基于芳基吡唑骨架的四元环状β‑内酰胺衍生物的超声合成与应用的制作方法
本发明涉及有机合成
技术领域:
,尤其涉及一种基于芳基吡唑骨架的四元环状β-内酰胺衍生物的超声波合成方法,还涉及该类化合物的应用。
背景技术:
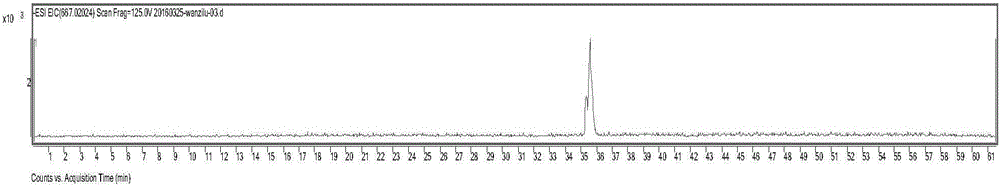
:芳基吡唑类化合物在当今农药发展中占有重要位置,氟虫腈(fipronil)是用于病虫害防治的极具代表性的芳基吡唑类杀虫剂,它能作用于GABA受体进而阻断氯离子通道,近年研究表明谷氨酸门控氯离子通道(GluCls)也是氟虫腈的一个潜在的昆虫特异性靶点。但氟虫腈对蜜蜂和水生生物毒性很大、对环境不友好制约了该药的广泛使用,为了研发出对靶点生物具有高选择性的高效低毒新型氟虫腈类杀虫剂,近年来对氟虫腈进行结构改造的研究课题受到国内外学者的广泛关注,各种各样的氟虫腈衍生物被合成出来。β-内酰胺类化合物是指分子中含有四元的β-内酰胺环状结构的一类化合物,这类化合物既可作为有机合成中间体,同时也是一种重要的生物活性物质,如青霉素、头孢霉素等在临床上被用作抗生素类药物使用,研究表明β-内酰胺环是β-内酰胺类抗生素保持生物活性的关键必需基团,β-内酰胺类抗生素的杀菌作用机制被认为是β-内酰胺类抗生素能够抑制细菌细胞壁的合成:其化学结构与粘肽转肽酶的作用底物D-丙氨酰-D-丙氨酸的末端有相似的空间结构,从而可与转肽酶的酶活性中心竞争性地共价结合,产生不可逆的抑制作用而使粘肽转肽酶失活,使其催化的转肽反应不能进行,从而阻碍细菌细胞壁的正常合成而导致细菌死亡。上世纪20年代,美国科学家Richard和Loomis首先发现超声波可以加速化学反应,但长期以来未能引起化学家们的重视,直到80年代中期大功率超声设备的普及和发展,超声波在化学工业中的应用迅速发展,并产生了新的交叉学科——声化学。近年来,国内外有关声化学的研究以及学术交流异常活跃,随着声化学的发展,超声波已被广泛应用于氧化反应、还原反应、加成反应、取代反应、缩合反应、水解反应等,几乎涉及有机化学的各个领域。基于以上考虑,通过活性结构拼接对氟虫腈的5位氨基进行结构修饰合成一系列氟虫腈类希夫碱化合物,进而在氟虫腈的芳基吡唑骨架中引入四元环状β-内酰胺结构,合成了一系列基于芳基吡唑骨架的四元环状β-内酰胺衍生物,通过活性结构剪裁实现了芳基吡唑类化合物和β-内酰胺类化合物的结合,基于芳基吡唑骨架的四元环状β-内酰胺衍生物中的内酰胺结构具有底物类似作用,吡唑环和四元β-内酰胺环具有π-π堆积作用,这些具有生物活性的结构的协同作用使得本发明所合成的基于芳基吡唑骨架的四元环状β-内酰胺衍生物具有优良的杀虫效果和抗菌防效;在合成方法上,目前合成四元β-内酰胺环的主要方法是通过烯酮和亚胺发生staudinger反应来合成,但是由于烯酮的化学性质非常活泼,尤其是单取代的烯酮,在制备上难度较大,这就限制了该方法的应用范围,本发明以酰氯作为烯酮前体,在碱的作用下原位生成反应活性极高的烯酮,此烯酮不经蒸馏分离直接用于下一步和氟虫腈类亚胺的反应,一锅合成了一系列基于芳基吡唑骨架的四元环状β-内酰胺衍生物,减少了中间步骤所带来的影响,对总的收率有所提高,另外将超声波辐射法应用到基于芳基吡唑骨架的四元环状β-内酰胺衍生物的合成中,超声波的空化作用使得C-X等特定振动频率的化学键更容易断裂,从而使得反应产率有进一步提高。技术实现要素:本发明通过活性结构拼接原理设计并提供了一类结构新颖的化合物——基于芳基吡唑骨架的四元环状β-内酰胺衍生物,通过对氟虫腈的5位氨基进行结构修饰合成一系列氟虫腈类希夫碱化合物,进而在氟虫腈的芳基吡唑骨架中引入四元环状β-内酰胺结构,从而实现芳基吡唑类化合物和β-内酰胺类化合物的结合,在合成过程中将超声波辐射法应用到基于芳基吡唑骨架的四元环状β-内酰胺衍生物的合成中,以酰氯作为烯酮前体,在碱的作用下原位生成反应活性极高的烯酮,此烯酮不经蒸馏分离直接用于下一步和氟虫腈类亚胺的反应,一锅合成了一系列基于芳基吡唑骨架的四元环状β-内酰胺衍生物,减少了中间步骤所带来的影响,以提高收率。基于以上考虑,本发明的三个发明目的为:第一个目的:提供一种基于芳基吡唑骨架的四元环状β-内酰胺衍生物,其结构式如通式(A)所示:所述结构通式(A)中:R1X为如下三种情况之一:(1)H;(2)卤素;(3)R1X中的X为SO或S,R1为-CN、C1~C4的烷基或C1~C4的卤代烷基;具体如下所示:R2为取代或未取代的苯基、萘基、蒽基、菲基、呋喃基、吡咯基、噻吩基、吡啶基、喹啉基、吲哚基,所述取代可以是单取代或多取代,取代的位置可以是苯环邻位、间位、对位,取代的基团包括烷基、烷氧基、羟基、卤素、硝基。R3、R4各自独立选自H、卤素、苯基、C1~C4的烷基、环烷基、C1~C4的卤代烷基、苯基取代的C1~C4烷基,R3和R4可相同或者不相同;优选的,所述R1X选自以下基团中的任意一种:H、—SOCF3以及—SOC2H5;优选的,所述R2为化合物R2-CHO中的基团R2,所述化合物R2-CHO选自以下化合物中的任意一种:R2-CHO为苯甲醛、4-甲氧基苯甲醛、2,3-二氯苯甲醛、2-氟-4-溴苯甲醛、2,5-二氟苯甲醛、2,6-二甲基苯甲醛、3-溴-4-羟基苯甲醛、4-氟苯甲醛、4-氯苯甲醛、3,5-二(三氟甲基)苯甲醛;2,4,6-三氟苯甲醛、1-甲氧基-2-萘甲醛、1-萘甲醛、2-羟基-1-萘甲醛;9-蒽甲醛;9-菲甲醛;2-呋喃甲醛、5-甲基-2-呋喃甲醛;2-吡咯甲醛、3-吡咯甲醛;2-噻吩甲醛;2-吡啶甲醛、2-溴-3-吡啶甲醛、2,6-二氯-3-吡啶甲醛;3-喹啉甲醛以及3-吲哚甲醛;最佳的,所述化合物R2-CHO选自以下化合物中的任意一种:苯甲醛、肉桂醛、4-氟苯甲醛、4-甲氧基苯甲醛以及4-羟基苯甲醛;优选的,R3、R4各自独立选自以下基团中的任意一种:-H、CH3-、Cl、以及取代或未取代的苯基;最佳的,R3、R4各自独立选自以下基团中的任意一种:-H、CH3-、以及苯基;第二个目的:提供一种易于高效制备、节能环保、生产成本相对较低的基于芳基吡唑骨架的四元环状β-内酰胺衍生物(A)的合成方法。一种结构式如通式(A)所示化合物的合成方法:通过对氟虫腈的5位氨基进行结构修饰合成一系列氟虫腈类希夫碱化合物(现有技术),进而在氮气保护下由氟虫腈类希夫碱酰氯和碱加溶剂在超声波辐射的条件下反应制备得到通式(A)所示化合物。更具体的,所述结构式如通式(A)所示化合物的合成方法为:将在氮气保护下向反应容器中加入氟虫腈类希夫碱碱和溶剂,然后将反应容器置于超声波反应器水槽中,在超声条件下向反应容器中滴加酰氯用溶剂稀释后的溶液,半小时滴完,然后继续超声反应3小时,分离纯化后得产物。反应装置如图4所示,其中超声波反应器可以调节频率、超声时间,快速称取一定量真空干燥过的氟虫腈类希夫碱并转移到圆底烧瓶中(此圆底烧瓶预先洗净、晾干并于105℃烘箱干燥2h后置干燥器中保存至室温),圆底烧瓶中间瓶口处连接恒压滴液漏斗,侧端瓶口和恒压滴液漏斗上端用翻口橡胶塞塞住,通过注射器针头和双排管将反应体系置换成氮气氛围,然后在翻口橡胶塞上插入连有塑料注射器的氮气球,通过注射器将溶于无水溶剂中的无水碱加入到反应体系中,最后将酰氯稀释于溶剂中并缓慢滴加到反应体系,分离纯化采用硅胶柱层析,其合成路线如下所示:所述氟虫腈类希夫碱的合成见:陈连清;周泉;万子露等.1-(2,6-二氯-4-三氟甲基)苯基-3-氰基-5-亚甲氨基吡唑型化合物及其微波消解合成法与应用.201610015077.5,公开号:CN105503731A。R1、R2、R3、R4如前所述;所述碱选自三乙胺、三丁胺、吡啶、N,N-二甲基乙酰胺、三苯基膦、碳酸钾以及氢化钠中的任意一种;所述溶剂选自苯、甲苯、二甲苯、氯苯、硝基苯、乙醚、二氯甲烷、三氯甲烷、二甲亚砜、四氢呋喃、乙二醇二乙醚以及N-环己基-2-吡咯烷酮中的任意一种;优选的,所述碱为三乙胺、吡啶或三丁胺。优选的,所述有机溶剂为苯、甲苯、乙醚、二氯甲烷、三氯甲烷以及四氢呋喃中的任意一种,最佳为苯;优选的,所述超声波辐射功率为100~150W,反应时间为2~4h。优选的,所述氟虫腈类希夫碱、酰氯和碱的摩尔比为1:2.2:2.5。第三个目的:提供一种上述基于芳基吡唑骨架的四元环状β-内酰胺衍生物在杀虫剂和抗菌药物中的应用。为实现本发明的第三个目的,本发明将制备得到的基于芳基吡唑骨架的四元环状β-内酰胺衍生物分别用于防治直翅目、缨翅目、同翅目、异翅目、鳞翅目、鞘翅目、膜翅目和双翅目类害虫及抗金黄色葡萄球菌、大肠埃希氏菌和普通变形杆菌试验,取得了良好的杀虫效果和抗菌效果。与现有技术相比,本发明的优点和有益效果如下:本发明在对氟虫腈的5位氨基进行结构修饰合成一系列氟虫腈类希夫碱化合物的基础上,进而在氟虫腈的芳基吡唑骨架中引入四元环状β-内酰胺结构,合成了一系列基于芳基吡唑骨架的四元环状β-内酰胺衍生物,实现了芳基吡唑类化合物和β-内酰胺类化合物的结合,提供了一类结构新颖的化合物;在合成方法上,目前合成四元β-内酰胺环的主要方法是通过烯酮和亚胺发生staudinger反应来合成,但是由于烯酮的化学性质非常活泼,尤其是单取代的烯酮,在制备上难度较大,这就限制了该方法的应用范围,本发明以酰氯作为烯酮前体,在碱的作用下生成反应活性极高的烯酮(Ⅰ),此烯酮不经蒸馏分离直接用于下一步和氟虫腈类亚胺的反应,一锅合成了一系列基于芳基吡唑骨架的四元环状β-内酰胺衍生物,减少了中间步骤所带来的影响,对总的收率有所提高,另外本发明将超声波辐射法应用到基于芳基吡唑骨架的四元环状β-内酰胺衍生物的合成中,对反应产率有进一步的提高。基于芳基吡唑骨架的四元环状β-内酰胺衍生物中的内酰胺结构具有底物类似作用,吡唑环和四元β-内酰胺环具有π-π堆积作用,这些具有生物活性的结构的协同作用使得本发明所合成的基于芳基吡唑骨架的四元环状β-内酰胺衍生物具有优良的生物活性,特别是在杀虫和抗菌方面表现出较高活性,因此具有非常大的开发应用价值。附图说明图1为化合物A2的总离子流图(TIC图);图2为化合物A2的提取离子流图(EIC图);图3为化合物A2的质谱图;图4为反应装置图。具体实施方式下面结合具体实施例对本发明作进一步的详细说明,但这些具体实施例不以任何方式限制本发明的保护范围。以下各实施例中所用的原料均为已知化合物,可在市场上购得,或可用本领域已知的方法合成。氟虫腈类希夫碱的合成见:陈连清;周泉;万子露等.1-(2,6-二氯-4-三氟甲基)苯基-3-氰基-5-亚甲氨基吡唑型化合物及其微波消解合成法与应用.201610015077.5,公开号:CN105503731A。实施例1、3、5、7、9和10中氟虫腈类希夫碱的合成方法见以上专利申请公开文本第0041-0043段;实施例2中氟虫腈类希夫碱的合成方法参考以上专利申请公开文本第0051-0053段的合成方法;实施例4中氟虫腈类希夫碱的合成方法类似于实施例1中氟虫腈类希夫碱的合成方法,可参考之,将其中反应物苯甲醛换作肉桂醛,其他条件如溶剂、催化剂、温度、投料摩尔比和分离纯化方法不变,即可得到1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-(3-苯基烯丙基)亚氨基-1H-吡唑;实施例6中氟虫腈类希夫碱的合成方法见以上专利申请公开文本第0053-0055段;实施例8中氟虫腈类希夫碱的合成方法见以上专利申请公开文本第0047-0049段。实施例1、化合物A1的合成从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.525g(1mmol)1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-苯基亚甲氨基-1H-吡唑,并用注射器吸取0.253g(2.5mmol,约0.25mL)无水三乙胺和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.34g(2.2mmol)苯乙酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去三乙胺盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得黄色油状化合物A1,产率:86.3%.1H-NMR(400MHz,DMSO-d6)7.4-7.65(m,5H,Ar-H),7.22-7.25(m,4H,Ar-H),7.15(d,1H,Ar-H),6.99(m,1H,Ar-H),6.94(m,1H,Ar-H),3.80(s,1H,COCH);3.62(s,1H,NCH);IRν(cm-1):3083,2258(νC≡N),1724(νC=O),1575,1497,1312(νC-F),761,685.实施例2、化合物A2的合成在从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.541g(1mmol)化合物1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-(4-羟基苯基)亚甲氨基-1H-吡唑,并用注射器吸取0.253g(2.5mmol,约0.25mL)无水三乙胺和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.34g(2.2mmol)苯乙酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去三乙胺盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得淡黄色固体A2,产率:76%.1H-NMR(400MHz,DMSO-d6)10.02(s,1H,OH),7.13-7.48(m,10H,Ar-H),6.61(d,1H,Ar-H),3.75(s,1H,COCH),3.21(s,1H,NCH).IRν(cm-1):3084,2252(νC≡N),1744(νC=O),1571,1498,1322(νC-F),762,681.实施例3、化合物A3的合成从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.525g(1mmol)1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-苯基亚甲氨基-1H-吡唑,并用注射器吸取0.253g(2.5mmol,约0.25mL)无水三乙胺和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.4g(2.2mmol)2-苯基丁酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去三乙胺盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得黄色油状化合物A3,产率:91%.1H-NMR(400MHz,DMSO-d6)7.51-6.88(m,12H,Ar-H),3.61(t,1H,NCH),1.95(q,2H,CH3CH2),1.05(t,3H,CH2CH3).IRν(cm-1):3091,2254(νC≡N),1727(νC=O),1574,1496,1312(νC-F),762,683.实施例4、化合物A4的合成从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.551g(1mmol)化合物1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-(3-苯基烯丙基)亚氨基-1H-吡唑,并用注射器吸取0.253g(2.5mmol,约0.25mL)无水三乙胺和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.34g(2.2mmol)苯乙酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去三乙胺盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:25)得黄棕色油状化合物A4,产率:82%.1H-NMR(400MHz,(CD3)2CO)7.18-7.50(m,13H,Ar-HandC=C-H),6.71(m,1H,Ar-H),3.79(s,1H,COCH),3.21(s,1H,NCH).IRν(cm-1):3083,2259(νC≡N),1734(νC=O),1575,1496,1311(νC-F),765,686.HPLC-MS见图1、2和3。实施例5、化合物A5的合成从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.525g(1mmol)1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-苯基亚甲氨基-1H-吡唑,并用注射器吸取0.198g(2.5mmol,约0.2mL)无水吡啶和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.204g(2.2mmol)丙酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去吡啶盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得黄色油状化合物A5,产率:85%.1H-NMR(400MHz,DMSO-d6)7.33-6.97(m,7H,Ar-H),3.76(s,1H,COCH);3.60(s,1H,NCH),1.23(d,3H,CHCH3).IRν(cm-1):3087,2258(νC≡N),1725(νC=O),1575,1497,1312(νC-F),763,687.实施例6、化合物A6的合成在从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.525g(1mmol)化合物1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-(4-氟苯基)亚甲氨基-1H-吡唑,并用注射器吸取0.253g(2.5mmol,约0.25mL)无水三乙胺和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.34g(2.2mmol)苯乙酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去三乙胺盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得淡黄色固体A6,产率:85%.1H-NMR(400MHz,DMSO-d6)7.13-7.48(m,10H,Ar-H),6.61(d,1H,Ar-H),3.75(s,1H,COCH),3.21(s,1H,NCH).IRν(cm-1):3084,2253(νC≡N),1754(νC=O),1575,1498,1312(νC-F),762,685.实施例7、化合物A7的合成从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.525g(1mmol)1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-苯基亚甲氨基-1H-吡唑,并用注射器吸取0.198g(2.5mmol,约0.2mL)无水吡啶和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.233g(2.2mmol)异丁酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去吡啶盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得黄色油状化合物A7,产率:88%.1H-NMR(400MHz,DMSO-d6)7.36-6.96(m,7H,Ar-H),3.62(s,1H,NCH),1.23(s,3H,CHCH3),1.29(s,3H,CHCH3).IRν(cm-1):3089,2251(νC≡N),1725(νC=O),1573,1497,1313(νC-F),761,681.实施例8、化合物A8的合成在从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.555g(1mmol)化合物1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-(4-甲氧基苯基)亚甲氨基-1H-吡唑,并用注射器吸取0.253g(2.5mmol,约0.25mL)无水三乙胺和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.34g(2.2mmol)苯乙酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去三乙胺盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得淡黄色固体A8,产率:78%.1H-NMR(400MHz,DMSO-d6)7.12-7.49(m,10H,Ar-H),6.62(d,1H,Ar-H),3.72(s,1H,COCH),3.58(m,3H,OCH3),3.22(s,1H,NCH).IRν(cm-1):3086,2254(νC≡N),1749(νC=O),1571,1492,1311(νC-F),762,681.实施例9、化合物A9的合成从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.525g(1mmol)1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-苯基亚甲氨基-1H-吡唑,并用注射器吸取0.198g(2.5mmol,约0.2mL)无水吡啶和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.506g(2.2mmol)二苯基乙酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去吡啶盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得黄色油状化合物A9,产率:93%.1H-NMR(400MHz,DMSO-d6)7.56-6.96(m,17H,Ar-H),3.61(s,1H,NCH),.IRν(cm-1):3081,2252(νC≡N),1735(νC=O),1573,1498,1312(νC-F),762,683.实施例10、化合物A10的合成从烘箱中取出干燥的反应用玻璃仪器和磁子,氮气保护下加入0.525g(1mmol)1-(2,6二氯-4-三氟甲基)苯基-3-氰基-4-三氟甲基亚磺酰基-5-苯基亚甲氨基-1H-吡唑,并用注射器吸取0.462g(2.5mmol,约0.46mL)无水三丁胺和4mL无水苯加入反应瓶中,将单口瓶放入KQ3200E型(功率150W)超声波水槽中反应,再将0.323g(2.2mmol)环己基甲酰氯的4mL苯溶液缓慢滴加到反应体系中,约半小时滴完,滴完后继续超声反应3h,反应结束后,抽滤除去三丁胺盐酸盐,旋干滤液后加入20mL乙酸乙酯,用15mL饱和碳酸钠水溶液洗两次,再用15mL饱和氯化钠水溶液洗一次,收集有机层,加入无水硫酸镁干燥过夜,次日抽滤除去无水硫酸镁,向滤液中加入2g硅胶粉旋干成粉末状,硅胶柱层析分离(干法上样,V乙酸乙酯:石油醚=1:20)得黄色油状化合物A10,产率:79%.1H-NMR(400MHz,DMSO-d6)7.37-6.98(m,1H,Ar-H),3.80(s,1H,NCH);1.62(m,4H,CCH2).1.44(m,6H,CCH2CH2).IRν(cm-1):3081,2251(νC≡N),1721(νC=O),1575,1494,1312(νC-F),761,689.对比例、化合物A1的合成在室温条件下分别或同时改变实施例1的反应方式,制备化合物A1,制备结果与实施例1的结果同时列于下表1中:表1不同反应条件下A1的合成及其产率注:对比例3中两步法先将烯酮分离出来,再用烯酮与亚胺反应生成目标产物。从对比例可以看出,反应时间和超声与否对反应产率均有影响,使用超声波辐射法与磁力搅拌相比产率有明显的提升,且一步法中的烯酮不经分离直接用于下一步反应所得到的产率高于两步法。实施例11、基于芳基吡唑骨架的四元环状β-内酰胺衍生物溶液的制备本实施例所制备的抗菌剂剂型为溶液剂,将供试化合物A1—A10分别溶于DMSO中,预配成0.1%(m/v)的浓度,然后用1%(m/v)醋酸蒸馏水溶液稀释到10,1,0.1mg/L3种浓度作为供试样品,阳性对照药诺氟沙星(NF),直接用1%(m/v)醋酸蒸馏水配制成10,1,0.1mg/L3种浓度,空白对照组为1%(m/v)醋酸溶液。所制备的稀释溶液剂备用于以下各实施例。实施例12、对金黄色葡萄球菌的抗菌活性评价对金黄色葡萄球菌的抗菌活性评价采用平皿试验法测定,使用实施例11制备的各化合物的稀释溶液剂,以牛肉膏蛋白胨作为培养基,接种后于37℃培养24h,观察、记录抑菌圈大小,并与阳性对照药诺氟沙星(NF)相比,依此评价供试化合物的抑菌活性高低,++表示高活性,+表示中等活性,-表示活性较弱。试验结果见表2。表2化合物A1~A10的抗金黄色葡萄球菌活性实施例13、对大肠埃希氏菌的抗菌活性评价对大肠埃希氏菌的抗菌活性评价采用平皿试验法测定,使用实施例11制备的各化合物的稀释溶液剂,以牛肉膏蛋白胨作为培养基,接种后于37℃培养24h,观察、记录抑菌圈大小,并与阳性对照药诺氟沙星(NF)相比,依此评价供试化合物的抑菌活性高低,++表示高活性,+表示中等活性,-表示活性较弱。试验结果见表3。表3化合物A1~A10的抗大肠埃希氏菌活性实施例14、对普通变形杆菌的抗菌活性评价对普通变形杆菌的抗菌活性评价采用平皿试验法测定,使用实施例11制备的各化合物的稀释溶液剂,以牛肉膏蛋白胨作为培养基,接种后于37℃培养24h,观察、记录抑菌圈大小,并与阳性对照药诺氟沙星(NF)相比,依此评价供试化合物的抑菌活性高低,++表示高活性,+表示中等活性,-表示活性较弱。试验结果见表4。表4化合物A1~A10的抗普通变形杆菌活性实施例15、化合物A1-A10悬浮剂的制备本实施例所制备的农药剂型为悬浮剂,以下所称“总质量”指“所制备的悬浮剂的总质量”。先将10份占总质量5%的表面活性剂萘磺酸钠甲醛缩合物分别稀释于10份占总质量5%适宜的防冻剂乙二醇中,并分别向该溶液中缓缓加入占总质量25%的水,在快速搅拌下分别向10组溶液中依次加入占总质量25%的实施例1-10制备的化合物A1~A10及占总质量5%的适宜助剂(防腐剂苯甲酸、消泡剂有机硅和增稠剂黄原胶),加完后对其进行研磨,最后加入占总质量35%的水。将制备得到的悬浮剂再加水稀释分别制备出化合物A1~A10所需浓度。所制备的稀释悬浮剂备用于以下各实施例。实施例16、对粘虫的生物活性评价按实施例15制备本发明提供的具有杀虫活性的基于芳基吡唑骨架的四元环状β-内酰胺衍生物的悬浮剂,用水稀释配成浓度100mg/L的农药溶液,选取20头3龄粘虫和10片一寸长的玉米叶片放于培养皿内进行定量喷洒,晾干后移入温室内正常饲养,24~72小时候统计存活和死亡数。实验重复3次,结果取平均值。活性相对于空白对照以百分比计,分为A、B、C、D四级,死亡率100%~90%为A级,死亡率90%~70~%为B级,死亡率70%~50%为C级,死亡率0%~50%为D级。测试结果见表5。表5A1~A10在测试浓度为100mg/L时对粘虫的活性化合物A1A2A3A4A5A6A7A8A9A10活性级别AAAAAABABA实施例17、对黑尾叶蝉的生物活性评价按实施例15制备本发明提供的具有杀虫活性的基于芳基吡唑骨架的四元环状β-内酰胺衍生物的悬浮剂,用水稀释配成浓度为160mg/L的农药溶液,选取二芯稻苗浸入药液中,5秒后取出晾干,置于大试管中,每管20株,然后引入20头或以上的黑尾叶蝉5龄若虫,管口用白色纱布包扎后置于室温条件下,24小时后检查存活和死亡虫数。实验重复3次。结果取平均值。活性相对于空白对照以百分比计,分为A、B、C、D四级,死亡率100%~90%为A级,死亡率90%~70%为B级,死亡率70%~50%为C级,死亡率0%~50%为D级。测试结果见表6。表6A1~A10在测试浓度为160mg/L时对黑尾叶蝉的活性化合物A1A2A3A4A5A6A7A8A9A10活性级别ABAAAABAAA实施例18:对尖音库蚊幼虫的生物活性评价按实施例15制备本发明提供的具有杀虫活性的基于芳基吡唑骨架的四元环状β-内酰胺衍生物的悬浮剂,用水稀释配成浓度为80mg/L的农药溶液,再加入10mL含有10头4龄蚊幼虫的饲养水。每天添加少许饲料,直到蚊幼虫全部化蛹或死亡。随时将化的蛹吸走。实验重复3次。结果取平均值。活性相对于空白对照以百分比计,分为A、B、C、D四级,死亡率100%~90%为A级,死亡率90%~70%为B级,死亡率70%~50%为C级,死亡率0%~50%为D级。测试结果见表7。表7化合物A1~A10在测试浓度为80mg/L时对尖音库蚊幼虫的活性实施例19、对小菜蛾的生物活性评价分别使用实施例15制备的10组化合物的40、80mg/L浓度的水稀释悬浮剂,为了验证其对小菜蛾的安全性是否提高,以小菜蛾为试虫,采用喷雾法测定了各化合物对小菜蛾的综合毒性(接触毒性和胃毒毒性),相关结果见表7。表7化合物A1-A10对小菜蛾的48h毒性考察结果从表5结果可知:氟虫腈对小菜蛾毒性较高,40mg/L时有4%的中毒率,而在80mg/L时中毒死亡率接近一半,达到46%;其主要表现为身体瘫软落地或死亡。化合物A1~A10则对小菜蛾的综合毒性相对较低,40mg/L处理时无中毒表现,在80mg/L处理时也无小菜蛾表现出中毒的症状。若定义中毒率不高于3%的处理质量浓度为安全质量浓度,则化合物A1~A10对小菜蛾的安全质量浓度至少为80mg/L,比氟虫腈(<40mg/L)至少提高1倍。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种4‑氨基安替比林产品的合成工艺及其试液的配备方法与制造工艺
- 一种吡唑酮生产工艺的制造方法与工艺
- 一类含呋喃的苯基联吡唑甲酰胺类衍生物及其制备方法和用图
- N’?5?(取代苯基)?2?呋喃甲酰基?5?丙基?1h?吡唑?3?甲酸乙酯类化合物及其制备方法和应用
- 吡唑啉酮化合物及其用图
- 5-(4-氟苯基)-N–吗啉基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(4-氯苯基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含醚性氧原子的全氟烷基取代吡唑环化合物及其制造方法
- 5-(4-氟苯基)-N–(4-氯苯乙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 含有色酮结构的吡唑类n‐对硝基苯基马来酰亚胺衍生物及其制备方法与应用
- 5-(4-氟苯基)-N,N-二甲基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 制备1-烷基-3-二氟甲基-5-氟-1h-吡唑-4-甲醛和1-烷基-3-二氟甲基-5-氟-1h-吡唑-4 ...的制作方法
- 三氟甲基吡唑基胍F<sub>1</sub>F<sub>0</sub>-ATP酶抑制剂及其治疗用图
- 3,5-二甲基-1h-吡唑-1-甲脒盐酸盐的制备方法
- 取代吡唑基吡唑衍生物及其作为除草剂的用图
- 取代吡唑基吡唑衍生物及其作为除草剂的用图
- 封端异氰酸酯、涂料组合物、粘接剂组合物和物品的制作方法
- 一种1-(3-甲基-1-苯基-1h-吡唑-5-基)哌嗪的制备方法
- 一种3-二氟甲基-1-甲基-1h-吡唑-4-羧酸乙酯的合成方法
- 一种1,3,5-三甲基-4-吡唑甲酸甲酯的合成方法