一种咪唑酮衍生物在制备抗登革热病毒的药物中的应用的制作方法
本发明涉及药学领域,更具体地,涉及一种咪唑酮衍生物在制备抗登革热病毒的药物中的应用。
背景技术:
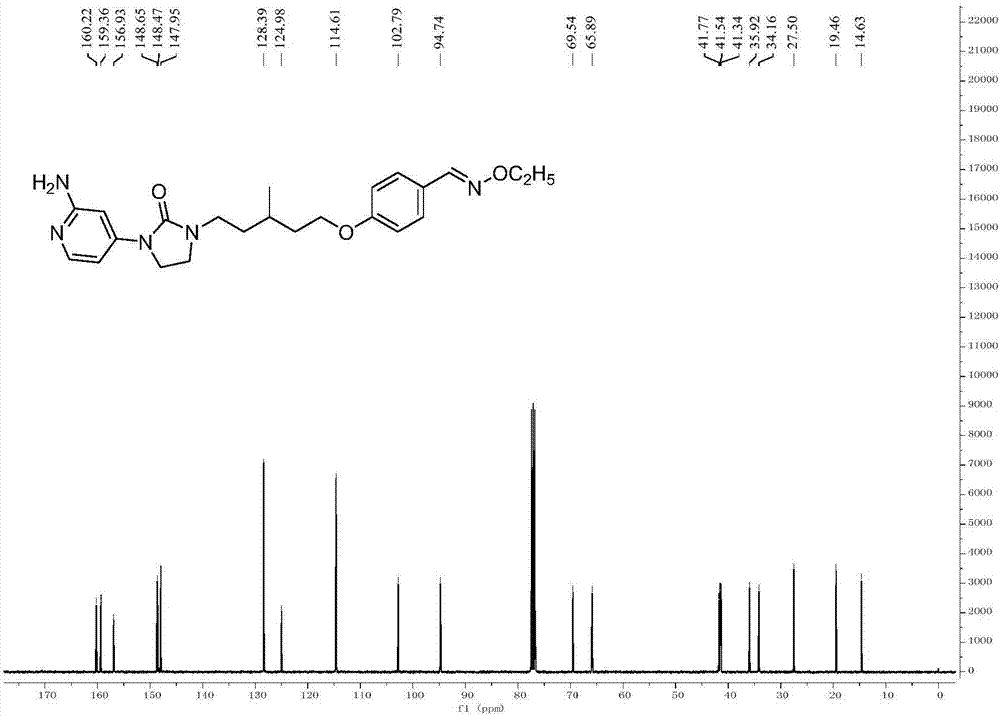
:登革热(denguefever)是由登革热病毒(denguevirus,denv)引起的一种急性传染病,最初发生于热带地区。denv是一种通过蚊子传播的黄病毒(flavivirus),据统计,其感染每年会导致5000万到1亿的登革热病例,全世界约有25亿人会受到这种病毒感染的威胁。感染denv后一般无明显症状,即隐性感染,仅部分患者出现程度不一的临床表现,轻者可表现为发热、呕吐头痛、关节痛、肌肉痛、皮疹及白细胞减少等,大多属于自限性,因denv可引起骨痛等特殊症状,登革热(denguefever,df)又被称为骨痛热;重度患者可引发df、登革出血热、登革休克综合征、器官衰竭、血浆渗出甚至死亡。二次感染异种denv后,即使再感染第三种甚至第四种血清型denv,出现有症状登革疾病的情况并不常见。然而,到目前为止,临床上还没有有效的药物来预防或治疗登革热病毒的感染。加之为了预防近些年来出现的生物恐怖分子会利用此病毒传播疾病,如何预防和治疗其感染再次引起了科学家们的关注。denv有四种病毒型:df-1、df-2、df-3和df-4,而且它们均有可能发生基因变异,这给相应的疫苗和药物的研发带来了很大的困难。截至2016年8月,仅有法国赛诺菲·巴斯德公司(sanofipasteur)研发的df疫苗dengvaxia(cyd-tdv)在墨西哥、菲律宾和巴西等多国陆续批准上市。截止到目前,仍没有全面应用于临床的抗登革病毒药。一般的抗病毒药物,如拉米呋定、达菲、小柴胡、板蓝根等仅可起到预防和部分治疗的作用。而且在针对登革热病毒疫苗研究过程中,绝大多数候选疫苗未能对四种血清型denv提供均衡的保护作用,导致部分研究仅进行到ⅰ或ⅱ期临床试验阶段即止步。因此,研发抗登革热病毒药物就变得更为迫切和重要。技术实现要素:针对现有技术中存在的问题,本发明提供了一种咪唑酮衍生物在制备抗登革热病毒的药物中的应用,所述咪唑酮衍生物的结构式为:其中,r1选自氨基、c1-3烷基氨基、c1-3酰基氨基、羟基、c1-3烷氧基、氢原子或氧原子;r2选自c1-3烷基、芳环、芳基杂环或氢原子;r3选自c1-3烷基、烷氧基或氢原子;r4选自o-烷基甲肟、o-烷基羟胺、噁二唑环、三氟甲基取代的噁二唑环、c1-5烷基取代的噁二唑环、c1-5烷基取代的咪唑环、c1-5烷基取代的三氮唑环、c1-5烷基取代的噻二唑环、n-羟基甲酰胺、n-烷氧基甲酰胺、氢原子、氰基或n-羟基脒基;x选自碳原子或氮原子;以及y选自c1-4烷基、c5-6环烷基或c1-3炔基。在上述应用中,x为氮原子,并且所述咪唑酮衍生物选自具有如下结构式的化合物:在上述应用中,所述咪唑酮衍生物选自具有如下结构式的化合物:在上述应用中,所述抗登革热病毒的药物包括:所述咪唑酮衍生物、所述咪唑酮衍生物在药学上可接受的盐、酯、水合物或它们的一种或多种的组合以及辅料。在上述应用中,所述咪唑酮衍生物在药学上可接受的盐为有机盐。在上述应用中,所述咪唑酮衍生物在药学上可接受的盐为甲磺酸盐、甲酸盐、乙酸盐、三氟乙酸盐、马来酸盐、酒石酸盐、琥珀酸盐、富马酸盐、柠檬酸盐、苯磺酸盐、对甲基苯磺酸盐、萘磺酸盐、乳酸盐或苯甲酸盐中的一种或者多种的组合。在上述应用中,述咪唑酮衍生物在药学上可接受的盐为无机盐。在上述应用中,所述咪唑酮衍生物在药学上可接受的盐为盐酸盐、氢溴酸盐、硫酸盐或磷酸盐中的一种或者多种的组合。在上述应用中,所述抗登革热病毒的药物的剂型选自片剂、胶囊剂、丸剂、栓剂、气雾剂、口服液体制剂、颗粒剂、散剂、注射剂、糖浆剂、酒剂、酊剂、露剂、膜剂或它们的组合。在上述应用中,所述抗登革热病毒的药物的给药方式包括:口服、注射、植入、外用、喷雾、吸入或它们的组合。本发明提供了一种可作为登革热病毒抑制剂的咪唑酮衍生物及该类衍生物药用盐,该类抑制剂在细胞水平上,对登革热病毒具有较高的选择性抑制作用,在制备治疗登革热病毒感药物上具有很好的前景。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍。图1示出了化合物7的氢核磁谱图;图2示出了化合物7的13c核磁谱图;图3示出了化合物7的质谱谱图;图4示出了化合物7的ec50值;以及图5示出了化合物7的cc50值。具体实施方式下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员所获得的所有其他实施例,都属于本发明保护的范围。以上仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。本发明挑选化合物7的合成过程作为具体实施例,来对本发明咪唑酮衍生物的具体合成路线进行说明,但实施例的选择并不意味着本发明的制备方法的任何限制。化合物7的具体合成过程如下:其制备方法包括以下步骤:(1)向带有冷凝管的三口瓶中加入4ml的1-氯-2-异氰酸乙酯和2g2-溴-4-氨基吡啶,体系于50℃下搅拌反应5h,tlc监测反应结束。将溶剂旋干,得到棕色油状物,硅胶柱纯化得1-(2-溴吡啶-4-基)-3-(2-氯乙基)脲,即中间体a(浅黄色固体,1.6g,收率50%)。(2)称取1g中间体a置于20ml干燥的dmf中,体系于冰浴下冷却,之后于搅拌状态下加入0.3g60%nah,加入完毕后于室温下搅拌反应2h。反应毕后,加少量水淬灭反应,将溶剂旋干,加入大量水搅拌,体系析出固体,将固体抽滤收集,水洗滤饼。滤饼干燥即为产物1-(2-溴吡啶-4-基)咪唑啉-2-酮,即中间体b(浅粉色粉末,0.7g,收率80%)。(3)称取2g中间体b及2.69gnan3加入到15ml的dmso中,然后在搅拌状态下加入1.2ml乙酸,并将反应液加热到120℃搅拌反应3h。反应结束后,将反应液降温至室温,并倾倒入冰水混合物中,再搅拌1h,有固体析出,抽滤,滤饼用水淋洗,收集滤饼并在红外干燥箱中干燥,得到1-(2-叠氮吡啶-4-基)咪唑啉-2-酮,即中间体c(棕色固体,1.45g,收率86%)。(4)称取8.1g中间体c置于100ml的无水dmf中,体系于冰浴下冷却,之后分批加入3.6g60%nah,加入完毕后,体系于冰浴下搅拌10min,滴加14.5g1,5-二溴-3-甲基戊烷,滴加完毕后,室温搅拌反应6h,tlc检测反应毕,将反应液倾倒入1000g的冰水混合物中,并搅拌1h,1h后过滤,滤饼依次用水和乙酸乙酯淋洗,收集固体在红外干燥箱中干燥,得1-(2-叠氮吡啶-4-基)-3-(5-溴-3-甲基戊烷)咪唑啉-2-酮,即中间体d(棕黄色粉末,8g,收率55%)。(5)称取8g中间体d溶于100ml乙醇中,搅拌下分别加入17.3gsncl2和3.2ml盐酸,反应加热至70℃搅拌反应6h,tlc检测反应毕,冷却至室温,旋除乙醇,加入200ml水,在搅拌状态下用10%的naoh水溶液调节ph=10并继续搅拌30min,过滤,水相用乙酸乙酯(100ml*3)萃取,滤饼用ea打浆多次,抽滤,直到滤液中无产物存在,合并收集乙酸乙酯相,用无水硫酸钠干燥。得1-(2-氨基吡啶-4-基)-3-(5-溴-3-甲基戊烷)咪唑啉-2-酮,即中间体e(白色粉末,5.3g,收率71.33%)。(6)称取5.3g中间体e以及4.27g中间体f和10.73g无水碳酸钾加入到50ml乙腈中,加热回流反应6h。tlc检测反应毕,将反应液趁热过滤,滤饼用dcm淋洗,合并有机相,旋干,硅胶柱分离纯化,得到1-(2-氨基吡啶-4-基)-3-(5-(4-((乙氧基亚胺)甲基)苯氧基)-3-甲基戊基)-咪唑啉-2-酮,即化合物7(白色固体,6.1g,收率92.3%)。化合物7的1h谱数据如下:1h-nmr(400mhz,cdcl3):δ8.06(s,1h),7.98(d,j=5.2hz,1h),7.54(d,j=8.2hz,2h),6.99–6.85(m,3h),6.78(d,j=4.6hz,1h),4.45(s,2h),4.24(q,j=6.8hz,2h),4.07(d,j=4.1hz,2h),3.75(t,j=7.4hz,2h),3.57–3.46(m,2h),3.45–3.29(m,2h),1.97–1.77(m,2h),1.71(dd,j=12.8,6.3hz,2h),1.48(dd,j=13.1,5.9hz,1h),1.36(t,j=6.9hz,3h),1.07(d,j=6.2hz,3h)。化合物7的相应的氢核磁谱图参见图1。化合物7的13c谱数据如下:13c-nmr(101mhz,cdcl3)δ160.22,159.36,156.93,148.65,148.47,147.95,128.39,124.98,114.61,102.79,94.74,69.54,65.89,41.77,41.54,41.34,35.92,34.16,27.50,19.46,14.63。化合物7的相应的13c核磁谱图参见图2。化合物7的质谱数据:对于c23h31n5o3,[m+h]+426.24,理论值426.40。化合物7的相应的质谱谱图参见图3。吡啶咪唑酮衍生物细胞水平的抗登革热病毒活性以及细胞毒性检测过程如下:a)bhk-21细胞的培养将在液氮中冻存的bhk-21细胞(购自atcc)37℃水浴锅中解冻后,加入到盛有12ml培养基的培养皿中,培养基成分为dmem(dulbecco'smodifiedeaglemedium)+10%胎牛血清(fetalbovineserum,fbs)+1%青霉素和链霉素(penicillin-streptomycin,ps),37℃,5%co2培养。b)dengue-gluc病毒的扩增待bhk-21细胞生长满度至90%时,加入dengue-gluc病毒(清华大学娄智勇老师惠赠)1ml,37℃,5%co2培养。72h后细胞开始发生病变,待细胞完全病变后收集上清,4000rpm离心10min,将分装分装后储存于-80℃。c)病毒滴度测定a549细胞(购自atcc)按照3×105个/ml接种于96孔板中,37℃,5%co2培养24h后,第一列3孔细胞中加入10倍稀释的病毒100μl,第二列3孔中加入20倍稀释的病毒100μl,第3列3孔中加入40倍稀释的病毒100μl,第4列3孔中加入50倍稀释的病毒100μl,培养48h后加入荧光底物进行数据测定,gluciferase读值在10,000-20,000之间的病毒滴度最为合适。d)a549细胞的培养将100μla549细胞按3×105个/ml接种于96孔板中,为防止培养基蒸发引起的边缘效应,96孔板外围一圈加入200μl磷酸盐缓冲液(phosphatebufferedsaline,pbs),37℃,5%co2培养24h。e)化合物的配制将化合物溶解在95%二甲基亚砜(dimethylsulfoxide,dmso)中,配制成10mm储液。用培养基dmem+10%fbs+1%ps将上述氨基咪唑酮化合物配制成100μm、20μm和4μm进行初步筛选,筛选过程如下f):。f)一块96孔板可以检测6个化合物,将100μl细胞中的培养基吸走,在96孔板的第2列bcd行3孔细胞中加入50μl100μm的第一种化合物,第3列bcd行3个孔中加入50μl20μm的化合物,第4列bcd行3个孔中加入50μl4μm的化合物,第5列bcd行3个孔中加入50μl100μm的第二种化合物,以此类推加到96孔板的第9列的3个孔,efg3行9列加入另3种化合物,第11列6个孔设为病毒对照和细胞对照。g)抑制剂与细胞先混合2h,然后用培养基将dengue-gluc病毒稀释20倍,然后每孔加入50μl病毒,总体积变为100μl,因此,抑制剂的浓度变为配制浓度的1/2,37℃,5%co2培养48h。h)细胞培养48h后,将细胞上清吸走,用pbs清洗两次细胞,加入1x细胞裂解液20μl,室温放置15min并慢慢晃动,直至细胞完全裂解,将细胞裂解物移至白色酶标板中,然后加入100μl海肾荧光素酶底物,于微孔板检测仪中读取数值,数值显示的是海肾荧光素酶与底物发生化学反应所产生的化学光,数值越小代表抑制剂的抑制效果越好。i)选取在10μm条件下抑制率50%以上的抑制剂进行确切的ec50测定。其方法如下:1)将100μla549细胞按3×105个/ml接种于96孔板中,外围用200μlpbs封闭,防止边缘效应,37℃,5%co2培养箱中培养24h。2)将筛选出中的小分子化合物10mm储液用培养基2倍梯度稀释成200μm-0.78μm9个浓度。3)将96孔板中的细胞上清吸走后,加入50μl上述浓度的抑制剂,同时设置病毒和细胞对照。4)加入抑制剂2h后,将dengue-gluc病毒稀释10倍,用排枪将50μl病毒加入96孔板中,37℃,5%co2培养箱中培养48h。5)将细胞上清吸走,用pbs清洗两次细胞,加入1x细胞裂解液20μl,室温放置15min并慢慢晃动,直至细胞完全裂解,将细胞裂解物移至白色酶标板中,然后加入100μl海肾荧光素酶底物,于微孔板检测仪中读取数值,并用graphpadprism中作图。j)氨基咪唑酮化合物细胞毒性(cc50)测定:1)100μlla549细胞按3×105个/ml接种于96孔板中,37℃,5%co2培养24h。2)将氨基咪唑酮化合物用培养基稀释成200μm、100μm、50μm、25μm、12.5μm、6.25μm、3.125μm、1.56μm和0.78μm9个浓度。3)将96孔板中细胞上清吸走,每孔加入100μl上述浓度的抑制剂,每个浓度重复3孔,第10个浓度为细胞对照。4)抑制剂作用24h后,用mtt法检测抑制剂的细胞毒性,每孔加入15μlmtt进行活细胞染色。5)4h后将上清吸去,并加入150μldmso充分溶解结晶物后在酶标仪490nm下读取荧光值。表1化合物编号ec50(μm)cc50(μm)74.25129.0198.215>50116.534>50121.861>50132.217>50184.71>50213.964>50在以上表1中示出了部分优选化合物的抑制活性数据。并且在图4和图5中分别示意出了化合物7的ec50值以及cc50值。从上述实验结果可以看出,通过抑制剂活性初步筛选出的吡啶咪唑酮衍生物及其药用盐进行细胞水平的抑制活性以及细胞毒性测试,发现对登革热病毒具有选择性抑制作用,其半数有效浓度ec50均小于10μm,具有较强的抗登革热病毒作用且细胞毒性较低,大部分化合物的cc50值均大于50μm。因此本发明专利中涉及的吡啶咪唑酮衍生物及其药用盐具有一定成药性,可以有效抑制登革热病毒的活性,在制备治疗登革热病毒感药物上具有很好的前景。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 苯并吲唑类衍生物及其制备方法、应用与制造工艺
- 一种二苯并环庚烷衍生物的制备方法与制造工艺
- Atropurpuran的苯并咪唑基和吗啉基衍生物组合物用于防治胰腺纤维化的制造方法与工艺
- 一种苯并咪唑衍生物镉配合物及其制备方法和应用
- 一种咪唑并吡啶巯乙酸类衍生物及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法
- 一种制备1,2?二甲基咪唑的方法
- 一种芬苯达唑纳米混悬剂及其制备方法
- 吡啶并咪唑类衍生物、其制备方法和应用
- 一种咪唑类衍生物化合物及其制备方法和发光器件的制作方法
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗红细胞低下性贫血药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗骨质疏松药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗急性痛风药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在预防或治疗胰腺纤维化药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗缺氧药物中的应用
- 适用于改进水分管理且提供抗微生物保护的包括衍生自直链四胺的季双咪唑啉化合物的 ...的制作方法