一种芽胞杆菌多基因叠加敲除方法与流程
本发明涉及微生物遗传工程领域,特别涉及一种芽胞杆菌多基因叠加敲除方法。
背景技术:
芽胞杆菌(Bacillus)是一类需氧或兼性厌氧的革兰氏阳性细菌,在一定条件下能产生抗逆性内生孢子,种类多样,作为益生菌的一种在工业、农业、食品及医疗等领域都有着重要的应用价值。枯草芽孢杆菌是芽胞杆菌属中研究较为详尽的外源基因表达宿主,但由于受自身分泌蛋白酶多、转化率低、构建的表达质粒不太稳定等因素影响,其应用受到一定的限制,其它种的芽胞杆菌同样存在这样的问题。
芽孢杆菌基因敲除技术是研究基因功能的重要手段,利用该技术进行分子生物学研究有利于加深对芽胞杆菌生物重要代谢和调控机制的认识。基因敲除(gene knockout)是一种反向的遗传学研究方法,通过一定途径从分子水平上将机体特定的基因失活或者缺失,对比表观性状的变化,可以对该基因的功能有所了解,该方法在生物学、医学等许多研究领域都具有极其重要的理论意义和实践意义。
实现基因敲除主要有两种方式:一是利用同源重组进行基因敲除,二是利用随机插入突变进行基因敲除。当前利用同源重组进行芽胞杆菌基因敲除已成为主流,同源重组的目的性强,重组过程具有高度特异性和保守性。目前同源重组法应用较为广泛,已成功应用于多种芽胞杆菌基因缺失突变株的构建。
为了开发高效的芽孢杆菌重组表达系统或对芽胞杆菌进行代谢工程改造,往往需要对多个基因(蛋白酶基因和其它调控相关基因)进行叠加敲除或敲入。通过传统的同源重组的敲除方法,连续敲除多个基因,最终得到多基因叠加敲除的菌株,目前已有一些成功的实例,如枯草芽胞杆菌多个蛋白酶缺陷性的菌株WB600,WB600BHM,WB700H,LB700,WB700NWB700NHM,WB800,WB800HM和WB800N等(Biochim Biophys Anta,2004,1694(1-3):299-310),也有地衣芽胞杆菌多基因成功敲除的案例(J Ind Microbiol Biotechnol,2015,42(2):287-95)。美国俄亥俄州立大学芽胞杆菌保藏中心(Bacillus Genetic Stock Center,简称BGSC)也收藏了许多枯草芽胞杆菌多基因缺失的菌株。然而,以上这些多基因缺陷性菌株都是一个个逐渐叠加敲除而成,每一个待敲除基因的上下游同源臂装载到一个敲除质粒中,待一个基因敲除成功后再转入第二个质粒。
现有的基因敲除过程操作繁琐,每个基因的敲除难度不一样,再加外源质粒在芽胞杆菌转化效率一般都比较低,因此需要耗费较长的时间才能获得多基因缺陷性菌株,一个基因敲除平均需要1个月左右的时间。此外,上述多基因敲除菌株的基因组中带有外源抗性片段,不利于其应用于食品和药品生产以及进一步遗传工程改造。
技术实现要素:
本发明要解决的技术问题是现有的多基因敲除方法效率低,还会在基因组DNA中留下筛选标记。
经过研究发现,现有的芽胞杆菌敲除方法,一个基因敲除成功后再转化入含有另外基因上下游同源臂的质粒,所以才会出现上述诸多问题,本发明提供了一种新的芽胞杆菌多基因叠加敲除方法,通过使用温敏型质粒,同时多线程将含有待敲除基因上下游同源臂的温敏型质粒转化进宿主菌,使得多个基因单交换的过程在差不多同一时间完成,利用基因组酶切后转化,实现不同基因单交换的菌株与上一轮的宿主菌发生整合,可以快速进行双交换的过程,从而提高敲除的效率。同时采用温敏型质粒实现无痕敲除过程也避免了宿主菌种引入外源抗性片段,保证了使用安全。
本发明提供的芽胞杆菌多基因叠加敲除方法,包括以下步骤:
(1)制备多种温敏型质粒,每种温敏型质粒含有温敏型复制子、待敲除基因的上下游同源臂片段,以及至少一个抗性基因;不同种温敏型质粒对应不同的待敲除目的基因;
(2)多种温敏型质粒同一时间转化由目标芽胞杆菌制备的感受态细胞A,通过抗性培养基筛选出阳性克隆子,对阳性克隆子进行升温至42~44℃高温培养迫使转化进感受态细胞A的质粒与目标芽胞杆菌基因组发生同源单交换以将质粒基因整合到目标芽孢杆菌基因组中;将发生同源单交换的菌株继续在42~44℃培养,每8~12h传代培养一次,至少传代两次,以剔除游离状态的质粒并促进整合后的基因发生同源双交换;挑取传代培养后的菌株在无抗性平板培养基培养繁殖,然后挑取繁殖获得的单菌落,分别点在有抗性和无抗性的培养基平板上进行培养,挑取在抗性培养基平板上不长而在无抗性培养基平板上生长的克隆子,进行PCR验证和测序验证,验证正确的即为成功敲除第1个目的基因的菌株;
其中,同一时间转化的多种温敏型质粒所含的抗性基因不同;
(3)步骤(2)获得的敲除第1个目的基因的菌株制备感受态细胞B,提取步骤(2)中其余成功发生同源单交换的菌株的基因组DNA,采用特定的限制性内切酶进行酶切,纯化酶切后的基因组DNA片段,同一时间电转化进感受态细胞B,筛选阳性克隆子;阳性克隆子升温到42~44℃的高温培养,迫使转入的基因组DNA片段与感受态细胞B的基因组发生同源单交换;将发生同源单交换的菌株继续在42~44℃培养,每8~12h传代培养一次,至少传代两次,以剔除游离状态的基因组DNA片段并促进整合后的基因发生同源双交换;挑取传代培养后的菌株在无抗性平板培养基培养繁殖,然后挑取繁殖获得的单菌落,分别点在有抗性和无抗性的培养基平板上进行培养,挑取在抗性培养基平板上不长而在无抗性培养基平板上生长的克隆子,进行PCR验证和测序验证,验证正确的即为成功叠加敲除第1个和第2个目的基因的菌株;
所述特定的限制性内切酶,仅能切割成功敲除的目的基因的上下游同源臂之间的序列;
(4)重复步骤(3)的过程,依次转化酶切处理过的步骤(2)余下成功发生同源单交换的菌株的基因组DNA,筛选,成功叠加敲除第1个、第2个和第3个目的基因的菌株,敲除的菌株作为下一轮叠加敲除的宿主菌,以此类推,最终获得多个基因敲除的菌株。
具体操作过程可以参见图1,图1是以4个基因为例的芽胞杆菌多基因叠加敲除过程示意图。
本发明采用温敏型质粒实现无痕敲除,不会留下筛选标记。原理是温敏型质粒上带有与基因组上待敲除基因的上下游基因同源臂时,转入到宿主菌后,由于温敏型复制子在升高温度培养时不能发生复制,容易丢失,在抗性培养基上培养时,只有温敏型质粒与基因组DNA发生同源单交换的菌株能存活;发生同源单交换的菌株在天然条件下有一定的概率发生同源双交换,在双交换的过程中留在基因组上的抗生素基因片段和其他载体片段会从基因组上消失,在高温和无抗性的培养条件下发生的概率大,发生同源双交换的菌株要么回复成野生型,要么形成无痕敲除的菌株,需要进行筛选;通过在抗性和无抗性培养基上分别培养,能够在抗性培养基上生长的,则说明其仍然携带有抗性基因,所以舍弃,筛选出只能在无抗性培养基上生长而不能在抗性培养基上生长的菌株,即为形成无痕敲除的菌株,再进行测序和验证即可确定。
将该菌株作为下一轮的宿主,转化入经过酶切处理的发生了单交换的基因组DNA,再进行上述高温培养,筛选,即可获得另外一个基因也成功敲除的菌株,依次类推,短时间内即可获得多基因成功敲除的菌株。
上述同源单交换的质粒种类可以根据需要进行调整,只需要保证同一时间进程内使用的不同种质粒内含有的抗性基因不同即可。例如多个待敲除基因分为两组在不同的时间段内进行,则两组之间的质粒可以有相同的抗性基因,但同一组内的质粒抗性基因应不同。
优选地,上述芽胞杆菌多基因叠加敲除方法中,所述抗性基因包括卡拉霉素抗性基因Kan,氯霉素抗性基因Cat,四环素抗性基因Tet和红霉素抗性基因Erm中的两种以上。
优选地,上述芽胞杆菌多基因叠加敲除方法中,所述待敲除基因的上下游同源臂片段序列长度在500~700bp之间。太短发生单交换和双交换的概率降低,太长敲除质粒较大,转化效率低,质粒不稳定。
优选地,上述芽胞杆菌多基因叠加敲除方法中,所述温敏型质粒为带有194ts温敏型复制子的大肠杆菌-芽胞杆菌穿梭载体。相较于普通温敏型质粒,该穿梭载体(质粒)在芽胞杆菌中的转化效率高,可以大大缩短转化时间。
优选地,上述芽胞杆菌多基因叠加敲除方法中,所述194ts温敏型复制子是以pE194质粒为模版,SEQ ID NO.1~2所示引物进行扩增获得。
优选地,上述芽胞杆菌多基因叠加敲除方法中,所述温敏型质粒的制备方法为:出发质粒进行进行全质粒PCR扩增,扩增产物经过DpnI过夜消化后回收,采用无缝克隆的方式对质粒和基因片段进行连接,转化大肠杆菌JM110,培养筛选阳性克隆子,经PCR验证和测序验证正确即可;
所述出发质粒为pWEBK15质粒、pWEBC26质粒或pHY300PLK质粒;所述基因片段包括194ts温敏型复制子、待敲除基因的上下游同源臂片段,以及抗性基因。
优选地,上述芽胞杆菌多基因叠加敲除方法中,所述芽胞杆菌为枯草芽胞杆菌(Bacillussubtilis)、地衣芽胞杆菌(Bacillus lincheniformis)、解淀粉芽胞杆菌(Bacillus amyloliquefaciens)、巨大芽胞杆菌(Bacillus megaterium)、短短小芽胞杆菌(Brevibacillus brevis)、短小芽胞杆菌(Bacillus pumilus)、嗜热芽胞杆菌(thermophilic bacillus)或苏云金芽胞杆菌(Bacillus thuringiensis)。
为了便于同行业的科研工作者理解本发明的内容,对本发明中的一些术语进行以下说明:
多基因叠加敲除:从原始宿主出发,先敲除其中一个待敲除基因,并以该菌株为宿主,将其它待敲除基因一个一个敲除,最后得到多基因敲除的菌株。
温敏型复制子:对温度敏感的复制子,在某一温度下可以复制。
无缝克隆:是不依赖于任何限制性内切酶和连接酶实现一个或多个目标DNA的片断插入到质粒上任何位点的方法。
温敏型质粒:含有温度敏感型复制元的质粒,在高温条件下无法复制,在温度高于某一值时可以自动丢失。
同源重组:是指发生在非姐妹染色单体之间或同一染色体上含有同源序列的DNA分子之间或分子之内的重新组合。
同源单交换:与基因组DNA有同源序列的DNA片段或质粒DNA,在细胞内与基因组DNA发生分子间的重新组合,插入到基因组DNA中某一段DNA片段中,发生同源单交换的菌株简称单交换菌株。
同源双交换:是指单交换菌株基因组之内同源重复DNA分子的重新组合,同源双交换的菌株可能是某个基因片段缺失的菌株,也有可能是回复成野生型的菌株。
基因无痕敲除:指敲除掉靶基因后,基因组上不引入任何外源DNA片段的方法。
本发明具有以下有益效果:
本发明将芽胞杆菌多个待敲除基因的上下游同源臂基因片段插入到温敏型质粒中,转化芽孢杆菌,通过高温(42~44℃)培养迫使芽胞杆菌中的敲除质粒与其基因组DNA发生同源单交换,高温传代培养筛选双交换的菌株。提取多个基因单交换成功菌株的基因组DNA,采用适当的限制性内切酶消化后,回收DNA片段,转化单基因成功实现无痕敲除的菌株,进行双交换筛选,叠加敲除成功的菌株作为下一轮叠加敲除的宿主菌,依次叠加最终实现芽胞杆菌多基因的敲除。
本发明采用温敏型敲除质粒可以实现芽胞杆菌基因的无痕敲除,采用限制性内切酶酶切的单交换菌株的基因组DNA转化芽胞杆菌不仅能提高转化的效率,还能有效避免宿主菌中已经敲除基因的回复,显著提高了芽胞杆菌中多基因叠加敲除的效率。
附图说明
图1是以4个基因为例的芽胞杆菌多基因叠加敲除过程示意图;
图2是温敏型基因敲除质粒pKan194ts的构建图谱;
图3是温敏型基因敲除质粒pCat194ts的构建图谱;
图4是温敏型基因敲除质粒pTet194ts的构建图谱;
图5是温敏型基因敲除质粒pAEm194ts的构建图谱。
具体实施方式
下面结合具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好的理解本发明并能予以实施,但所举实施例不作为对本发明的限定。本发明中具体选用芽胞杆菌Bacillus subtilis CICC10073的8个基因(aprE、bpr、epr、mpr、NprB、NprE、vpr和WprA)为例来进一步解释本发明的方法。
本发明中用到的芽胞杆菌菌株Bacillus subtilis CICC10073购买自CICC(中国工业微生物菌种保藏管理中心,http://www.china-cicc.org/);本发明所用到的原始质粒pE194和pMUTIN-GFP+质粒购买自BGSC(Bacillus Genetic Stock center,http://www.bgsc.org/);pHY300PLK质粒和大肠杆菌JM110购买自Takara公司(http://www.takara.com.cn/);pWEBK15载体和pWEBC26质粒按照专利CN201410430501.3公开的方法构建;
pET-28a(+)、pUC57、pWB980均购自杭州宝赛生物科技有限公司,大肠杆菌DH5α购自湖北晶茂生物技术有限公司。
本发明中用到的所有的Phusion DNA聚合酶和限制性内切酶等分子生物学试剂从Thermofisher公司购买(http://www.thermofisher.com/cn/);无缝克隆试剂盒购自南京诺唯赞生物科技有限公司(http://www.vazyme.com/);其它常用生化试剂均是市售分析纯。PCR产物回收和胶回收DNA的方法均采用omega公司的试剂盒的方法。
表1扩增引物与测序引物
实例1 温敏型敲除质粒pKan194ts的构建
1、以pE194质粒为模版,以引物F1和R1(引物见表1)扩增载体温敏型复制子194ts片段;PCR体系为:10×PCRBuffer 5μL,2mM dNTPS 5μL,25mM MgSO4 5μL,10μM primer F/R各1.5μL,模版DNA 0.5μL,KOD-Plus-Neo 1μL,dH2O 32.5μL;PCR反应条件如下:94℃,30个循环(98℃ 10s,58℃ 30s,68℃ 1.5min),68℃ 5min,4℃保温。后续实验中PCR体系和反应条件均参照以上条件配置和设定,退火温度和延伸时间根据实际情况而调整,其它参数不变。
2、参考CN201410430501.3专利文献中的方法构建pWEBK15质粒,具体构建方法如下:
提取pET-28a(+)载体质粒和pUC57质粒。以pET-28a(+)质粒DNA为模版,以引物P1(5’-CGAGATATCATGAGC CATATTCAACGGGA-3’)和P2(5’-CCCACATGTCAGGTGGCACTTTTCGGG GA-3’)扩增pET-28a(+)载体上的Kan基因片段。EcoR V和Pci I双酶切Kan基因片段和pUC57质粒,PCR产物回收产物,连接,转化大肠杆菌DH5α,测序正确的中间载体命名为pUCKan;
提取pUCKan质粒,EcoR I和EcoR V双酶切pUCKan质粒后回收大片段;以枯草芽胞杆菌BS168基因组DNA为模版,以引物P3(5’-CCGGAATTCACACAGGGATAAAATCGGCG-3’)和P4(5’-CGAGATATCTATGCGCT GCATCTCC TCAC-3’)扩增pEB1启动子;EcoR I和EcoR V双酶切pEB1,用PCR产物回收试剂盒回收酶切后的DNA片段,分别再与双酶切后的pUCKan质粒片段连接,转化大肠杆菌DH5α,涂布到含有50μg/mL卡拉霉素的LB固体培养基平板上筛选,最终测序成功的中间载体命名为pUCKanEB1;
提取pUCKanEB1质粒DNA,以其为模版,以磷酸化处理的引物P5(5’-ACCTGACGTCTAAGAAACCA-3’)和P6(5’-GTGAGTTTTCGTTCCACTGA-3’)进行扩增,扩增产物经胶回收试剂盒纯化后,采用平末端连接的条件,在22℃连接2h,转化大肠杆菌DH5α,涂布到含有50μg/mL卡拉霉素的LB固体培养基平板上筛选,最终测序成功的中间载体命名为pKanEB8;
以pWB980质粒DNA为模版,以P7(5’-CCGGAATTCGAGCTCAGCA TTAT-3’)和P8(5’-CTGGACGTCAGCATCTAATCTTCAACAAAC-3’)为引物扩增基因片段psr。提取pKanEB8质粒DNA,用EcoR I和Aat II双酶切psr和pKanEB8质粒,回收大片段,连接,转化大肠杆菌DH5α,涂布到含有50μg/mL卡拉霉素的LB固体培养基平板上筛选,最终测序成功的载体命名为pWEBK15。
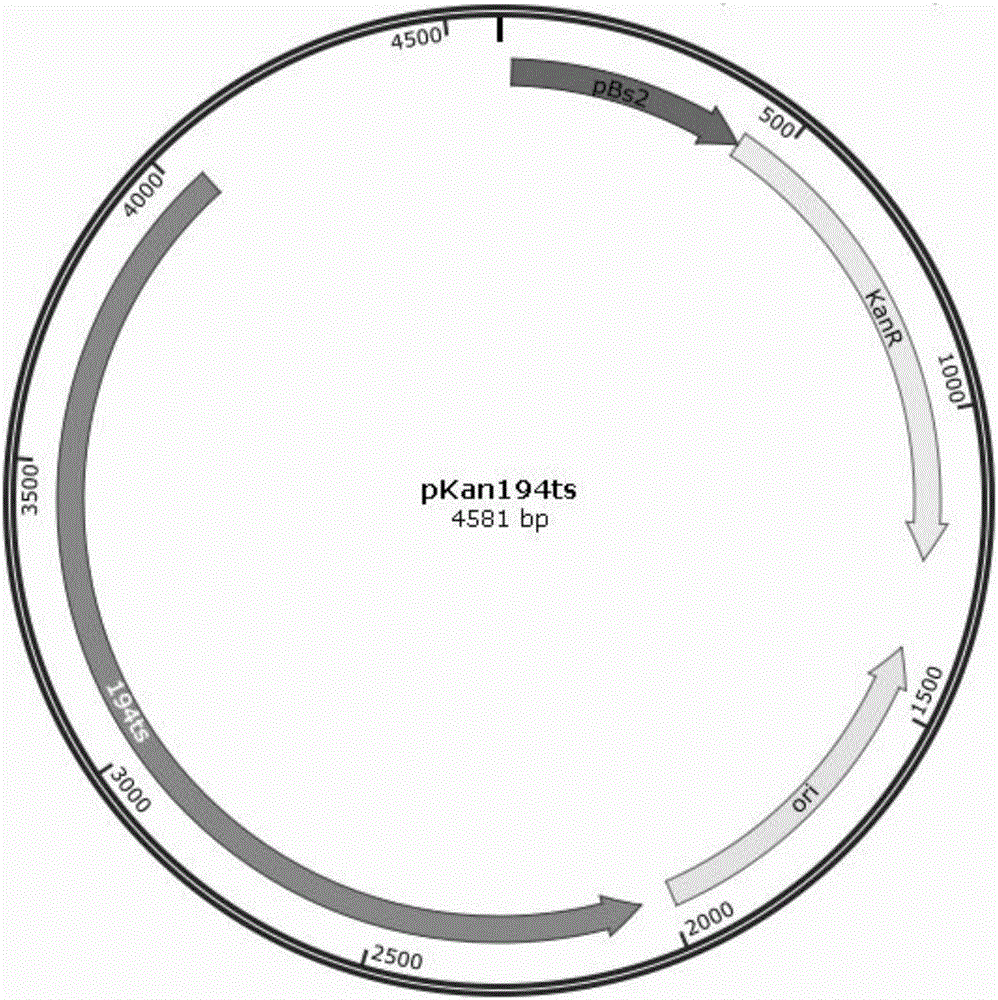
3、以pWEBK15质粒为模版,使用上下游引物F2和R2进行全质粒PCR扩增,扩增产物在37℃经DpnI过夜消化后回收,采用无缝克隆试剂盒的操作说明对质粒片段和卡拉霉素抗性基因Kan的DNA片段进行连接,转化大肠杆菌JM110,涂布在50μg/ml卡拉霉素抗性LB平板(酵母提取物5g/L,蛋白胨10g/L,NaCl 10g/L,琼脂20g/L)上,置30℃培养20h后筛选克隆子,用引物R3和P1进行PCR验证和测序验证,测序成功的载体命名pKan194ts质粒,质粒图谱参见图2。
实例2 温敏型敲除质粒pCat194ts的构建
1、以pE194质粒为模版,以引物F1和R1扩增载体温敏型复制子194ts片段。
2、参考CN201410430501.3专利文献中的方法构建pWEBC26质粒。构建方法如下:
提取提取pWEBK15质粒,EcoR V和Pci I双酶切,DNA胶回收试剂盒回收1500bp左右的大片段。以地衣芽孢杆菌ATCC14580的基因组DNA为模版,以合成的P9(CGAGATATCATGAATTTTCAAACAATCGAGC)和P10(CCCACATGTACAGAAAGTTTGTTGAGAGC)为引物扩增氯霉素抗性基因Cat,EcoR V和Pci I双酶切,PCR产物纯化试剂盒纯化酶切片段。EcoR V和Pci I双酶切的载体片段和Cat基因片段,连接,转化大肠杆菌DH5α,涂布到含有10μg/mL氯霉素的LB固体培养基平板上筛选,阳性克隆子提取质粒后进行菌液PCR验证和测序验证,最终测序成功的载体命名为pWEBC22。
提取枯草芽孢杆菌BS168基因组DNA为模版,以引物P11(CCGGAATTCCGCGTCCAGTTAAGAGCA)和P12(CGAGATATCGAAATGATCCTCCACAAA)扩展启动子pEB5序列,EcoR I和EcoR V双酶切pEB5扩增片段和pWEBC22质粒,PCR产物纯化试剂盒回收大片段,连接,转化大肠杆菌DH5α,涂布到含有10μg/mL氯霉素的LB固体培养基平板上筛选,阳性克隆子提取质粒后进行菌液PCR验证和测序验证,最终测序成功的载体命名为pWEBC26。
以pWEBC26质粒基因组为模版,使用上下游引物F2和R2进行全质粒PCR扩增,扩增产物在37℃经Dpn I过夜消化后回收,采用无缝克隆试剂盒的操作说明对载体片段和基因片段进行连接,转化大肠杆菌JM110,涂布在10μg/ml氯霉素抗性LB平板上,置30℃培养20h后筛选克隆子,用引物R3和P1进行PCR验证和测序验证,测序成功的载体命名为pCat194ts,质粒图谱参见图3。
实例3 温敏型敲除质粒pTet194ts的构建
以pHY300PLK质粒为模版,以引物F4和R4扩增四环素抗性基因Tet;以质粒pKan194ts为模版,使用上下游引物F5和R5进行全质粒PCR扩增,扩增产物在37℃经Dpn I过夜消化后回收,采用无缝克隆试剂盒的操作说明对载体片段和基因片段进行连接,转化大肠杆菌JM110,涂布在10μg/ml四环素抗性LB平板上,置30℃培养20h后筛选克隆子,用引物R3和P1进行PCR验证和测序验证,测序成功的载体命名为pTet194ts,质粒图谱参见图4。
实例4 温敏型敲除质粒pAEm194ts的构建
以pE194质粒为模版,以引物F6和R6扩增载体温敏型复制子194ts片段;以质粒pMUTIN-GFP+为模版,使用上下游引物F7和R7进行全质粒PCR扩增,扩增产物在37℃经DpnI过夜消化后回收,采用无缝克隆试剂盒的操作说明对载体片段和基因片段进行连接,转化大肠杆菌JM110,涂布在300μg/ml红霉素抗性LB平板上,置30℃培养20h后筛选克隆子,用引物P2和P3进行PCR验证和测序验证,测序成功的载体命名为pAEm194ts质粒,质粒图谱参见图5。
实例5 枯草芽孢杆菌基因敲除质粒的构建
采用CTAB法(十六烷基三甲基溴化铵)提取芽胞杆菌的基因组DNA作为基因扩增的模板(微生物学报,2006,46(1):7-12.);以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的aprE-F1/R1为引物扩增aprE基因的上游同源臂aprE-Up,以表1中的aprE-F2/R2为引物扩增aprE基因的下游同源臂aprE-Down;以pKan194ts质粒为模版,以引物F3和R3扩增载体片段,载体片段的扩增产物在37℃经Dpn I过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在50μg/ml卡拉霉素抗性平板上筛选阳性克隆子,用引物aprE-F1和aprE-R2进行测序验证,测序成功的载体命名为pKan194-aprE。
以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的bpr-F1/R1为引物扩增bpr基因的上游同源臂bpr-Up,以表1中的bpr-F2/R2为引物扩增bpr基因的下游同源臂bpr-Down;以pKan194ts质粒为模版,以引物F3和R3扩增载体片段,载体片段的扩增产物在37℃经DpnI过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在50μg/ml卡拉霉素抗性平板上筛选阳性克隆子,用引物bpr-F1和bpr-R2进行测序验证,测序成功的载体命名为pKan194-bpr。
以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的epr-F1/R1为引物扩增epr基因的上游同源臂epr-Up,以表1中的epr-F2/R2为引物扩增epr基因的下游同源臂epr-Down;以pCat194ts质粒为模版,以表1中的F3和R3扩增载体片段,载体片段的扩增产物在37℃经DpnI过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在5μg/ml氯霉素抗性平板上筛选阳性克隆子,用引物epr-F1和epr-R2进行测序验证,测序成功的载体命名为pCat194-epr。
以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的mpr-F1/R1为引物扩增mpr基因的上游同源臂mpr-Up,以表1中的mpr-F2/R2为引物扩增mpr基因的下游同源臂mpr-Down;以pCat194ts质粒为模版,以引物F3和R3扩增载体片段,载体片段的扩增产物在37℃经DpnI过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在5μg/ml氯霉素抗性平板上筛选阳性克隆子,用引物mpr-F1和mpr-R2进行测序验证,测序成功的载体命名为pCat194-mpr。
以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的NprB-F1/R1为引物扩增NprB基因的上游同源臂NprB-Up,以表1中的NprB-F2/R2为引物扩增NprB基因的下游同源臂NprB-Down;以pTet194ts质粒为模版,以表1中的F3和R3扩增载体片段,载体片段的扩增产物在37℃经DpnI过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在5μg/ml四环素抗性平板上筛选阳性克隆子,用引物NprB-F1和NprB-R2进行测序验证,测序成功的载体命名为pTet194-NprB。
以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的NprE-F1/R1为引物扩增NprE基因的上游同源臂NprE-Up,以表1中的NprE-F2/R2为引物扩增NprE基因的下游同源臂NprE-Down;以pTet194ts质粒为模版,以表1中的F3和R3扩增载体片段,载体片段的扩增产物在37℃经DpnI过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在5μg/ml四环素抗性平板上筛选阳性克隆子,用引物NprE-F1和NprE-R2进行测序验证,测序成功的载体命名为pTet194-NprE。
以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的vpr-F1/R1为引物扩增vpr基因的上游同源臂vpr-Up,以表1中的vpr-F2/R2为引物扩增vpr基因的下游同源臂vpr-Down;以pAEm194ts质粒为模版,以表1中的F3和R3扩增载体片段,载体片段的扩增产物在37℃经DpnI过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在5μg/ml四环素抗性平板上筛选阳性克隆子,用引物vpr-F1和vpr-R2进行测序验证,测序成功的载体命名为pAEm194-vpr。
以枯草芽胞杆菌CICC10073基因组DNA为模版,以表1中的WprA-F1/R1为引物扩增WprA基因的上游同源臂WprA-Up,以表1中的WprA-F2/R2为引物扩增WprA基因的下游同源臂WprA-Down;以pAEm194ts质粒为模版,以表1中的F3和R3扩增载体片段,载体片段的扩增产物在37℃经DpnI过夜消化后回收,按照无缝克隆试剂盒的操作说明对载体片段和两个基因片段进行连接,转化大肠杆菌JM110,涂布在5μg/ml四环素抗性平板上筛选阳性克隆子,用引物WprA-F1和WprA-R2进行测序验证,测序成功的载体命名为pAEm194-WprA。
实例6 枯草芽孢杆菌单基因敲除菌株的构建
提取上述构建的质粒pKan194-aprE,pKan194-bpr,pCat194-epr,pCat194-mpr,pTet194-NprB,pTet194-NprE,pAEm194-vpr和pAEm194-WprA,将其电转化到枯草芽胞杆菌CICC10073菌株中,感受态细胞制备和电转化方法参照以下文献:J Microbiol Meth.1999,34(3):183-191;转化的细胞根据抗性涂布到LB固体抗性平板(卡拉霉素20μg/ml,氯霉素10μg/ml,四环素10μg/ml,红霉素5μg/ml)上30℃培养16h,PCR验证阳性克隆子,将阳性克隆子接种到LB液体培养基中于44℃培养,每8~12h转接0.2mL菌液到20mL新鲜LB培养基中传代培养,从传代第2次开始取样,在抗性平板上划线,利用敲除基因上下游同源臂的外侧引物和待敲除基因敲除载体中的引物对单菌落进行菌落PCR扩增,验证单交换菌株;将发生单交换的菌株接种到20mL新鲜LB培养基中于42℃传代培养继续培养,从传代第2次开始取样,将菌液稀释10-6涂布无抗性LB平板置于37℃培养8~10h,将无抗平板上的单菌落进行标记,用灭菌后的牙签挑取到抗性平板上37℃培养8~10h,将抗性平板上不长的克隆子挑取出来做菌落PCR验证和测序验证,正确的克隆子即是单基因成功敲除的菌株。
aprE基因用引物aprE-F3和aprE-R2验证上游单交换,用引物aprE-F1和aprE-R3验证下游单交换,双交换用引物aprE-F1和aprE-R2进行验证。bpr基因用引物bpr-F3和bpr-R2验证上游单交换,用引物bpr-F1和bpr-R3验证下游单交换,双交换用引物bpr-F1和bpr-R2进行验证。epr基因用引物epr-F3和epr-R2验证上游单交换,用引物epr-F1和epr-R3验证下游单交换,双交换用引物epr-F1和epr-R2进行验证。mpr基因用引物mpr-F3和mpr-R2验证上游单交换,用引物mpr-F1和mpr-R3验证下游单交换,双交换用引物mpr-F1和mpr-R2进行验证。NprB基因用引物NprB-F3和NprB-R2验证上游单交换,用引物NprB-F1和NprB-R3验证下游单交换,双交换用引物NprB-F1和NprB-R2进行验证。NprE基因用引物NprE-F3和NprE-R2验证上游单交换,用引物NprE-F1和NprE-R3验证下游单交换,双交换用引物NprE-F1和NprE-R2进行验证。vpr基因用引物Vpr-F3和Vpr-R2验证上游单交换,用引物Vpr-F1和Vpr-R3验证下游单交换,双交换用引物Vpr-F1和Vpr-R2进行验证。WprA基因用引物WprA-F3和WprA-R2验证上游单交换,用引物WprA-F1和WprA-R3验证下游单交换,双交换用引物WprA-F1和WprA-R2进行验证。
通过以上方法先后筛选到8个基因的单交换菌株,先筛选到aprE基因无痕敲除的菌株CICC10073(ΔaprE),后续以此菌株为宿主菌进行多基因叠加敲除。
实例7 多基因叠加敲除
提取NprE,NprB,bpr,epr,mpr,vpr和WprA基因单交换菌株的基因组DNA,胶回收纯化基因组DNA。以菌株CICC10073(ΔaprE)为宿主菌,将epr,mpr,NprE,WprA和NprB基因发生单交换菌株的基因组DNA用NcoI进行酶切消化,柱回收DNA片段,转化CICC10073(ΔaprE),涂布在不同抗性的LB平板上(氯霉素10μg/ml,四环素10μg/ml,红霉素5μg/ml)上37℃培养12h,PCR验证筛选第2个位点发生单交换且第一个位点(aprE基因)没有发生回复的阳性克隆子。将发生单交换的菌株接种到20mL新鲜LB培养基中42℃传代继续培养,从传代第2次开始取样,将菌液稀释10-6涂布无抗性LB平板置于37℃培养8~10h,将无抗平板上的单菌落进行标记,用灭菌后的牙签挑取到抗性平板上37℃培养8~10h,将抗性平板上不长的克隆子挑取出来做菌落PCR验证和测序验证,正确的克隆子即是第2个基因成功叠加敲除的菌株。通过以上方法先筛选到epr基因无痕敲除的菌株CICC10073(ΔaprE epr),后续以此菌株为宿主菌进行多基因叠加敲除。
mpr和NprE基因发生单交换菌株的基因组DNA用NcoI和BamHI进行酶切消化,柱回收DNA片段,转化CICC10073(ΔaprE epr),涂布在不同抗性的LB平板上(氯霉素10μg/ml,四环素10μg/ml)上37℃培养12h,PCR验证筛选第3个位点发生单交换且aprE和epr基因没有发生回复的阳性克隆子。采用以上方法进行双交换筛选,筛选到NprE基因无痕敲除的菌株CICC10073(ΔaprE epr NprE),后续以此菌株为宿主菌进行多基因叠加敲除。
mpr基因发生单交换菌株的基因组DNA用NcoI,BamHI和NdeI进行酶切消化,NprB基因发生单交换菌株的基因组DNA用NcoI和Kpn I进行酶切消化,柱回收DNA片段,转化CICC10073(ΔaprE epr),涂布在氯霉素和四环素抗性的LB平板上37℃培养12h,PCR验证筛选第4个位点发生单交换且aprE,epr和NprE基因没有发生回复的阳性克隆子。采用以上方法进行双交换筛选,筛选到mpr基因无痕敲除的菌株CICC10073(ΔaprE epr NprE mpr),后续以此菌株为宿主菌进行多基因叠加敲除。
NprB基因发生单交换菌株的基因组DNA用NcoI,Kpn I和BstX I进行酶切消化,柱回收DNA片段,转化CICC10073(ΔaprE epr NprE mpr),涂布在四环素抗性的LB平板上37℃培养12h,PCR验证筛选第5个位点发生单交换且aprE,epr,NprE和mpr基因没有发生回复的阳性克隆子。采用以上方法进行双交换筛选,筛选到NprB基因无痕敲除的菌株CICC10073(ΔaprE epr NprE mprNprB),后续以此菌株为宿主菌进行多基因叠加敲除。
WprA基因发生单交换菌株的基因组DNA用NcoI,KpnI和XhoI进行酶切消化,柱回收DNA片段,转化CICC10073(ΔaprE epr NprE mpr NprB),涂布在红霉素抗性的LB平板上37℃培养12h,PCR验证筛选第6个位点发生单交换且aprE,epr,NprE,mpr和NprB基因没有发生回复的阳性克隆子。采用以上方法进行双交换筛选,筛选到WprA基因无痕敲除的菌株CICC10073(ΔaprE epr NprE mpr NprB WprA),后续以此菌株为宿主菌进行多基因叠加敲除。
vpr基因发生单交换菌株的基因组DNA用BstXI,SacI,SacII和XhoI进行酶切消化,柱回收DNA片段,转化CICC10073(ΔaprE epr NprE mpr NprB WprA),涂布在红霉素抗性的LB平板上37℃培养12h,PCR验证筛选第7个位点发生单交换且aprE,epr,NprE,mpr,NprB和WprA基因没有发生回复的阳性克隆子。采用以上方法进行双交换筛选,筛选到vpr基因无痕敲除的菌株CICC10073(ΔaprE epr NprE mpr NprB WprA vpr),后续以此菌株为宿主菌进行多基因叠加敲除。
bpr基因发生单交换菌株的基因组DNA用BamHI,Age I和XhoI进行酶切消化,柱回收DNA片段,转化CICC10073(ΔaprE epr NprE mpr NprB WprA vpr),涂布在红霉素抗性的LB平板上37℃培养12h,PCR验证筛选第8个位点发生单交换且aprE,epr,NprE,mpr,NprB,WprA和vpr基因没有发生回复的阳性克隆子。采用以上方法进行双交换筛选,筛选到bpr基因无痕敲除的菌株CICC10073(ΔaprE epr NprE mpr NprB WprA vpr bpr)。
利用本发明的方法,用了8周左右的时间成功敲除枯草芽孢杆菌8个蛋白酶的基因,与传统的基于温敏型质粒的敲除方法(约需8个月时间)相比,敲除效率显著提高。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!