一种高产丙酮酸的大肠杆菌基因工程菌及其构建方法和应用与流程
本发明属于微生物基因工程技术领域,具体地,本发明涉及一种高产丙酮酸的大肠杆菌基因工程菌及其构建方法和应用。
背景技术:
丙酮酸(Pyruvic acid),浅黄色至黄色透明液体,是生物体基本代谢中间产物之一,广泛应用于食品添加剂、农药、医药等领域。可作为前体,酶法合成L-色氨酸、L-酪氨酸、D/L-丙氨酸和L-二羟基苯丙氨酸等,同时在减肥、增强耐力、降低胆固醇等人类健康领域有应用潜力。发酵法生产丙酮酸的研究开始于二十世纪90年代,日本、美国等发达国家在菌株选育、工程菌株构建、分离纯化等方面开展了一系列的工作,我国的相关研究主要由江南大学和中国科学院微生物研究所等单位开展,也取得了不错的进展。
减弱丙酮酸的降解途径是实现丙酮酸积累的主要思路。目前发酵菌株研究最多的是光滑球拟酵母(Torulopsis glabuata)和大肠杆菌(Escherichia coli)基因工程菌。日本学者Yonehara等筛选出一株多种维生素营养缺陷型光滑球拟酵母T.glabuata IFO 005,经过诱变后,丙酮酸得率可达到0.52-0.54g/g[JP,patent 2000078996]。采用分批补料方式培养63小时,丙酮酸浓度达到67.8g/L[J.Ferment.Bioeng.,1996,82(5):475-479]。刘立民等在高浓度NaCl溶液中驯化得到一株高盐耐受菌株T.glabuata RS23,培养82小时,丙酮酸浓度可达到94.3g/L,葡萄糖转化率达到0.635g/g[Biotech.Bioeng.,2007,97(4):825-832]。
与采用筛选、驯化手段改造酵母相比,产丙酮酸大肠杆菌构建主要采用基因工程技术,通过直接将丙酮酸降解途径失活来实现。Ingram等以E.coli W3110为出发菌株,通过敲除ldhA、poxB、ackA、pflB、adhE、frdBC、sucAB、atpFH基因,在阻断乙酸合成,减弱TCA代谢流的同时,增强糖酵解途径的代谢强度,实现菌体生物量和CO2产生量降低,积累丙酮酸达到66g/L,转化率和生产强度分别达到0.75g/g、1.2g/Lh[PNAS,2004,101(8),2235-2240]。Eiteman等采用类似的思路,通过敲除aceEF、pfl、poxB、pps、ldhA、atpFH、arcA基因,批次补料培养44h,丙酮酸浓度达到90g/L,转化率和生产强度分别达到0.68g/g、2.1g/Lh[Appl.Environ.Microbiol.,2008,74(21),6649-6655]。
中国科学院过程工程研究所通过调控脂肪酸代谢途径,增强糖酵解途径的同时,降低TCA循环强度,得到丙酮酸生产菌株YP01,发酵48小时获得69g/L的丙酮酸。但在发酵过程中仍有较大量的副产物酸生成,主要包括丁二酸、富马酸、酮戊二酸等,副产物酸的总浓度达到丙酮酸浓度的20%-30%。大量副产物的生成不仅降低丙酮酸的产率,也会大幅度的提高后续的纯化成本。分析副产物酸的组成后发现,TCA途径中的中间代谢物是副产物的主要成分,因此降低TCA途径的代谢强度从而减少副产物酸的积累是菌株改造的重点。
在已报道的文献中,多种方法可以降低TCA途径的代谢强度。例如敲除ATP合成酶基因atpFH,使ATP的生成与跨膜电子传递解偶联,降低ATP的生成量,减弱TCA途径强度。还比如敲除sucA基因,使酮戊二酸转化为丁二酸乙酰辅酶A的反应弱化,从而弱化TCA途径。这些途径可以降低TCA途径中副产物酸的产量,但改造菌株的生长都会受到严重影响,降低丙酮酸的生产速率。
技术实现要素:
本发明的一个目的是提供可以高产丙酮酸的大肠杆菌基因工程菌。
本发明的另一个目的是提供上述高产丙酮酸的大肠杆菌基因工程的构建方法。
本发明的再一个目的是提供上述高产丙酮酸的大肠杆菌基因工程菌的应用。
本发明的再一个目的是提供一种生产丙酮酸的方法。
本发明提供的大肠杆菌(Escherichia coli)基因工程菌YP02,其菌种保藏编号为CGMCC No.12463。
本发明的上述大肠杆菌基因工程菌YP02的构建方法,包括以下步骤:
1)在出发大肠杆菌中弱化磷酸烯醇式丙酮酸羧化酶(ppc)的活性,得到重组大肠杆菌,记作重组大肠杆菌Ⅰ;
2)所述重组大肠杆菌Ⅰ中弱化柠檬酸合酶(gltA)的活性,得到重组大肠杆菌,记作重组大肠杆菌Ⅱ,即大肠杆菌基因工程菌YP02。所述大肠杆菌基因工程菌YP02保存时间:2016年5月18日,保存单位:中国微生物菌种保藏管理委员会普通微生物生物中心(CGMCC),保藏编号:CGMCC No.12463。
所述出发大肠杆菌为已经使丙酮酸氧化酶、乙酸激酶和乙酰磷酸化酶失活的大肠杆菌,命名为YPⅠ。所述出发大肠杆菌优选使用现有的大肠杆菌K-12MG1655。
所述弱化磷酸烯醇式丙酮酸羧化酶的活性是使所述磷酸烯醇式丙酮酸羧化酶编码基因的起始密码子由ATG变为GTG;所述使所述磷酸烯醇式丙酮酸羧化酶编码基因的启始密码子替换的方法包括以下步骤:向所述出发大肠杆菌中导入序列表中SEQ ID NO.3所示DNA;
SEQ ID NO.3:
所述序列表中SEQ ID NO:3所示DNA与磷酸烯醇式丙酮酸羧化酶编码基因发生同源重组;
所述弱化柠檬酸合酶的活性包括使所述柠檬酸合酶基因编码基因的启始密码子由ATG变为GTG;所述使所述柠檬酸合酶编码基因的启始密码子替换的方法包括以下步骤:向所述重组大肠杆菌Ⅰ中导入序列表中SEQ ID NO.6所示DNA;
SEQ ID NO.6:
所述序列表中SEQ ID NO:6所示DNA与所述出发大肠杆菌中的柠檬酸合酶编码基因片段发生同源重组。
本发明还提供了一种重组大肠杆菌,即上述的重组大肠杆菌Ⅰ,所述重组大肠杆菌由替换出发大肠杆菌中的磷酸烯醇式丙酮酸羧化酶基因的起始密码子ATG为GTG获得;其中,所述出发大肠杆菌为已经使丙酮酸氧化酶、乙酸激酶和乙酰磷酸化酶失活的大肠杆菌。
本发明还提供了上述大肠杆菌基因工程菌YP02的应用,尤其是其在生产丙酮酸中的应用。
本发明提供的生产丙酮酸的方法,包括以下步骤:发酵上述大肠杆菌重组基因工程菌YP02,生产得到丙酮酸,其中,
所述发酵温度为35-38℃;
所述发酵体系的pH值为6.5-7.5
所述发酵时间为20小时-60小时;
所述发酵接种量的体积百分比为0.1%-10%;
所述发酵培养基中的碳源可以是下述的一种或几种:葡萄糖、木糖、淀粉;
所述发酵培养基中的氮源可以是下述的一种或几种:酵母膏、蛋白胨、玉米浆、糖蜜、氨水、铵盐、尿素。
本发明所构建的大肠杆菌基因工程菌菌株YP02,在好氧条件下,利用无机盐培养基发酵36小时,可将89g/L葡萄糖转化生成61g/L丙酮酸,菌体的生长不受影响,副产物酸浓度明显降低,酮戊二酸为2.5g/L,富马酸为0.4g/L,柠檬酸为0.6g/L。
本发明以已经阻断乙酸主要合成途径的大肠杆菌为出发菌株,通过起始密码子的弱化,降低进入TCA循环的碳流,减慢TCA途径的整体代谢速率,达到积累丙酮酸的目的。本发明中构建的大肠杆菌基因工程菌发酵副产物明显降低,丙酮酸的转化率提高。
附图说明
图1为大肠杆菌合成丙酮酸代谢途径改造示意图。
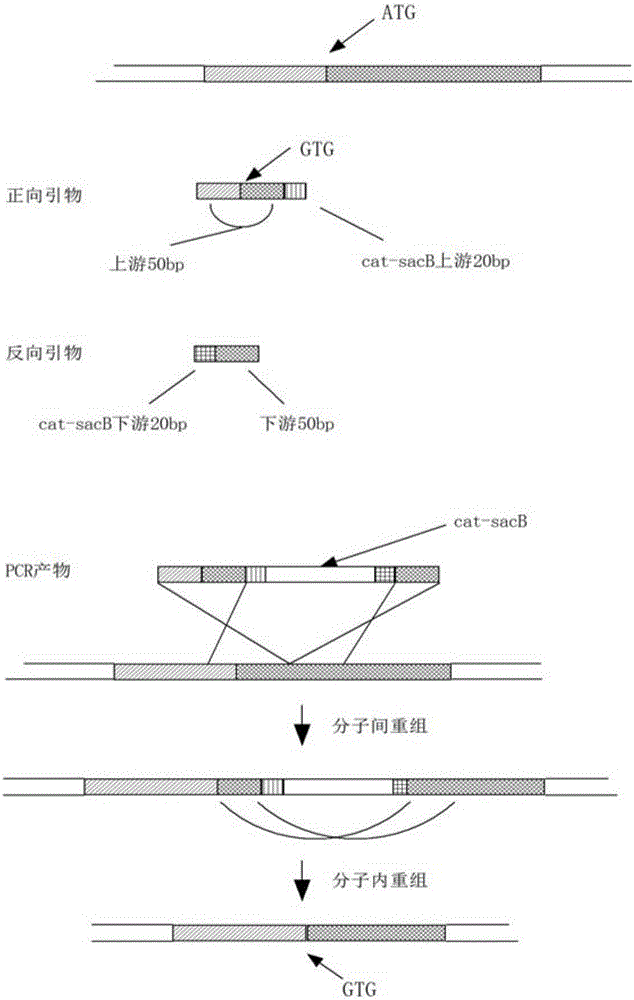
图2为本发明大肠杆菌基因工程菌YP02构建方法中起始密码子替换的操作步骤示意图。
图3为本发明大肠杆菌基因工程菌YP02与出发菌株的主要发酵副产物的对比。
本发明的大肠杆菌基因工程菌YP02(Escherichia coli)已于2016年5月18日保藏于中国微生物菌种保藏管理委员会普通微生物中心(北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,100101),保藏编号为CGMCC NO.12463。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法,具体可参照《分子克隆实验指南》(第三版)J.萨姆布鲁克一书中所列的具体方法进行,或者按照试剂盒和产品说明书进行;下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
本发明的大肠杆菌改造示意图如图1所示。
实施例1大肠杆菌基因工程菌Ⅰ的构建
如图2所示,磷酸烯醇式丙酮酸羧化酶基因(ppc)起始密码子由ATG替换为GTG,构建大肠杆菌基因工程菌Ⅰ。启动密码子替换采用两步同源重组的方法。
大肠杆菌基因工程菌Ⅰ的构建的步骤如下。
第一步,采用热激转化法将pKD46质粒转入出发菌株大肠杆菌YPⅠ中,得到重组大肠杆菌A。具体操作为:吸取1μL pKD46质粒加入感受态细胞中混合均匀,静止5min后置于42℃水浴锅热激90s,冰水浴3min。随后加入800μL无抗生素的LB培养基,在30℃,140rpm孵育1h,涂布于Amp抗性平板,30℃静止培养16h至单菌落长出。
第二步,NCBI数据库中查到ppc基因的DNA序列,以此为依据设计引物ppc-F/ppc-R,其中ppc-F中包括含ppc起始密码子ATG的50bp(片段1)、其下游50bp(片段2)及cat-sacB的上游25bp(片段3),其中,将ATG更改为GTG;ppc-R包括片段2的50bp及cat-sacB的下游25bp,以质粒p EASY-cat-sacB为模版进行PCR扩增。引物序列为:
ppc-F(SEQ ID NO.1):
GGGCTATCAAACGATAAGATGGGGTGTCTGGGGTAATATGAACGAACAATATTCCGCATTGCGTAGTAATGTCAGTATGCTCGGCAAAGTGCTGGGAGAATCCTGGTGTCCCTGTTGATA
ppc-R(SEQ ID NO.2):
TTCTCCCAGCACTTTGCCGAGCATACTGACATTACTACGCAATGCGGAATATAGATACATCAGAGCTTTTACGAG
扩增体系为:
PCR程序为:98℃预变性2min,(98℃热变性10s,55℃退火20s,72℃延伸2min)35cycles,72℃5min。得到含有ppc-F/ppc-R和cat-sacB序列的DNA片段,记作DNA片段Ⅰ,其基因序列如序列表中SEQ ID NO.3所示。
第三步,DNA片段Ⅰ用于第一轮重组。将DNA片段Ⅰ电转至重组大肠杆菌A。电转条件为:首先准备重组大肠杆菌A的电转化感受态细胞;将50μL感受态细胞置于冰上,加入50ng DNA片段Ⅰ,冰上放置2分钟,转移至0.2em的Bio-Rad电击杯,电击参数为2.5kv。电击后迅速将1mL LB培养基转移至电击杯中,吹打5次后转移至试管中,200rpm,30℃孵育2小时。取200μL菌液涂在含有氨苄青霉素和氯霉素的LB平板上,过夜培养后,挑选5个单菌落进行PCR验证。挑选一个正确的单菌落,将其命名为重组大肠杆菌B。
第四步,重组大肠杆菌B的自身同源重组,删除cat-sacB基因片段。具体操作为:将重组大肠杆菌B活化于LB液体培养基,过夜培养;取1mL培养物接种于10mL含有蔗糖的LB液体培养基,过夜培养;取一环菌液划线培养于LB固体培养基,过夜培养;每个菌株挑取400个单克隆点对点划线于含有氯霉素和不含氯霉素的固体培养基,过夜培养;PCR检测失去氯霉素抗性的菌落,测定序列验证正确的即为大肠杆菌基因工程菌Ⅰ。
实施例2大肠杆菌基因工程菌Ⅱ的构建
将大肠杆菌基因工程菌Ⅰ的柠檬酸合酶基因(gltA)的起始密码子ATG替换为GTG,构建大肠杆菌基因工程菌Ⅱ。
如图2所示,启动密码子替换采用两步同源重组的方法。
第二步,NCBI数据库中查到gltA基因的DNA序列,以此为依据设计引物gltA-F/gltA-R,其中gltA-F中包括含gltA起始密码子ATG的50bp(片段1)、其下游50bp(片段2)及cat-sacB的上游25bp(片段3),其中,将ATG更改为GTG;gltA-R包括片段2的50bp及cat-sacB的下游25bp,以质粒p EASY-cat-sacB为模版进行PCR扩增。引物序列为:
gltA-F(SEQ ID NO:4):
AGTCTTACGCAATAAGGCGCTAAGGAGACCTTAAGTGGCTGATACAAAAGCAAAACTCACCCTCAACGGGGATACAGCTGTTGAACTGGATGTGCTGAAATCCTGGTGTCCCTGTTGATA
gltA-R(SEQ ID NO:5):
TTTCAGCACATCCAGTTCAACAGCTGTATCCCCGTTGAGGGTGAGTTTTGATAGATACATCAGAGCTTTTACGAG
扩增体系和PCR程序同实例1,得到含有gltA-F/gltA-R和cat-sacB序列的DNA片段,记作DNA片段III,其基因序列如序列表中SEQ ID NO.6所示。
第二步,DNA片段III用于第一轮重组。将DNA片段III电转至重组大肠杆菌Ⅰ。电转条件为:首先准备重组大肠杆菌Ⅰ的电转化感受态细胞;将50μL感受态细胞置于冰上,加入50ng DNA片段III,冰上放置2分钟,转移至0.2em的Bio-Rad电击杯,电击参数为2.5kv。电击后迅速将1mL LB培养基转移至电击杯中,吹打5次后转移至试管中,200rpm,30℃孵育2小时。取200μL菌液涂在含有氨苄青霉素和氯霉素的LB平板上,过夜培养后,挑选5个单菌落进行PCR验证。挑选一个正确的单菌落,将其命名为重组大肠杆菌C。
第三步,重组大肠杆菌C的自身同源重组,删除cat-sacB基因片段。具体操作为:将重组大肠杆菌C活化于LB液体培养基,过夜培养;取1mL培养物接种于10mL含有蔗糖的LB液体培养基,过夜培养;取一环菌液划线培养于LB固体培养基,过夜培养;每个菌株挑取400个单克隆点对点划线于含有氯霉素和不含氯霉素的固体培养基,过夜培养;PCR检测失去氯霉素抗性的菌落,测定序列验证正确的即为大肠杆菌基因工程菌Ⅱ,也即大肠杆菌基因工程菌YP02。
实施例3用大肠杆菌基因工程菌YP02生产丙酮酸
种子培养基和发酵培养基含以下成分(除葡萄糖外):
大量元素(mmol/L):(NH4)2HPO4 19.92,NH4H2PO4 7.56,KCl 2.00,MgSO4·7H2O1.50;
微量元素(μmol/l):FeCl3·6H2O 8.88,CoCl2·6H2O 1.26,CuCl2·2H2O 0.88,
ZnCl2 2.20,Na2MoO4·2H2O 1.24,H3BO3 1.21,MnCl2·4H2O 2.50;
以大肠杆菌基因工程菌株YP02生产丙酮酸,包括以下步骤:
(1)种子培养:在250mL三角瓶中加入种子培养基100mL,加入葡萄糖1wt%,115℃灭菌30min。冷却后将菌株YP02按照1%(v/v)的接种量接种于种子培养基,在pH=7.0、37℃和200rpm的条件下培养12小时得到种子液。
(2)发酵培养:5L发酵罐发酵培养基体积为2L,121℃灭菌20min,添加已高温灭菌的葡萄糖母液,使发酵罐中葡萄糖浓度为5wt%。将步骤1中种子按照5%(v/v)的接种量接种于发酵培养基,在pH=7.0、37℃和400rpm的条件下培养24小时,添加葡萄糖母液,使发酵罐中葡萄糖总浓度达到10wt%,培养48小时后得到发酵液。
分析方法:使用安捷伦(1200型)高效液相色谱对发酵液中的成分进行定量分析,发酵液中的葡萄糖和有机酸浓度测定采用伯乐公司的Aminex HPX-87H有机酸分析柱分析。结果如图3所示,对照为出发菌株大肠杆菌YPⅠ在同等条件下的发酵结果。
由图3可以看出:大肠杆菌基因工程菌菌株YP02,在好氧条件下,利用无机盐培养基发酵36小时,可将89g/L葡萄糖转化生成65g/L丙酮酸,转化率达到0.73g/g葡萄糖,而且副产物酸浓度相对于大肠杆菌YPⅠ明显降低,α-酮戊二酸为2.5g/L,富马酸为0.4g/L,柠檬酸为0.6g/L。
- 还没有人留言评论。精彩留言会获得点赞!