一种应用于丝状真菌中特异性沉默基因的干扰质粒pHX‑RNAi的制作方法
本发明涉及一种用于真菌中基因改造的载体,尤其涉及一种在丝状真菌中对难敲除的特定基因进行单基因或多基因沉默的载体,即一种应用于丝状真菌中特异性沉默基因的干扰质粒pHX-RNAi,属于基因工程领域。
背景技术:
RNA干扰(RNA interference,RNAi)是指由双链RNA(double-stranded RNA,dsRNA)或MicroRNA((miRNA)诱发的转录后基因沉默机制,它通过机体切割dsRNA使mRNA降解进而影响目的基因表达(Fire A,Xu S,Montgomery MK,Kostas SA,Driver SE,et al.1998.Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans.Nature 391:806-811.)。RNAi技术可以特异性关闭特定基因的表达,因此该技术被广泛用于探索基因功能和医疗研究领域。
RNAi广泛存在于真核生物中,它能特异性使靶标基因沉默,在动植物中该技术已发展成为一种新型的基因治疗方法;在真菌中,基因沉默更多是通过同源重组进行基因敲除的方式来实现的。但是由于丝状真菌中存在多核和易发生非同源重组的现象,传统的基因敲除手段并不能很好地发挥作用,特别是研究一些致死基因时,必须使基因部分沉默来分析其功能,在这种情况下,与难度高、操作复杂、周期长的基因敲除等传统的基因改造手段相比,RNAi技术更具有特殊的意义。
RNAi的发生需要生成dsRNA,目前生成dsRNA主要有两种策略,一种是体外合成dsRNA然后直接导入体内,这种方法在动物中经常被使用,但在真菌中这一技术应用还不广泛,并且干扰效果不稳定;另一种策略就是通过在基因组上插入一段序列,使其转录后形成双链或发卡结构的RNA,从而起到干扰目的基因的作用(Liu H,Cottrell TR,Pierini LM,Goldman WE,Doering TL.2002.RNA interference in the pathogenic fungus Cryptococcus neoformans.Genetics 160(2):463-470.)。Nakayashiki等应用第二种策略构建了在子囊真菌的通用表达载体psilent-1(Nakayashiki H,Hanada S,Nguyen BQ,Kadotani N,Tosa Y,et al.2005.RNA silencing as a tool for exploring gene function in ascomycete fungi.Fungal Genet Biol 42(4):275-283.),在Magnaporthe oryzae和Colletotrichum lagenarium中对增强绿色荧光蛋白eGFP等基因进行干扰,发挥了较好的作用。psilent-1质粒上含有功能元件的多拷贝现象(两拷贝的trpC的启动子及终止子),实际应用时很可能会导致在青霉中的作用发挥不稳定。
技术实现要素:
针对丝状真菌中有些基因的沉默不能直接用敲除的手段实现的现状,本发明要解决的问题是提供一种应用于丝状真菌中特异性沉默基因的干扰质粒pHX-RNAi。
本发明所述的应用于丝状真菌中特异性沉默基因的干扰质粒pHX-RNAi包括一个完整的潮霉素抗性基因表达盒,一个草酸青霉来源的启动子,一个草酸青霉来源的内含子结构,该质粒的核苷酸序列如SEQ ID No.1所示。
本发明所述的干扰质粒pHX-RNAi应用于丝状真菌草酸青霉菌中对一些很难敲除的基因进行单基因和多基因的表达沉默。
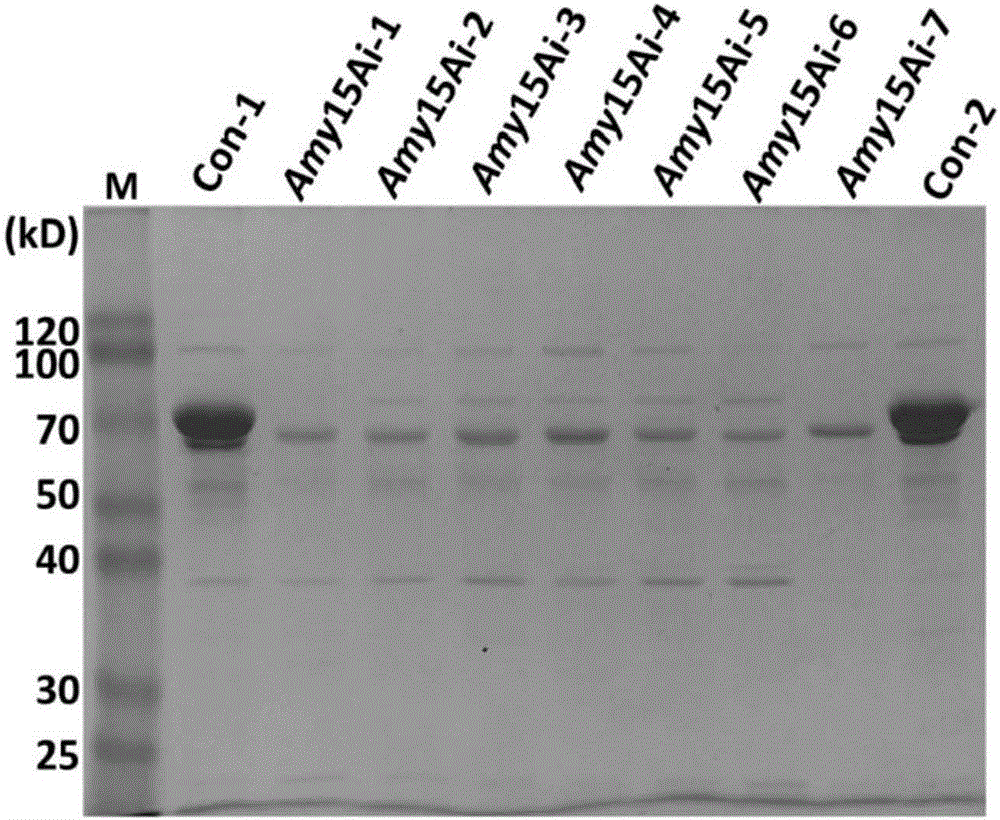
以淀粉酶基因amy15A(GenBank:EPS34453)为报告基因进行单基因打靶,蛋白电泳显示其表达产淀粉酶Amy15A的量明显减少;RT-qPCR对其mRNA水平进行检测,发现与原始菌株相比,干扰菌株amy15A对应的mRNA减少了超过90%。结果证明干扰菌株中的Amy15A的表达受到了严重的抑制,而且这种抑制是通过减少该基因mRNA的量实现的。
同时,以淀粉酶基因amy15A和外切纤维素酶基因cel7A-2(GenBank:ACV95805)为报告基因进行双基因打靶,蛋白电泳显示其表达产淀粉酶Amy15A和纤维二糖水解酶CBHI的量均有明显减少;酶活测定显示,与出发株相比,干扰菌株的胞外淀粉酶活力以及外切纤维素酶活力均有所降低;RT-qPCR对其mRNA水平进行检测结果也显示,与原始菌株相比,双基因干扰菌株相应的应的mRNA明显下调,表明pHX-RNAi可以通过一步操作沉默多个基因。
本发明所述的应用于丝状真菌的基因沉默的RNAi载体pHX-RNAi具体构建步骤为:
(1)将潮霉素抗性hph表达盒hph通过HindIII/SphI两个限制性酶切位点连入出发载体pUC19,得到质粒K-hph;
(2)终止子TtrpC通过SacI/EcoRI连接到载体K-hph,得到质粒K-hph-ter;
(3)将启动子Ppgm片段通过SphI/PstI连接到质粒K-hph-ter中,得到表达载体K-hph-PpgmC;
(4)将内含子片段和上述K-hph-PpgmC质粒通过限制性内切酶PstI和SacI进行连接,得到重组质粒命名为pHX-RNAi(附图1)。
本发明所述的pHX-RNAi用于沉默丝状真菌特定的基因,所述一类优选沉默草酸青霉(Penicillium oxalicum)114-2中淀粉酶基因amy15A,其具体操作为:
(1)amy15A干扰载体pHX-amyi的构建:用PCR得到amy15A-片段连接到干扰质粒pHX-RNAi得到重组质粒pHX-amyi(-);然后将amy15Ai+与质粒pHX-amyi(-)连接经筛选得到amy15A干扰载体pHX-amyi。
(2)草酸青霉单基因干扰菌株114-Amy15Ai的构建:将amy15A干扰载体pHX-amyi通过原生质体转化的方法转化入草酸青霉114-2中,pHX-amyi整合到草酸青霉114-2基因组上并转录出amy15A基因相关发卡结构dsRNA,从而降解淀粉酶amy15A转录出的mRNA,使amy15A沉默,获得草酸青霉单基因干扰菌株,并命名为114-Amy15Ai。
本发明所述的pHX-RNAi用于沉默单基因amy15A构建的草酸青霉单基因干扰株114-Amy15Ai的具体鉴定方法为:
(1)菌种选择:选择草酸青霉(Penicillium oxalicum)114-Amy15Ai;
(2)平板培养:将上述重组菌草酸青霉114-Amy15Ai接种于含有质量百分比为1.5~2.0%的琼脂的固体基本培养基平板上,30℃条件下,静置培养48h;
(3)种子培养:将步骤(4)培养的菌株,在无菌条件下用接种环接1~2环分生孢子于100毫升并加有2%(质量百分比)葡萄糖为碳源的液体基本培养基中,30℃条件下,150~250转/分钟,pH5.0~5.8,摇床振荡培养16~24小时,制得种子液;
(4)产酶培养:以质量体积比为10%的接种量,将步骤(5)的种子液接种于100毫升并加有终浓度2%淀粉或1%微晶纤维素或2%麦麸纤维素为碳源的液体产酶培养基中,pH5.0~5.8,30℃条件下,150~250转/分钟振荡培养24~96h,制得发酵培养菌液;
(5)收集发酵培养菌液:发酵结束后,将发酵液在10,000转/分钟的转速下离心10分钟,收集上清液,分别取上清液跑蛋白电泳胶(SDS-PAGE)分析蛋白条带,并检测其上清液中相应酶活力;
(6)分别收集培养12-48h的菌体,提取RNA,做RT-qPCR分析相应基因mRNA的量。
上述的应用中,步骤(3)、(4)中所述振荡培养,优选220转/分钟。
上述的应用中,步骤(4)所述的振荡培养,优选培养时间为48小时。
上述的应用中,步骤(4)所述的pH优选为pH5.6。
本发明所述的pHX-RNAi同样可用于丝状真菌多个特定基因的基因,所述应用优选是将草酸青霉淀粉酶基因amy15A和cel7A-2在草酸青霉(Penicillium oxalicum)114-2中同时沉默,其具体构建及验证方法是:
(1)双基因amy15A/cel7A-2干扰载体pHX-iac的构建:将融合PCR得到iac-片段连接到干扰质粒pHX-RNAi得到重组质粒pHX-iac(-);然后将iac+与质粒pHX-iac(-)连接经筛选得到双基因干扰载体pHX-iac。
(2)草酸青霉单基因干扰菌株114-Amy15Ai的构建
将pHX-iac通过原生质体转化的方法转化入草酸青霉114-2中,pHX-iac整合到草酸青霉114-2基因组上并转录出amy15A和cel7A-2基因相关发卡结构dsRNA,从而降解淀粉酶amy15A和cel7A-2转录出的mRNA,使amy15A和cel7A-2沉默,获得草酸青霉双基因干扰菌株,并命名为114-iac。
本发明所述的pHX-RNAi用于沉默多基因构建的草酸青霉双基因干扰株114-ia的具体鉴定方法参照单基因干扰菌株114-Amy15Ai的鉴定方法。
本发明提供的应用于丝状真菌中特异性沉默基因的干扰质粒pHX-RNAi应用RNAi原理,能在丝状真菌中特异性沉默目的基因,并且可以进行单基因或多基因操作。本发明以草酸青霉中淀粉酶基因amy15A为例,证实了干扰质粒pHX-RNAi的高效稳定性,并以amy15A和cel7A-2双基因干扰为例,验证了载体pHX-RNAi用于多基因沉默的优势。确定本发明的干扰载体pHX-RNAi在丝状真菌中对单基因和多基因的沉默均具有显著效果。SDS-PAGE,酶活测定以及RT-qPCR检测结果表明RNA干扰在转录后水平发挥了显著且稳定的作用,说明pHX-RNAi在丝状真菌中可以得到有效的应用。
考虑到丝状真菌中同源重组效率低等问题,传统的基因敲除不易实施,通过pHX-RNAi可实现特定基因高效稳定地沉默。且由于RNAi是一种非彻底性沉默基因的方法,可以通过控制启动元件来控制基因沉默的力度,因此本发明提供的载体pHX-RNA具有良好的应用前景。
附图说明
图1干扰载体pHX-RNAi图谱。
图2使用本发明沉默淀粉酶基因amy15A的干扰菌株114-Amy15Ai发酵液上清32μl进行蛋白电泳胶分析(SDS-PAGE)电泳图。
图3使用本发明沉默淀粉酶基因amy15A的干扰菌株114-Amy15Ai中amy15A基因在纤维素和淀粉诱导条件下的转录情况。
图4使用本发明沉默双基因amy15A和cel7A-2的干扰菌株114-iac i发酵液上清32μl进行蛋白电泳胶分析(SDS-PAGE)电泳图。
图5使用本发明构建的双基因干扰菌株114-iac在不同碳源诱导下得到的发酵液酶活。
图6使用本发明构建的双基因干扰菌株114-iac中amy15A和cel7A-2基因在不同碳源诱导条件下的转录情况。
具体实施方式
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
一般性说明:
微生物来源
实施例中的出发菌株草酸青霉(Penicillium oxalicum)CGMCC No.5302是申请人课题组先前专利申请时保藏的菌株,该菌株已于2011年9月28日保藏于中国微生物菌种保藏管理委员会普通微生物中心(CGMCC)。申请人课题组于2013年公布了草酸青霉基因组序列(Liu,G.,et al.2013.Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens.PLoS One,8(2),e55185.)。
培养基
所述基本培养基(Vogel’s)盐组分(1L):Na3Citrate·2H20,125g,KH2PO4,250g,NH4NO3,100g,MgSO4·7H20,10g,CaCl2·2H20,5g,微量元素溶液5ml,生物素溶液2.5ml,121℃灭菌30min。
所述微量元素溶液(100ml):Citric acid·H2O 5g,ZnSO4·7H2O 5g,Fe(NH4)2(SO4)2·6H2O 1g,CuSO4·5H2O 0.25g,MnSO4·H2O 0.05g,H3BO3 0.05g,Na2MoO4·2H2O 0.05g。
所述生物素溶液:5.0mg溶于50ml无菌水中。
所述转化培养基:基本培养基中加2%的葡萄糖为碳源,1M D-山梨醇为原生质体的渗透压稳定剂。基本培养基中根据转化外源片段遗传筛选标记基因的不同,分别加入终浓度200μg/ml的潮霉素B(EMRESCO,产品编号2012C098,抗性基因hph)、终浓度300ng/ml的硫胺吡啶(Sigma,产品编号P0256,抗性基因ptra)或终浓度1.6mg/ml的草胺磷(PESTANAL,产品编号278-636-5,抗性基因bar),115℃灭菌30min,pH 5.6。
所述分单孢培养基:基本培养基中加2%的葡萄糖为碳源,pH 5.6。,115℃灭菌30min,冷却后根据转化外源片段中遗传筛选标记基因加入终浓度200μg/ml的潮霉素B(hph),
所述发酵产酶培养基:基本培养基盐组分中,添加2%的淀粉为碳源,pH 5.6。
洗分生孢子生理盐水:0.9%的NaCl和0.05%的吐温-80水溶液,121℃灭菌30min。
pSilent-1质粒采用如下论文中记载的方法制备(Nakayashiki,H et al.2005.RNA silencing as a tool for exploring gene function in ascomycete fungi.Fungal Genetics and Biology,42(4),275-283.)。
大肠杆菌转化及转化子验证
(1)大肠杆菌DH5α感受态细胞购自北京全式金生物有限公司,转化步骤如下:
①取100μL在-80℃保存的大肠杆菌感受态细胞置于冰上,加入10μL连接液,用枪尖搅动混匀,冰上放置30min。
②42℃热击90s,冰浴2min后,加入700μL LB液体培养基,37℃温育1h。
③将培养液低速离心1min,弃600μL上清液,吹吸混匀菌体,分别将50μL和150μL菌体涂布于含有0.1%氨苄青霉素的LB固体平板上,37℃培养12h。
④将LB固体平板上长出的大肠杆菌单菌落用无菌的小枪尖挑到含氨苄的LB液体培养基中,37℃摇床培养6h。
(2)载体构建过程中的验证步骤如下:
①菌落PCR验证:以转化子菌液为模板,扩增插入载体的目的基因,通过琼脂糖凝胶电泳判断载体中是否含有目的基因。
②双酶切验证:提取菌落PCR验证正确的转化子质粒,提取方法参照质粒小提试剂盒说明书。将转化子质粒用合适的限制性内切酶进行双酶切,酶切体系(10μL)为:0.5μL限制性内切酶1,0.5μL限制性内切酶2,1μL 10×Buffer,5μL质粒,3μL ddH2O;酶切条件为37℃,3h。酶切产物通过琼脂糖凝胶电泳观察目的基因是否正确得连到载体上。
③测序验证:将菌落PCR和双酶切验证均正确的转化子送到测序公司(华大基因)进行测序,比对测序结果与基因组上的序列一致无突变,即为正确的目的基因表达载体。
草酸青霉的转化方案
(1)原生质体的制备
①取新鲜生孢的草酸青霉平板或斜面,用无菌的生理盐水(0.9%NaCl,0.05%Tween),将分生孢子洗下,制备草酸青霉分生孢子悬液。
②在Vogel’s基本培养基(2%葡萄糖为碳源)平板上盖一层玻璃纸,并用玻璃棒铺平,在玻璃纸上加入50-100μl孢子悬液,涂布均匀。做8-10个同样处理,30℃培养12-15h。
③加0.1g裂解酶(Lysing Enzymes from Trichoderma harzianum,Sigma产品编号L1412)到20ml溶液1中(溶液1:1.2M sorbitol,0.1M KH2PO4,pH 5.6),轻轻摇均,并用0.2μm滤器过滤除菌到培养皿或试管中。
④吸取2-3ml裂解酶溶液到无菌培养皿中,在溶液表面覆盖一层长有草酸青霉菌体的玻璃纸,然后再加2-3ml裂解酶液,按此顺序依次叠放6-7层,在两层玻璃纸之间不能留存大的气泡。放置培养皿于30℃培养箱中,为使酶液在培养皿中分布均匀,酶解期间可轻轻晃动培养皿1-2次。
⑤酶解约90min后,用镊子挑取玻璃纸,用移液枪吸取溶液冲掉玻璃纸上残留的菌丝体,大的菌丝团块可用移液枪(枪尖剪掉)缓慢吹打。
⑥准备带有玻璃棉的漏斗,并滴加数滴溶液1到玻璃棉使其紧贴漏斗内壁。用玻璃棉漏斗过滤原生质体悬液到50ml离心管中,并置于冰上,然后用数毫升溶液1冲洗玻璃棉,过滤液的总体积不超过35ml。利用水平转子2000rpm,4℃离心10min后,小心弃掉上清液,并用4ml冰冷的溶液2(溶液2:1M sorbitol 18.22g;50mmol/L CaCl2;10mM/L TrisHCl,pH 7.5)重悬原生质体,然后离心4℃,2000rpm,离心10min,小心弃掉上清液,并用0.5-1.0ml冰冷的溶液2重悬原生质体,置原生质体悬液于冰上。
⑦原生质体的质量控制(显微镜观察):a、在载玻片上滴加3μl原生质体悬液,在盖玻片旁滴加3μl无菌水,原生质体细胞内因渗透压高于细胞外水的渗透压吸水胀裂,而分生孢子则不受影响;b、通过血球计数板小室计算原生质体数量(几个小格即可)。原生质体终浓度应大于108,如果原生质体数量太高,可用溶液2稀释到5×108。
z=n×(400/f)×105,n=原生质体计数数量,f=计数的小格数量。
(2)草酸青霉原生质体的转化
①分装两管100μl草酸青霉原生质体悬液,加入10μl的质粒pHX-amyi,并加入50μl PEG溶液(25%PEG 6000,50mmol/L CaCl2,10mmol/L TrisHCl,pH 7.5),混匀转化体系,并在冰上放置20min。转化用DNA片段应纯化,DNA浓度应不少于1μg/μl。
②加1ml PEG(室温),轻轻混匀,室温放置5min,然后加入4ml溶液2,轻轻混匀。
③吸取0.2-1ml转化体系混合液到4ml预保温(55℃)的上层转化培养基中,轻轻混匀,并立即倒入铺有下层转化培养基的平板上,等培养基凝固后,置30℃培养,其中转化培养基的上、下层均加有终浓度300ng/ml的潮霉素作为筛选转化子的抗性试剂。
④在转化培养基上培养3-4天后,用接种针挑取转化子到分单孢培养基,分单孢培养基中加入终浓度400μg/ml的潮霉素B作为转化子的筛选试剂,30℃培养2-3天,长出分生孢子,用无菌的生理盐水将分生孢子洗下,制成孢子悬液,并将其在含有Triton-X100以及潮霉素B抗性的的分单孢培养基平板上划线培养,以消除转化子异核体的影响,获得单基因以及双基因干扰重组草酸青霉,分别命名为草酸青霉114-Amy15Ai。
草酸青霉染色体DNA大量提取:
(1)将草酸青霉1×106数量的分生孢子接种于100ml的Vogel’s盐基本培养基中,碳源为2%的葡萄糖,200rpm,30℃培养48h。
(2)将菌悬液用真空抽滤泵除去培养基,并用0.09M NaCl溶液洗涤一次,转移抽干的菌丝体至研钵中,加液氮冷冻并将菌丝体研磨至粉末状。
(3)把菌丝体粉末以质量体积比1∶5(菌丝重∶抽提液)加入到抽提液缓冲液(200mM Tris-HCl、pH 8.5,250mM NaCl,25mM EDTA,2%SDS)中,混匀,65℃保温30min。
(4)加入等体积的酚∶氯仿∶异戊醇(25∶24∶1)溶液至样品中,颠倒混匀,12,000rpm室温离心10min。
(5)缓慢吸取上层水相至另一离心管中,加入0.6倍体积异丙醇,颠倒混匀,-20℃放置20min,12,000rpm,4℃,10min,收集沉淀。
(6)以70%乙醇洗涤沉淀一次,12,000rpm,2min,然后,真空干燥,用适量ddH2O完全溶解沉淀。
(7)加入终浓度0.1μg/μl的RNAase,37℃温育1h。
(8)再加入等体积苯酚/氯仿抽提一次,12,000rpm,20min。
(9)将上清转移至另一离心管中,加0.1倍体积3M NaAc(pH 4.8)和0.6倍体积异丙醇,于-20℃放置20min,12,000rpm,4℃,10min,收集沉淀。
(10)加70%乙醇,洗涤一次,待乙醇挥发完全,用适量ddH2O溶解,制得草酸青霉基因组DNA。
草酸青霉RNA提取方案
将培养后的菌体洗涤抽滤后,用液氮研磨成粉状,然后用RNAiso试剂提取总RNA。
(1)将用无菌水洗涤二次并抽滤好的菌丝放入高温灭菌的研钵中,加入液氮后研磨成粉末。
(2)将研磨好的少量样品(50mg为宜)迅速转移至已加入1ml预冷的RNAiso试剂的1.5ml EP并立即混合,至匀浆不粘且无颗粒即可。然后室温静置5min,以使核蛋白完全分解。4℃,10000rpm,离心10min,上层清液移至另一离心管(约800μl),弃沉淀。
(3)加入上清体积0.2倍的氯仿,震荡15s充分混匀后于室温静置5min,4℃,10000rpm,离心15min。使混合物分为三层,RNA存在于最上层的水样中,吸取约300-400μl上层另一新离心管中。
(4)向上层水样中加入2倍体积的异丙醇,将管中液体轻轻混匀,室温放置10min后于4℃,10000rpm,离心10min,弃上清,RNA沉于管底。
(5)加1ml预冷的75%乙醇洗涤沉淀2次,温和震荡离心管,4℃不超过10000rpm,离心10min,尽量弃去上清。室温放置晾干5-10min。
(6)加入适量(50-100μl)DEPC处理的双蒸水溶解RNA,可在55-60℃放置10min助溶。
实施例1干扰载体pHX-RNAi的构建
(1)干扰载体pHX-RNAi中元件克隆
以草酸青霉114-2基因组为模板,扩增得到带有相应酶切位点序列的PpgmC以p-silent为模板扩增hph和TtrpC,引物序列如下。
PpgmC上游引物:Ppgm-F(SphI):ACATGCATGCCCGACTCATTACATACCTCCA
PpgmC下游引物:Ppgm-R(PstI):AACTGCAGGAGGTGGTGAAGGTTGGATTTG
TtrpC上游引物:TtrpC-F(SacI):CGGAGCTCACTTAACGTTACTGAAATCAT
TtrpC下游引物:TtrpC-R(EcoRI):CGGAATTCCTAGAGCGGCCGCAACCCAG
Hph上游引物:Hphs-F(HindⅢ):CCCAAGCTTCGACCTTAACTGATATTGAA
Hph下游引物:Hphs-R(SphI):ACATGCATGCCAACCCAGGGCTGGTGACGG
(2)质粒K-hph的构建
将潮霉素抗性hph表达盒hph通过HindⅢ/SphI两个限制性酶切位点连入出发载体pUC19,转化大肠杆菌DH5α,筛选得到正确连接质粒K-hph
(3)质粒K-hph-ter的构建
将终止子TtrpC通过SacI/EcoRI连接到载体K-hph,得到质粒K-hph-ter
(4)表达质粒K-hph-PpgmC的构建
将启动子Ppgm片段通过SphI/PstI连接到质粒K-hph-ter中,得到表达载体K-hph-PpgmC。
(5)干扰质粒pHX-RNAi构建
将合成的内含子片段和(4)中构建的K-hph-PpgmC质粒通过限制性内切酶PstI和SacI进行连接,得到重组质粒命名为pHX-RNAi。干扰质粒pHX-RNA图谱如图1所示。该质粒的核苷酸序列如SEQ ID No.1所示。
实施例2单基因干扰质粒pHX-amyi的构建
(1)片段的克隆
以114-2基因组为模板,扩增得到带有相应酶切位点的amy15A序列作为干扰片段,其中iamyF1(PstI)/iamyR1(BamHI)用来扩增amy15Ai+,iamyF2(XbaI)/iamyR2(SacI)用来扩增amy15Ai-,引物序列如下。
iamyF1(PstI):AACTGCAGTGAAAAATGACCCTCTAATGAA
iamyR1(BamHI):CGCGGATCCCCAAGCACCGGTGAAGGCG
iamyR2(SacI):CGGAGCTCCCAAGCACCGGTGAAGGCG
iamyF2(XbaI):GCTCTAGATGAAAAATGACCCTCTAATGAA
PCR扩增产物凝胶电泳后切胶并按照试剂盒说明书进行纯化回收。amy15A干扰片段的核苷酸序列如SEQ ID No.2和SEQ ID No.3所示.
(2)amy15A干扰质粒pHX-amyi的构建
用限制性内切酶XbaI和SacI双酶切amy15A-片段和干扰质粒pHX-RNAi并连接酶切后的载体和片段,转化大肠杆菌感受态细胞DH5α,筛选验证得到正确的重组质粒pHX-amyi(-),然后用PstI和BamHI酶切后连接pHX-amyi(-)和amy15Ai+,转化DH5α得到大肠杆菌转化子,扩大培养并提取质粒,通过酶切和质粒凝胶电泳验证重组质粒,正确的即为pHX-amyi。
实施例3草酸青霉单基因干扰菌株114-Amy15Ai的构建及验证
(1)草酸青霉单基因干扰菌株114-Amy15Ai的构建:将干扰载体pHX-amyi使用原生质体转化的方法转入草酸青霉114-2中,将得到的转化子进行传代分纯,在传代培养基中分纯5代。
(2)SDS-PAGE验证干扰菌株的干扰效果
将草酸青霉出发菌株114-2以及草酸青霉单基因干扰菌株114-Amy15Ai的孢子悬液接种于基本培养基(50ml),碳源为2%葡萄糖,200rpm,30℃培养24h,分别将得到的菌丝真空抽滤,各称取0.5g的湿菌丝体转接到装有100ml 2%淀粉为碳源培养基的三角瓶中,200rpm,30℃培养72h,分别取菌体培养液样品,然后将在淀粉诱导培养48h后的发酵液10000rpm离心5min,吸取样品上清液25ul,然后在制备好的12.5%蛋白分离胶上样。首先以90V电压跑30min,然后140V电压直至上样buffer跑出分离胶底部。结果见图2。
(3)RT-qPCR检测干扰菌株amy15A和cel7A-2的mRNA
将出发菌株(对照)和转化菌株在1%(v/w)的葡萄糖液体培养基中摇瓶培养24h后抽滤得到菌丝,分别转接等量(0.5g)的菌丝于淀粉培养基、纤维素培养基和麦麸纤维素培养基中,30℃200rpm摇床培养,分别在培养12h、24h、36h及48h抽滤后取样,用液氮将菌丝研磨成粉状,然后用RNAiso试剂提取总RNA。将提取的总RNA参照试剂盒PrimeScriptTMRT Reagent Kit with gDNA Eraser(Perfect Real Time)(TaKaRa,Japan)说明书的操作反转录成cDNA,以此作为模板,进行RT-qPCR。分别从amy15A基因的干扰片段内部(amy15Ai-F1/amy15Ai-R1)、amy15A基因中干扰片段外部(amy15Ai-F2/amy15Ai-R2)以及cel7A-2基因中(cel7A-qF/cel7A-qR)设计引物,同时用看家基因actin基因为内参设计引物(act-qF/act-qR),按照RT-qPCR标准曲线绘制方法绘制相关基因的标准曲线。
反应体系为10μl SYBR Premix Ex TaqTM,0.8μl引物1,0.8μl引物2,1.0μl模板,7.4μl ddH2O;RT-qPCR反应程序:95℃2min;95℃30s,61℃30s,读板,40cycles;61℃5min,65~95℃溶解曲线。
RT-qPCR所用引物序列如下所示。
amy15Ai-qF1:TACTCTCAAAGCCCTTGTGGAT
amy15Ai-qR1:TTGAACTTGGGCTCACCGAG
amy15Ai-qF2:GGTCGGTTCTATTTCTCAGCTCG
amy15Ai-qR2:ACTTGGCAGGGACGGTGTAGG
act-qF:CTCCATCCAGGCCGTTCTG
act-qR:CATGAGGTAGTCGGTCAAGTCAC
根据标准曲线来计算amy15A相对于actin的表达量,结果如图3。
实施例4双基因干扰质粒pHX-iac的构建
(1)片段的克隆
以114-2基因组为模板,扩增得到带有相应酶切位点序列,amy15A以及cel7A-2的片段。其中iamy(-iCBH)F1/iamy(-iCBH)(BamHI)R1用来扩增amy15A+,iamy(-iCBH)(Xbal)F2/iamy(-iCBH)R2用来扩增amy15A-,newicbhIF1(pstI)/newicbhIR1(BamHI)用来扩增cel7A+,newicbhIF2(xbaI)/newicbhIR2(SacI)用来扩增cel7A-;回收纯化出相应片段后,用融合PCR的方法分别将amy15A+/cel7A+融合,并将amy15A-/cel7A-片段融合,用newicbhIF1(pstI)/newicbhIR2(SacI)作为巢式引物,得到的片段分别命名为iac+和iac-,具体引物序列如下。
iamy(-iCBH)F1:CACTGGTAACACCTGGGACGTGAAAAATGACCCTCTAATGAA
iamy(-iCBH)(BamHI)R1:CGCGGATCCCCAAGCACCGGTGAAGGCG
iamy(-iCBH)(XbaI)F2:5’-GCTCTAGACCAAGCACCGGTGAAGGCG-3’
iamy(-iCBH)R2:CTTGCCGTTCTTGTCGTGAATGAAAAATGACCCTCTAATGAA
newicbhIF2(xbaI):GC TCTAGA TTCACGACAAGAACGGCAAG
newicbhIR2(SacI):tCC GAGCTC GCGTCCATGTCGACGAAGTA
newicbhIF1(pstI):AACTGCAG GCAAGGCAGGTTGGAAACAT
newicbhIR1(BamHI):CGC GGATCC CACTGGTAACACCTGGGACG
PCR扩增产物凝胶电泳后切胶并按照试剂盒说明书进行纯化回收,iac+核苷酸序列如SEQ ID No.4所示,iac-核苷酸序列如EQ ID No.5所示.
(2)pHX-iac的构建
用限制性内切酶XbaI和SacI双酶切位点连接iac-片段和干扰质粒pHX-RNAi,转化大肠杆菌感受态细胞DH5α,筛选得到正确的pHX-iac(-);然后用PstI和BamHI酶切后连接pHX-iac(-)和iac+,转化DH5α得到大肠杆菌转化子,扩大培养并提取质粒,通过酶切和质粒凝胶电泳验证重组质粒,正确的即为双基因干扰载体pHX-iac。
实施例5草酸青霉双基因干扰菌株114-iac的构建及验证
(1)使用原生质体转化的方法,将pHX-iac转化入草酸青霉114-2中,得到转化子后进行传代分纯,在传代培养基中分纯5代.
(2)SDS-PAGE验证干扰菌株的干扰效果:具体实施方法同实施例3(2),结果见图4。
(3)淀粉酶活和纤维素外切酶活测定:将新鲜孢子接入葡萄糖培养基中,30℃,200rpm摇床培养24h,抽滤得到菌丝,分别转接等量(0.5g)菌丝到纤维素培养基、淀粉培养基和麦麸纤维素培养基中,摇床培养48h后,将发酵液离心取上清,按照文献中方法测定三种碳源诱导条件下淀粉酶活和纤维素外切酶活(刘国栋,2012木质纤维素降解真菌斜卧青霉的比较基因组学与功能基因组学研究.In.:山东大学,pp.)结果见图5。
(4)RT-qPCR检测干扰菌株amy15A和cel7A-2的mRNA:具体实施方法同实施例3(3),结果见图6。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于治疗HBV感染和相关病症的三特异性结合分子的制造方法与工艺
- 对磺胺类药物具有特异性的固相微萃取纤维的制备方法与流程
- 一种检测猪初乳中抗PEDV特异性IgA抗体的夹心ELISA试剂盒的制造方法与工艺
- 用于施用CD19xCD3双特异性抗体的给药方案的制造方法与工艺
- 双特异性抗‑VEGF/抗‑ANG‑2抗体及其在治疗眼血管疾病中的应用的制造方法与工艺
- 特异性人EWS‑FLI1融合蛋白抗体的制备方法及在制备尤文肉瘤诊断抗体试剂的应用与制造工艺
- 双特异性四价抗体及其制造和使用方法与制造工艺
- 利用特异性挥发物筛选昆虫相关嗅觉蛋白的方法与制造工艺
- 使用与表面偶联的单特异性四聚抗体复合物组合物从样品中分离出靶实体的方法与制造工艺
- 抗-WT1/HLA双特异性抗体的制造方法与工艺