裂解法,可从培养物中获取的降解产物达十多个峰(图4),在hplc色谱图中具有较好的分离度。经过3天(72h)培养后,在降解产物中测定到检测到降解产物邻苯二甲酸、水杨酸和4-菲羧酸的存在(如图4所示)。色谱图中,芘、邻苯二甲酸、水杨酸的峰面积积分分别为715022、135155、43150,分别对应的代入图3中芘、邻苯二甲酸、水杨酸标准曲线公式中换算可得,10ml培养体系中:
芘的浓度(mgl-1)=715022/160679/5(提取10ml培养体系中代谢产物最后甲醇定容为2ml,实质浓缩了5倍;下同)=0.89mgl-1;
降解芘的摩尔含量(μmol)=[(50-0.89)mgl-1/1000]×(10ml/1000)]/202.25×106=2.43μmol;
邻苯二甲酸的浓度(mgl-1)=135155/27194/5=0.99mgl-1;
邻苯二甲酸的摩尔含量(μmol)=[0.99mgl-1/1000×(10ml/1000)]/166.13×106=0.060μmol;
水杨酸的浓度(mgl-1)=43150/11476/5=0.75mgl-1;
水杨酸的摩尔含量(μmol)=[0.75mgl-1/1000×(10ml/1000)]/138.12×106=0.054μmol。
以上数据表明,mycobacteriumsp.wy6可将2.43μmol的芘同时通过1-羟基-2-萘甲酸→邻苯二甲酸以及1-羟基-2-萘甲酸→水杨酸的途径进行降解,并还通过4-菲羧酸途径降解。3天培养中所消耗的芘为2.43μmol,而所获得的通过1-羟基-2-萘甲酸途径的下游降解产物邻苯二甲酸含量为0.060μmol,水杨酸含量为0.054μmol,通过邻苯二甲酸和水杨酸途径降解的芘仅占总量的4.69[(0.060μmol+0.054μmol)/2.43μmol=4.69%],可能该途径仅为其中很小一部分,也可能菌株也利用邻苯二甲酸、水杨酸,从而积累量较少。所推导mycobacteriumsp.wy6对芘的降解途径如图5所示。
验证实验1:
直接将实施例1培养72小时所得的“培养所得物”按照目前本行业所公认的检测结果最正确的气相色谱-质谱联用仪(gaschromatography-massspectrometer,gc-ms)(型号:agilent6890n/5975b)法进行检测。
样品获取及提取方法与实施例1保持一致,最后定容至2ml正己烷中,后进行两种衍生化反应:
①硅烷化:吸取100μl正己烷溶解样品,添加100μlbstfa:tmcs(99:1)硅烷化试剂(n,o-双三甲硅基三氟乙酰胺-三甲基氯硅烷,纯度>99.5%,sigma-aldrich)进行衍生化反应,于60℃条件下衍生反应30min后,gc-ms上机测定。
②甲基化:吸取180μl正己烷溶解样品,添加20μl(三甲基硅烷基)重氮甲烷正己烷溶液[(trimethylsily)diazomethane2.0m己烷溶液,麦克林]进行衍生化反应,于60℃条件下衍生反应30min后,gc-ms上机测定。
检测条件为:
色谱柱为hp-5ms(30m×0.25mm×0.25μm),载气为氦气,流速为1.0mlmin-1,进样量1.0μl不分流,ms扫描范围为50-500amu,进样口温度280℃,离子源温度230℃,四级杆温度150℃,传输线温度280℃,溶剂延迟6.2min,升温程序:初始温度100℃保持1min,然后以5℃min-1升至200℃保持5min,再以10℃min-1升至280℃,保持10min。
所得结果为:通过衍生化后,4-菲羧酸、邻苯二甲酸、水杨酸均可被测得,三种物质对应质谱图如图6所示,且gc-ms定量结果与hplc-ms定量结果无显著差异。
实施例2、提取massiliasp.wg5(cctccab2015362)以菲为唯一碳源的降解产物,依次进行如下步骤:
1)吸取200μl25%甘油保存菌种于20mllb培养基中,在28℃130rpm条件下培养12h;吸取200μl活化菌液转入新的20mllb培养基中,在28℃130rpm条件下培养14~16h获得菌液。
2)在4℃5000rpm条件下将菌液离心10min,获得降解菌;将获得菌体采用无菌水涡旋,将菌体重悬于无菌水中;再次离心,重复该步骤两次,最后将菌体重悬于无菌水中获得菌悬液;
3)调节菌悬液在紫外分光光度计od600nm=1.0(此时菌体浓度约为5×108cfuml-1);
4)吸取1mlod600nm=1.0菌液加入9ml含100mgl-1菲的矿质培养基中,于28℃130rpm条件下培养。
5)每12h采样一次。按上述“二、降解产物提取”中方法对降解产物进行提取降解产物。
6)按照“三、hplc-ms分离鉴定”中条件设置仪器参数,对所提取降解产物进行定性定量分析。
通过48h的培养后提取所得的降解产物经hplc-ms上机检测,在降解产物中测定到检测到降解产物邻苯二甲酸和1-羟基-2-萘甲酸的存在(如图7所示)。色谱图中,菲、邻苯二甲酸、1-羟基-2-萘甲酸的峰面积积分分别为5416946、625462、983338,分别对应的代入图3中菲、邻苯二甲酸、1-羟基-2-萘甲酸标准曲线公式中换算可得。10ml培养体系中:
菲的浓度(mgl-1)=5416946/873701/5(提取10ml培养体系中代谢产物最后甲醇定容为2ml,实质浓缩了5倍;下同)=1.24mgl-1;
降解菲的摩尔含量(μmol)=[(100-1.24)mgl-1/1000×(10ml/1000)]/178.23×106=5.54μmol;
邻苯二甲酸的浓度(mgl-1)=625462/27194/5=4.60mgl-1;
邻苯二甲酸的摩尔含量(μmol)=[4.60mgl-1/1000]×(10ml/1000)]/166.13×106=0.28μmol;
1-羟基-2-萘甲酸的浓度(mgl-1)=983338/504276/5=0.39mgl-1;
1-羟基-2-萘甲酸的摩尔含量(μmol)=[(0.39mgl-1/1000)×(10ml/1000)]/188.18×106=0.021μmol。
在48h时,碳源菲已几乎消耗殆尽,但继续培养至72h后,样品中邻苯二甲酸的浓度并未降低,表明massiliasp.wg5不可代谢利用邻苯二甲酸,与此同时,当massiliasp.wg5以邻苯二甲酸为唯一碳源时,菌体不生长,邻苯二甲酸浓度也未降低。即通过48h的培养,在10ml培养体系中,massiliasp.wg5可将5.54μmol菲通过1-羟基-2萘甲酸→邻苯二甲酸的途径对其进行降解,产生0.28μmol的邻苯二甲酸,通过邻苯二甲酸途径代谢的菲仅占总量的4.5%(0.28μmol/5.54μmol=4.5%)。
因massiliasp.wg5不可利用邻苯二甲酸为碳源生长,为探究其利用1-羟基-2-萘甲酸生成邻苯二甲酸的比例,以1-羟基-2-萘甲酸(100mgl-1)为唯一碳源对massiliasp.wg5进行培养。
7)重复1)-3)步骤。
8)吸取1mlod600nm=1.0菌液加入9ml含100mgl-11-羟基-2-萘甲酸的矿质培养基中,于28℃130rpm条件下培养。
9)每12h采样一次。按上述“二、降解产物提取”中方法对降解产物进行提取降解产物。
10)按照“三、hplc-ms分离鉴定”中条件设置仪器参数,对所提取降解产物进行定性定量分析。通过72h的培养后提取所得的降解产物经hplc-ms上机检测,色谱图中,1-羟基-2-萘甲酸和邻苯二甲酸、的峰面积分别为428635和239715,分别对应的代入图3中1-羟基-2-萘甲酸和邻苯二甲酸标准曲线公式中换算可得,10ml培养体系中:
1-羟基-2-萘甲酸的浓度(mgl-1)=428635/504276/5(提取10ml培养体系中代谢产物最后甲醇定容为2ml,实质浓缩了5倍)=0.17mgl-1;
降解1-羟基-2-萘甲酸的摩尔含量(μmol)=[(100-0.17)mgl-1/1000×(10ml/1000)]/188.18×106=5.31μmol;
邻苯二甲酸的浓度(mgl-1)=239715/27194/5×10(提取10ml培养体系中代谢产物最后甲醇定容为2ml,实质浓缩了5倍;后因浓度过高稀释10倍上机测定)=17.63mgl-1;
邻苯二甲酸的摩尔含量(μmol)=[(17.63mgl-1/1000)×(10ml/1000)/166.13×106=1.06μmol;
即通过72h的培养,在10ml培养体系中,massiliasp.wg5可将5.31μmol的1-羟基-2-萘甲酸通过邻苯二甲酸途径进行降解,产生1.06μmol的邻苯二甲酸。由此可得,通过邻苯二甲酸途径代谢的1-羟基-2-萘甲酸占其总量的20%(1.06μmol/5.31μmol≈20%)。
通过实施例2可得,通过碱裂解法获取的降解产物达十多个峰(图7),目前解析出的降解产物有1-羟基-2-萘甲酸和邻苯二甲酸,在对降解产物定性定量的同时,将其降解途径与已有报道降解途径进行比对发现,仅有约乎25%的菲通过1-羟基-2-萘甲酸途径降解,其余75%的菲的降解途径不通过水杨酸途径,也不通过2,2’-联苯二羧酸途径,可能存在其他未报道的新途径,从而使得其降解速率较高。辅助massiliasp.wg5对1-羟基-2-萘甲酸的降解实验可推断,当生成0.28μmol的邻苯二甲酸时,实际有1.38μmol的1-羟基-2-萘甲酸生成,即通过1-羟基-2-萘甲酸降解的菲只有~25%(1.38μmol÷5.54μmol≈25%)。推测massiliasp.wg5对菲的降解途径如图8所示。
验证实验2:
直接将实施例2培养48小时所得的“培养所得物”按照目前本行业所公认的检测结果最正确的气相色谱-质谱联用仪(gaschromatography-massspectrometer,gc-ms)(型号:agilent6890n/5975b)法进行检测。
样品获取及提取方法与实施例1保持一致,最后定容至2ml正己烷中,后进行衍生化反应:吸取100μl正己烷溶解样品,添加100μlbstfa:tmcs(99:1)硅烷化试剂(n,o-双三甲硅基三氟乙酰胺-三甲基氯硅烷,纯度>99.5%,sigma-aldrich)进行衍生化反应,于60℃条件下衍生反应30min后,gc-ms上机测定。
检测条件为:
色谱柱为hp-5ms(30m×0.25mm×0.25μm),载气为氦气,流速为1.0mlmin-1,进样量不分流1.0μl,ms扫描范围为50-500amu,进样口温度280℃,离子源温度230℃,四级杆温度150℃,传输线温度280℃,溶剂延迟6.2min,升温程序:初始温度100℃保持1min,然后以5℃min-1升至200℃保持5min,再以10℃min-1升至280℃,保持10min。
所得结果为:通过衍生化后,1-羟基-2-萘甲酸、邻苯二甲酸均可被测得,两种物质对应质谱图如图9所示,且gc-ms定量结果与hplc-ms定量结果无显著差异。
对比实验1-1、实验设置与实施例1保持一致,但降解产物提取方法如下,其余等同于实施例1:
1)培养4天后,将以芘为唯一碳源的培养物用6moll-1hcl酸化至ph=2.0。
2)加入10ml乙酸乙酯后超声处理10min。
3)离心使得有机相与水相分离,收集上层有机相。
4)氮吹至乙酸乙酯完全挥发后,用乙腈定容至2ml,上机测定。
所得结果为:仅能测定到所添加污染物芘(浓度为50mgl-1),实施例1中所测得降解产物4-菲羧酸、邻苯二甲酸、水杨酸均未测得。
对比实验1-2、将实施例1步骤3)的“hplc上机检测”改成如同常规技术所述,具体如下表5:
流动相a为甲醇,b为水。
表5
其余等同于实施例1。所得结果为:水杨酸与邻苯二甲酸无法很好的分离,且1-羟基-2-萘甲酸与3-羟基-2-萘甲酸峰形不好,拖尾严重,均无法进行很好的定量。
对比实验2-1、实验设置与实施例2保持一致,但降解产物提取方法如下,其余等同于实施例2:
1)培养48h后,将以菲为唯一碳源的培养物离心,将上清液转移至新的50ml棕色玻璃离心管中。
2)用6moll-1hcl酸化至ph=2.0。
2)加入10ml乙酸乙酯后超声处理10min。
3)离心使得有机相与水相分离,收集上层有机相至新的50ml棕色玻璃离心管中。
4)再次添加10ml乙酸乙酯后超声处理,离心,收集上层有机相至3)中棕色离心管中。
5)重复添加10ml乙酸乙酯后超声处理,离心,收集上层有机相至3)中棕色离心管中。
6)氮吹至乙酸乙酯完全挥发后,用乙腈定容至2ml,上机测定。
所得结果为:仅能测定到所添加污染物菲,实施例2中所测得降解产物1-羟基-2-萘甲酸、邻苯二甲酸均未测得。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例,显然不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!