一种基于CRISPR的DNA检测和分型方法及其应用与流程
本发明属于生物技术领域,具体一种基于crispr的dna检测和分型方法及其应用。
背景技术:
对于基础研究、各种检测及诊断应用,dna检测和基因分型一直很重要。因此,dna检测和基因分型技术一直受到广泛关注,从而促进了该类技术发展。简而言之,主要有三类dna检测和基因分型技术被广泛应用。第一种是基于聚合酶链反应(pcr)的各种技术。pcr是最常用的dna检测和基因分型技术。基于pcr的dna检测和基因分型主要依赖于特异性引物的设计和多重pcr扩增。pcr检测可以通过传统pcr(tpcr),定量pcr(qpcr)和最近开发的数字pcr来实现。因为具有明显的优点,如实时检测和高灵敏度,q-pcr在几乎所有的研究、检测和诊断实验室中得到高度普及。现在已经开发出更准确的数字pcr,作为临床检测工具,具有很大的潜力和优势。然而,pcr技术在用于区分高度相关的基因型时,要受到多重扩增和高度特异性引物的限制。除pcr技术外,dna微阵列等多种dna杂交技术也被广泛用于检测和分型dna。然而,由于其昂贵的设备,复杂的检测流程和不可避免的非特异性杂交,dna微阵列技术不能像pcr一样成为常规dna检测和基因分型工具。dna测序是另一种有效的dna检测和基因分型技术。特别是随着下一代测序(ngs)技术的出现,诸如illuminanovaseq等ngs平台的dna测序工具越来越多。然而,由于需要昂贵的设备和化学试剂,它们仍然不能像pcr一样用于常规研究,检测和诊断。因此,相比之下,如果克服了引物设计的限制,pcr仍然是最方便、经济高效的dna检测和基因分型的平台。
ishino等人于1987年首次在大肠杆菌(e.coli)的基因组中发现了成簇的规律间隔的短回文重复序列,并由jansen等人在2002年定义为crispr(clusteredregularlyinterspacedshortpalindromicrepeat)。crispr系统有三种不同类型(i,ii和iii型)。i型和iii型系统需要多种cas蛋白互相作用才能发挥正常功能,因此要比ii型复杂很多。在ii型系统中,只需要一种蛋白(cas9),cas9与引导rna(grna)结合后能够特异地识别和切割双链dna(dsdna)。cas9是ii型系统的标志蛋白,在反式激活crrna(tracrrna)和crisprrna(crrna)的引导下发挥作用。tracrrna能够激活cas9核酸酶,crrna特异地与靶dna的20个核苷酸序列互补。因此crrna决定了crispr-cas9系统的特异性。将tracrrna和crrna整合成一个rna即单导向rna(sgrna)后,极大地简化了ii型crispr系统的应用。cas9介导的位点特异性切割依赖于sgrna和pam(protospaceradjacentmotif)。如果目标dna中有pam,在sgrna的引导下,cas9在pam上游三个碱基处切割靶dna。目前,由于简便和高效,crispr-cas9系统已被许多研究者广泛应用于基因组编辑领域。另外,dcas9(deadcas9)是由cas9改造而成,其失去了核酸酶活性,但保留基因转录激活结构域(ad)或抑制结构域(id),dcas9(deadcas9)作为一种新的人工转录因子已被广泛应用于内源性基因表达调控。
尽管cas9/sgrna已广泛应用于基因编辑和调控,但很少应用于核酸检测。凭借高特异性的dna切割能力(能够区分单碱基),cas9/sgrna在dna检测和分型上有具有很大的潜力。最近,crispr-cas9系统已被用于检测zika病毒并且能够对美国和非洲zika病毒进行分型。基于crispr的高特异性,crispr-cas9在区分病毒株时可以达到单碱基的分辨率,可以在单碱基水平上对直系同源的细菌和病毒进行分型检测。最近crispr系统(iii型的cas13a/c2c2)已经应用于zika病毒的检测并且具有超高灵敏度(病毒颗粒的量低至2am)。这些研究表明,crispr系统用于开发核酸检测技术时具有很大的潜力和优势。然而,在报道的基于cas9的dna检测中,cas9/sgrna系统尚未直接用于检测和分型基因组dna。
hpv是双链dna病毒,与子宫颈癌,肛门癌和其他癌症的发病机制密切相关。大约有100种不同变异类型的hpv。根据致癌能力的不同,hpv分为高危型hpv(hrhpv)和低危型hpv(lrhpv)。世界上最常见的hrhpv是hpv16和hpv18,它们导致了大约70%的宫颈癌。其他hrhpv包括hpv31、33、35、39、45、51、52、56、58、59、68、82等。lrhpv包括hpv6、11、40、42、43、44、61、81等。因其丰富的dna多态性,hpv是研究dna检测及分型技术良好的实验材料。因此,本发明以hpvdna为材料,进行本发明方法的论证。
聚合酶链反应(pcr)作为最常用的核酸检测方法之一,被几乎所有的生物学、检测和诊断相关的实验室作为基本的核酸检测工具。在传统pcr(tpcr)的基础上,衍生了定量pcr(qpcr),并被广泛应用于dna检测和诊断。另外,最近开发出的数字pcr(dpcr)已经显示出其作为临床检测工具的巨大潜力和优势。因此,crispr和pcr技术的结合为开发出新的核酸检测和分型技术提供了新的机会。这些技术同时具备了crispr技术的高特异性和pcr技术的高灵敏度,在dna检测和基因分型上更有优势。
技术实现要素:
发明目的:针对现有技术存在的问题,本发明提供一种基于crispr的dna检测和分型方法,该方法被命名为carp,即crispr辅助反向pcr(crispr-assistantreversepcr)或者cas9/sgrna相关的反向pcr。该方法为一种快速,便宜和灵敏的基于crispr的pcr方法,可以有效地对目标dna进行特异性检测和分型。
技术方案:为了实现上述目的,如本发明所述一种基于crispr的dna检测和分型方法,其特征在于,包括如下步骤:
(1)用一对通用引物pcr扩增靶dna;
(2)用cas9/sgrna切割扩增后的靶dna;
(3)用dna连接酶连接被切割的靶dna;
(4)用pcr扩增连接后的靶dna。
其中,步骤(1)所述通用引物为可将含有靶dna序列的dna片段从待检测dna样品中pcr扩增出来的一对引物;该对引物是单一序列或简并序列。
步骤(2)所述cas9/sgrna切割扩增后的靶dna为利用cas9核酸酶与靶向目的dna的一对sgrna混合,形成两个cas9/sgrna复合物;该复合物在sgrna的引导靶向目的dna,使cas9/sgrna复合物与目的dna结合并在cas9的作用下发生目的dna的双链切割。
步骤(3)所述dna连接酶连接被切割的靶dna为利用dna连接酶处理步骤(2)产生的dna,产生分子内或分子间的平末端dna连接。作为优选,其中所使用的的dna连接酶为t4dna连接酶及其他具有类似功能的酶。
步骤(4)所述用pcr扩增连接后的靶dna为使用一对在靶dna上呈反向的pcr引物;该对反向的pcr引物在靶dna未发生cas9/sgrna切割时和之后步骤(3)的连接时,不能扩增靶dna,而在靶dna发生cas9/sgrna切割并经步骤(3)连接后,则可扩增靶dna。
作为优选,步骤(1)和步骤(4)所述pcr包括普通pcr、定量pcr或数字pcr。
所述检测和分型方法,在检测高拷贝靶dna或仅用于含量丰富的靶dna分型检测时,通过cas9/sgrna直接切割高拷贝靶dna或含量丰富的靶dna进行检测,不需要用一对通用引物pcr扩增靶dna。
其中,步骤(2)所述cas9可以替换成与cas9类似其他crispr相关核酸酶,如cpf1;所述sgrna可以替换成与其他crispr相关核酸酶匹配的引导rna,如cpf1匹配的引导rna。当使用cpf1等非平末端双链dna断裂的crispr核酸酶时,可通过末端加工如延伸补齐等,使切割产物成为平末端,以便连接;或使用与crispr核酸酶产生的序列已知的粘性末端匹配的接头dna将切割产物连接起来。
本发明基于crispr的dna检测和分型方法在双链dna相关生物检测中的应用;具体的基于crispr的dna检测和分型方法应用在人乳头状瘤病毒双链dna生物检测中;如人类乳头状瘤病毒(hpv)dna的检测及分型。
其中,所述hpv病毒包括hpv16和hpv18两种高危型hpv病毒。尤其是,人类乳头状瘤病毒(hpv)的l1和e6-e7基因的各种基因型等。本发明只是用hpv作为一种实验材料来验证carp方法的可行性。该方法也可用于检测其他dna。本发明采用hpvdna作为carp检测的dna靶标。结果表明carp可以检测和分型hpvdna。发现carp可以在少至0.002ng宫颈癌细胞系gdna中检测到hpv16和hpv18dna。
在本发明的carp检测中,cas9分别和两种sgrna结合后切割靶dna。然后dna连接酶连接切割产物,最后连接产物用于pcr扩增。所用的sgrna对靶dna具有特异性。另外,针对目标dna设计一对反向引物,这一对引物方向相反不能直接用于扩增靶dna。当靶dna被cas9/sgrna切割,然后在分子间连接成线性或分子内连接成环状dna后,反向引物就会变为正常引物,就可以通过pcr扩增靶dna。
由于人类乳头状瘤病毒(hpv)的l1基因已广泛用于检测和鉴别hpv亚型。在本发明中,首先设计了两对sgrna和两对反向pcr引物,用于检测hpv16和hpv18的l1基因。用这些sgrna和引物通过基于tpcr和qpcr的carp方法检测hpv16和hpv18的l1基因。结果表明,carp可以在9种hpv亚型中特异性地检测hpv16和hpv18的l1基因,显示了crispr系统用于dna分型时所具有的巨大潜力和可行性。然而,据报道,hpv基因在整合到宿主细胞基因组的过程中,l1dna会有缺失,这就会导致hpv检测遗漏。例如,在使用hpvl1区域通用引物(my09/my11)检测56例浸润性宫颈癌活检样本,发现与hpve6-e7型引物检测相比,l1区域丢失了多达23个样本。使用my09/my11引物在15,774例患者样本中进行的另一个hpv检测中,有10.9%的样本(522个)丢失。在随访中,发现使用my09/my11pcr鉴定为阴性的409例患者中有104例(25.4%)发展为cin2+。据报道,hpv18的l1/e1区域更容易丢失。几乎所有hpv18阳性宫颈癌基因组中只整合了hpv18的基因,hpv16阳性宫颈癌基因组中hpv16基因在所有整合基因中的比例≤60%。这些研究一起表明,在hpv检测中使用my09/11引物进行l1区域的pcr扩增可能导致严重的错误检测。因此,hpv检测越来越依赖于致癌基因e6/e7,因为e6/e7在整合后不会缺失。这可以防止错误检测。因此,相对于l1基因,e6/e7基因可以用作更可靠的hpv检测目标。
本发明中,针对两种高危型hpv,即hpv16和hpv18,设计了两对靶向e6/e7基因的sgrna;运用这些sgrna,用本发明提出的方法检测了三种人宫颈癌细胞系hela、siha和c-33a中的hpv16和hpv18dna。同时设计了两对sgrna和反向引物,用于检测这些人宫颈癌细胞系中的hpv16和e6/e7基因。结果表明,分别在hela和siha细胞中成功检测到hpv18和16;然而,在c-33a细胞中没有检测到两种hpv。这与hela是hpv18阳性细胞,siha是hpv16阳性细胞和c-33a是hpv阴性细胞的事实相符合。
本发明中,在检测人宫颈癌细胞中的hpv16和18时,用了两轮pcr。第一轮qpcr用l1基因通用引物my09/my11和e6-e7基因通用引物e67-6f/7r进行扩增。用cas9/sgrna切割qpcr产物,并用t4dna连接酶连接切割产物。然后用反向引物进行第二轮qpcr。因此,通过第一轮pcr扩增hpvdna,可以快速判断样品是否有hpv感染。“切割和连接”步骤用于hpv分型。第二轮qpcr用于显示分型结果。qpcr扩增的高灵敏度可以保证检测下限足够低。由第一轮qpcr扩增产生的靶dna足够用于随后的分型检测。carp检测使用q-pcr有助于增加检测下限,缩短检测时间,使carp适用于临床检测。
由于cas9核酸内切酶具有大量的脱靶结合位点,能够在一些错配的位置进行切割。脱靶是crispr-cas9系统应用于全基因组范围内的一个瓶颈,特别是用于基因治疗和临床应用。虽然cas9在全基因组范围内有许多脱靶点,但在小dna片段上应该具有非常少或没有脱靶位点。本发明为了确保carp方法的特异性,用pcr从大的dna片段或gdna扩增出小的dna片段,然后使用一对sgrna来切割靶dna。结果表明,通过carp从9个hpv亚型和2种含有hpvgene的宫颈癌细胞株中鉴定了hpv16和hpv18。此外,cas9具有足够高的切割效率,以避免检测信号强度的过度损失,并且pcr的扩增效率很高。因此carp在检测dna时具有足够的特异性和灵敏性。
有益效果:与现有技术相比,本发名具有如下优点:
本发明开发了一种基于crispr的dna检测和分型方法,该方法被命名为carp,代表基于cas9/sgrna的反向pcr。该方法为一种快速,便宜和灵敏的基于crispr的pcr方法,可以有效地对目标dna进行特异性检测和分型。本发明通过在9个hpv亚型中检测两种高危hpv(hpv16和hpv18)的l1基因验证了该方法。在两个hpv阳性宫颈癌细胞系(hela和siha)中检测两种高风险hpv(hpv16和hpv18)的e6/e7基因,再次验证了该方法。通过检测7个非靶向hpv亚型(45、40、35、26、11和6)中两种高危hpv的l1基因和一个hpv阴性子宫颈中两种高危hpv的e6/e7基因癌细胞系(c-33a),显示carp检测的高特异性。另外,在检测中由于对dna进行了第一轮pcr扩增,所以该方法具有高灵敏度。总之,本发明开发了一种具有高特异性和灵敏度的dna检测新方法carp。carp的全部检测过程可以通过常用而普遍的核酸检测设备定量pcr仪在3小时内完成。
本发明利用了crispr技术对dna的特异性识别切割特性,可以简单、快速、灵敏地对目标dna进行特异性检测和分型,成功避免了目前核酸检测和分型领域中核酸杂交和特异性pcr引物设计等关键瓶颈问题。
附图说明
图1为carp检测及分型dna分子的原理及流程示意图;其中carp检测由三个步骤组成:(1)cas9核酸内切酶切割靶dna,一对sgrna(sgrnaa和b)与cas9核酸酶结合后切割靶dna;(2)切割后的dna由t4dna连接酶连接(分子间和分子内连接);(3)连接后的dna用pcr扩增;在本发明中,运用carp检测hpv16和hpv18的l1和e6/e7基因来进行carp检测技术可行性的论证,在pcr扩增步骤,分别使用了传统pcr(tpcr)和定量pcr(qpcr);
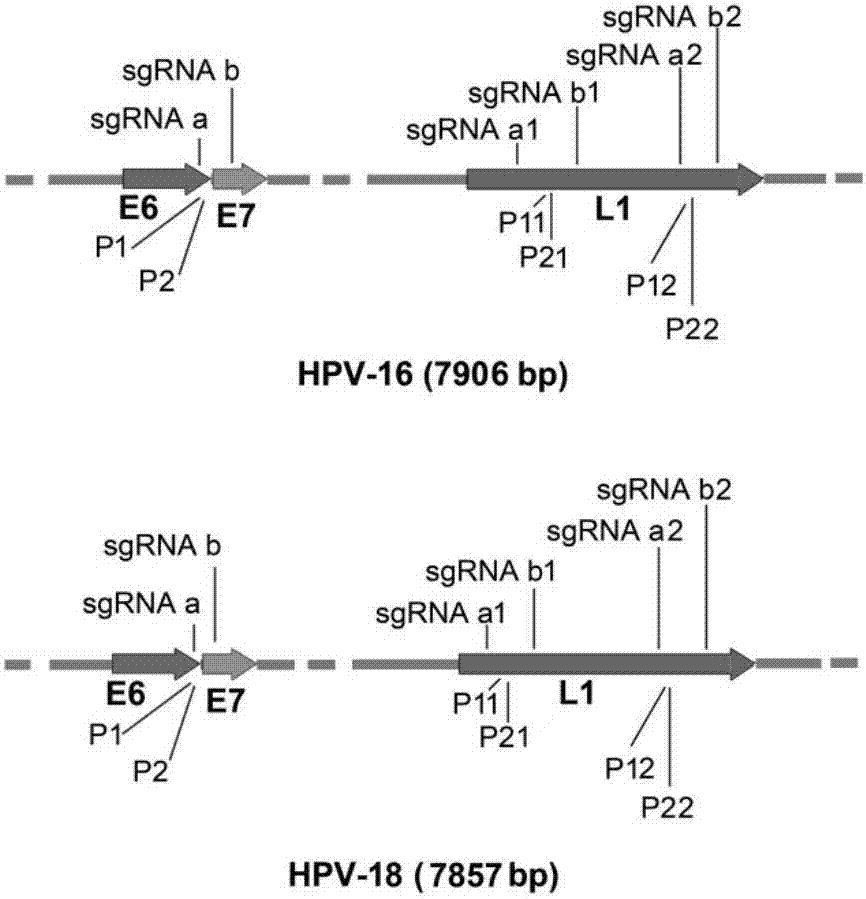
图2为本发明中检测的目标dnahpv16和hpv18的l1和e6/e7基因,以及针对这些基因设计的反向pcr引物、sgrna的位置;
图3为用cas9/sgrna切割和carp检测hpv16和hpv18l1基因;其中(a)cas9/sgrna切割hpv16和hpv18l1基因,hpv16或18l1基因的特异性sgrna与cas9结合后切割hpv16或hpv18l1基因;(b)用tpcr进行hpv16和18l1基因的carp检测;(c)用tpcr检测hpv16和18l1基因carp的特异性;在图a、b和c中列出了各泳道中的反应产物;
图4为carp检测hpvl1基因的灵敏度;其中(a)使用tpcr检测各种量的hpv16l1基因;(b和c)使用qpcr检测各种量的hpv16l1基因;最终的qpcr产物也进行了琼脂糖凝胶电泳检测(c);(d)使用qpcr检测各种量的hpv18l1基因;
图5为使用基于tpcr的carp在9种hpv亚型中检测hpv16或18l1基因;其中(a)通过carp在9种hpv亚型中检测hpv16l1基因;(b)通过carp在9种hpv亚型中检测hpv18l1基因;
图6为使用基于qpcr的carp在9个hpv亚型中检测hpv16或18l1基因;其中(a)在9个hpv亚型中检测hpv16l1基因;(b)在9个hpv亚型中检测hpv18l1基因;最终的qpcr产物也用琼脂糖凝胶电泳检测;
图7为用carp检测细胞中的hpv18和16e6-e7和l1基因;其中(a)用qpcr1扩增三个宫颈癌细胞中的hpve6-e7和l1基因;(b和c)用carp检测三种宫颈癌细胞中的hpv16和hpv18e6-e7(b)和l1(c)基因;(d)用carp检测各种量的helagdna中的hpv18e6-e7和l1基因。
具体实施方式
以下结合附图和实施例对本发明作进一步说明。
实施例1
carp检测及分型dna分子的原理及流程示意图如图1所示。carp检测由三个步骤组成:(1)cas9核酸内切酶切割靶dna。一对sgrna(sgrnaa和b)与cas9核酸酶结合后切割靶dna;(2)切割后的dna由t4dna连接酶连接(分子间和分子内连接);(3)连接后的dna用pcr扩增。在本发明中,运用carp检测hpv16和hpv18的l1和e6/e7基因来进行carp检测技术可行性的论证。在pcr扩增步骤,分别使用了传统pcr(tpcr)和定量pcr(qpcr)。
本发明中各实施例检测的目标dnahpv16和hpv18的l1和e6/e7基因,以及针对这些基因设计的反向pcr引物、sgrna的位置如图2所示。该示意图2有有助于理解本发明实例2~5中的实验。
实施例2用cas9/sgrna切割hpv16和hpv18l1基因
实验方法:
制备sgrna:根据t7聚合酶(newenglandbiolabs)的使用说明,用t7聚合酶通过体外转录合成sgrna。使用表1(seqidno.1-27)中列出的寡核苷酸经过三次pcr扩增出sgrna的dna模板。用f1和r(7个循环)进行第一次pcr。用第一次pcr的产物作为模板,以f2和sgr作为引物进行第二次pcr(30个循环);用第二pcr的产物作为模板,以f3和sgr作为引物进行第三次pcr(30个循环)。第三次pcr的产物纯化后作为体外转录的模板。然后将纯化后的sgrna模板用t7rna聚合酶(newenglandbiolabs)在37℃孵育过夜进行体外转录。将体外转录的rna与trizol溶液混合,然后用氯仿和异丙醇依次萃取,用乙醇沉淀。将纯化的rna溶解在无rnase的ddh2o中,并通过光谱法进行定量。实施例2制备的sgrna还用于本发明中的实施例3-5的实验。
用pcr制备hpvl1基因片段:通过pcr扩增制备hpv16、18、26、33、35、40、45、6和11的l1基因片段。用hpv16、18、26、33、35、40、45、6和11的质粒dna作为模板pcr扩增出全长的l1基因片段。pcr引物m13f和m13r为通用引物(表2;seqidno.28-29)。m13f和m13r在质粒l1基因两边的序列上。pcr反应(20μl):10μl2×预混taq(takara),500nmm13f,500nmm13r和10ng质粒dna。pcr程序如下:95℃5分钟;30个循环:95℃20秒,60℃30秒和72℃90秒;72℃5分钟。用1.5%琼脂糖凝胶对pcr产物进行电泳检测,用axyprepdna凝胶提取试剂盒(axygen)回收l1片段。纯化的l1片段用nanodrop(thermo)定量。然后将纯化的l1片段作为验证carp检测的底物dna。实例1制备的hpvl1基因片段还用于本发明中的实施例2和实施例3实验。
cas9/sgrna切割hpv16and18l1基因:重组cas9蛋白购自newenglandbiolabs(neb)。cas9切割反应(30μl):1×cas9核酸酶反应缓冲液,1μmcas9核酸酶(neb),300nmsgrnaa(16l1a1或18l1a1;表1)和300nmsgrnab(16l1b1或18l1b1;表1)。首先将cas9反应液在25℃温育10分钟。然后向cas9反应液中加入200ng底物dna(纯化的l1片段),并在37℃下孵育20分钟。最后,cas9在65℃灭活10分钟。用2%琼脂糖凝胶进行电泳检测。
实验结果:
cas9与一对sgrna(sgrnaa和b)结合后切割hpv16和18l1基因。未切割的hpv16和18l1基因用作对照。将含有两个切割位点的dna(m13f/m13rpcr扩增产物)切成几个较短的dna片段(图3a)。切割20分钟后,在琼脂糖凝胶中几乎看不到完整的dna底物(l1基因片段)(图3a)(即未被切的l1基因片段),这表明cas9的切割效率很高,这有利于提高检测灵敏度。
表1用于制备sgrna的体外转录模板的寡核苷酸
表2.用于pcr扩增的寡核苷酸
实施例3用carp检测hpv16和18l1基因
实验方法:
sgrna及hpvl1基因片段的制备同实施例1。
cas9/sgrna切割hpv16and18l1基因:重组cas9蛋白购自newenglandbiolabs(neb)。cas9切割反应(30μl):1×cas9核酸酶反应缓冲液,1μmcas9核酸酶(neb),300nmsgrnaa(16l1a1或18l1a1;表1)和300nmsgrnab(16l1b1或18l1b1;表1)。首先将cas9反应液在25℃温育10分钟。然后向cas9反应液中加入200nghpvl1基因片段(底物dna),并在37℃下孵育20分钟。最后,cas9在65℃灭活10分钟。用2%琼脂糖凝胶进行电泳检测。
连接反应体系(15μl):1×t4连接酶缓冲液,5ut4dna连接酶(thermo)和5μlhpv16和hpv18l1基因的cas9酶切产物(如上所述)。在22℃下温育20分钟。
tpcr检测:传统的pcr(tpcr)反应(20μl)由10μl2×预混合taq(takara),500nm16l1p11(或18l1p11)、500nm16l1p21(或18l1p21)和1μl连接产物组成。pcr程序:95℃5分钟;30个循环:95℃20秒,60℃20s和72℃30s;72℃5分钟。反应产物用2%琼脂糖凝胶进行电泳检测。
qpcr检测:用qpcr检测hpv16和18l1基因,将各种量的纯化的hpv16和18l1片段(200、20、2、0.2、0.02、0.002、0ng)进行切割和连接。qpcr反应(20μl)由10μl2×sybrgreenmastermix(yeasen),500nm16l1p11(或18l1p11),500nm16l1p21(或18l1p21)和1μl连接产物组成。pcr程序:95℃10分钟,45个循环:95℃15秒和60℃1分钟。反应在实时pcr装置steponeplus(abi)上进行。
实验结果:
hpv16和18l1基因被cas9切割后,用t4dna连接酶连接切割产物。然后用tpcr检测连接产物。结果表明,carp可以用于检测hpv16和18l1基因(图3b)。然而,如果基因未切割或切割产物未连接,则pcr会失败(图3b)。如果基因没有被切割和连接,两个引物会向相反的方向延伸,靶dna不能被扩增(图3b)。一旦dna被切割并连接后,反向引物就会转变成普通的引物,从而进行靶dna的pcr扩增(图3b)。这些结果表明carp方法是可行的。为了初步验证carp检测的特异性,用hpv16和18的sgrna交叉作用于hpv16和18l1基因。结果表明,hpv16和18l1基因仅能被自身的sgrna识别并被cas9切割(图3c),验证了所设计的sgrna以及carp检测的特异性。除了sgrna之外,反向pcr引物也有助于carp检测特异性。值得注意的是,pcr产物主要集中在两个sgrna目标位点之间预期大小的扩增条带上,表明连接步骤的主要产物是环状dna。
实施例4carp检测hpv16和18l1基因的灵敏度
实验方法:
sgrna及hpvl1基因片段的制备同实施例1。
cas9消化反应(30μl)由1×cas9核酸酶反应缓冲液,1μmcas9核酸酶(neb)和300nmsgrnaa(16l1a1)和sgrnab(16l1b1)组成。首先在25℃下将反应液温育10分钟。然后向反应液加入不同用量hpv16l1基因片段(200、20、2、0.2、0.02、0ng),并在37℃下孵育20分钟。然后cas9在65℃灭活10分钟。连接反应(15μl)由1×t4连接酶缓冲液,5ut4dna连接酶(thermo)和5μlcas9酶切产物组成。将连接反应液在22℃下孵育20分钟。tpcr反应(20μl)由10μl2×premixtaq(takara),500nm16l1p11、500nm16l1p21和1μl连接产物组成。pcr程序:95℃5分钟;30秒循环:95℃20秒,60℃20秒和72℃30秒;72℃5分钟。用2%琼脂糖凝胶进行电泳检测。
实验结果:
为了探讨carp的灵敏度,用cas9/sgrna分别切割200、20、2、0.2、0.02、0nghpv16l1dna,然后用t4dna连接酶连接和tpcr扩增。结果表明,carp可以检测到少至0.02ng的dna(图4a),表明carp检测具有很高的灵敏度。因为qpcr检测在应用中优于tpcr检测,因此又探讨了基于qpcr的carp检测的灵敏度。同样,用carp检测hpv16l1基因。结果,获得了低至0.002ng的灵敏度和200至0ng的检测范围(图4b)。为了进一步验证qpcr结果的可靠性,用琼脂糖凝胶电泳检测了hpv16l1的最终qpcr产物(图4c),表明qpcr产物的电泳结果与tpcr几乎一致(图4a)。比较tpcr,使用qpcr后carp检测的灵敏度得到提高,检测时间进一步缩短。用qpcr检测hpv18l1基因,得到了与tpcr类似的检测范围(图4c)。应该注意的是,tpcr产物只有一个在两个sgrna的靶位点之间预期大小的带(图4a),表明在各种carp检测中连接反应的主要产物是的环状dna。这也表明cas9/sgrna切割的高效率。与tpcr相比,qpcr产生了片段更大的dna(图4c),表明qpcr的高扩增效率和在carp连接反应中存在的分子间连接。
实施例5用carp检测9个hpv亚型中的hpv16或18l1基因
实验方法:
sgrna及hpvl1基因片段的制备同实施例1。
hpv16或18的sgrnaa(16l1a1或18l1a1)和sgrnab(16l1b1或18l1b1)与cas9核酸酶结合后切割9种hpvs亚型(高危型:16、18、26、33、35、40、45;低危型:6、11)的l1基因片段(200ng)。然后将cas9灭活,并将切割产物用t4dna连接酶连接。cas9消化反应及t4dna连接酶连接反应组分、反应条件同实施例3。连接产物通过tpcr和qpcr扩增分别检测连接产物。tpcr和qpcr的反应组分、反应条件同实施例3。
实验结果:
为了进一步验证carp的特异性。用一对特异于hpv16或18的sgrna结合cas9核酸酶后切割9个亚型的hpvs(高危型:16、18、26、33、35、40、45;低危型:6、11)(200ng)l1基因,然后将切割产物连接并用于tpcr扩增。pcr产物用琼脂糖凝胶电泳检测。结果表明,carp在九种hpv亚型中成功区分出了hpv16或hpv18(图5)。另外,进一步用基于qpcr的carp在9个hpv亚型中检测了hpv16和hpv18。也成功鉴定了hpv16和hpv18(图6)。这些结果再次证实了hpv16和hpv18l1的sgrna只对它们自身起作用。因此,carp可用于特异地检测hpv16和hpv18。
实施例6用carp检测宫颈癌细胞中的hpvl1和e6-e7基因
实验方法:
sgrna制备同实施例1。
首先对l1和e6-e7基因进行qpcr扩增。pcr反应(20μl):10μl2×sybrgreenmastermix(yeasen),500nmmy09或e67-6f,500nmmy11或e67-7r以及各种量(见附图)的宫颈癌细胞gdna。pcr程序:95℃10分钟;35个循环:95℃15秒、58.5℃30秒和72℃45秒。我们将这一轮的qpcr命名为qpcr1。
cas9切割反应(30μl)由1×cas9核酸酶反应缓冲液,1μmcas9核酸酶(neb),300nmsgrnaa(用于l1基因的16l1a2和18l1a2;用于e6-e7基因的16e6a和18e6a;表1)和sgrnab(用于l1基因的16l1b2和18l1b2;用于e6-e7基因的16e7b和18e7b;表1)组成。首先在25℃下将反应液温育10分钟。然后向反应液中加入qpcr1产物(5μl),并在37℃下孵育20分钟进行cas9切割,然后在65℃下孵育10分失活cas9。将切割产物(5μl)与1×t4连接酶缓冲液和5ut4dna连接酶混合,并在22℃下孵育20分钟。qpcr反应(20μl)由10μl2×sybrgreenmastermix(yeasen),500nm引物p1(用于l1基因的16l1p12和18l1p12;用于e6-e7基因的16e6p1和18e6p1;表2),500nm引物p2(用于l1基因的16l1p22和18l1p22;用于e6-e7基因的16e6p2和18e6p2;表2)和1μl的连接产物组成。pcr程序:95℃10分钟,40个循环:95℃15秒和60℃1分钟。反应在实时pcr装置steponeplus(abi)上进行。此轮pcr命名为qpcr2。qpcr1和qpcr2中使用的引物列于表2。
实验结果:
为了检测宫颈癌细胞中的hpve6-e7基因,首先设计并合成了一对通用引物e67-6f和e67-7r,用于扩增各种hpv的e6-e7基因。此外还设计了一对特异于hpv16和hpv18e6-e7基因的sgrna,其中一个sgrna针对e6基因,另一个针对e7基因。
为了用carp检测含有的hpve6-e7基因的gdna,首先使用一对通用引物e67-6f和e67-7r通过qpcr扩增来自三个宫颈癌细胞(hela,siha和c-33a)的200nggdna。结果表明,hela和siha细胞的gdna中有e6-e7基因,这两种细胞均为hpv阳性细胞(图7a)。然而,没有从c-33agdna扩增出hpve6-e7基因,表明该细胞是hpv阴性细胞(图7a)。这个第一轮qpcr被命名为qpcr1。然后将针对hpv16或18设计的sgrna与cas9核酸酶结合以切割qpcr1产物,将酶切产物用t4dna连接酶连接。最后,使用一对反向pcr引物,用qpcr扩增连接产物。结果表明,hpv16e6-e7基因存在于siha细胞中,hpv18e6-e7基因存在于hela细胞中(图7b)。然而,在c-33agdna中没有发现hpv16和hpv18e6-e7基因。这与hela是hpv18阳性细胞,siha是hpv16阳性细胞,c-33a是hpv阴性细胞的报道相符合。第二轮qpcr被命名为qpcr2。
使用相同的反应流程,也用三个相同的宫颈癌细胞检测了hpvl1基因。结果表明,使用通用引物my09/my11进行qpcr1,仅在hela和siha细胞的gdna中发现了hpvl1基因(图7a)。qpcr2检测也表明hpv16l1基因存在于siha细胞中,hpv18l1基因存在于hela细胞中(图7c)。qpcr1和qpcr2都在c-33a细胞中没有发现hpvl1基因。这些结果与hpve6-e7基因的carp检测结果相符合,表明carp检测的可靠性。
最后,探讨了carp方法的检测灵敏度。为此,使用不同量的helagdna作为模板用qpcr1扩增了l1和e6-e7基因。将qpcr1产物用cas9/sgrna切割并用t4dna连接酶连接。使用反向引物通过qpcr2扩增qpcr1的连接产物(1μl),用于检测hpv18(图7d)。基于qpcr的carp可以检测到少至0.002ng的gdna。这些数据表明,通过carp可以敏感地,特异性地检测宫颈癌细胞gdna中的hpv。
序列表
<110>东南大学
<120>一种基于crispr的dna检测和分型方法及其应用
<160>45
<170>siposequencelisting1.0
<210>1
<211>53
<212>dna
<213>人工序列(artificialsequence)
<400>1
gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttg53
<210>2
<211>52
<212>dna
<213>人工序列(artificialsequence)
<400>2
aaaaaaaagcaccgactcggtgccactttttcaagttgataacggactagcc52
<210>3
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>3
aaaaaaaagcaccgactcggtgccactttttc32
<210>4
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>4
actgtgtttattaactctaagttttagagctagaaatagcaag43
<210>5
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>5
ttctaatacgactcactatagactgtgtttattaactctaag42
<210>6
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>6
aaaccaaacttattggggtcgttttagagctagaaatagcaag43
<210>7
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>7
ttctaatacgactcactatagaaaccaaacttattggggtc41
<210>8
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>8
aaaccaaatttatttgggtcgttttagagctagaaatagcaag43
<210>9
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>9
ttctaatacgactcactatagaaaccaaatttatttgggtc41
<210>10
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>10
agatatacggtattgtcactgttttagagctagaaatagcaag43
<210>11
<211>42
<212>dna
<213>人工序列(artificialsequence)
<400>11
ttctaatacgactcactatagagatatacggtattgtcactg42
<210>12
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>12
ggagtacctacgacatgggggttttagagctagaaatagcaag43
<210>13
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>13
ttctaatacgactcactatagggagtacctacgacatgggg41
<210>14
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>14
ggatcttctttaggtgctgggttttagagctagaaatagcaag43
<210>15
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>15
ttctaatacgactcactatagggatcttctttaggtgctgg41
<210>16
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>16
gcatcatattgcccaggtacgttttagagctagaaatagcaag43
<210>17
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>17
ttctaatacgactcactataggcatcatattgcccaggtac41
<210>18
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>18
tgttgctattacctgtcaaagttttagagctagaaatagcaag43
<210>19
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>19
ttctaatacgactcactatagtgttgctattacctgtcaaa41
<210>20
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>20
gattccataatataaggggtgttttagagctagaaatagcaag43
<210>21
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>21
ttctaatacgactcactataggattccataatataaggggt41
<210>22
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>22
gaggaggaggatgaaatagagttttagagctagaaatagcaag43
<210>23
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>23
ttctaatacgactcactataggaggaggaggatgaaataga41
<210>24
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>24
gtgctgcaaccgagcacgacgttttagagctagaaatagcaag43
<210>25
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>25
ttctaatacgactcactataggtgctgcaaccgagcacgac41
<210>26
<211>43
<212>dna
<213>人工序列(artificialsequence)
<400>26
cgagcaattaagcgactcaggttttagagctagaaatagcaag43
<210>27
<211>41
<212>dna
<213>人工序列(artificialsequence)
<400>27
ttctaatacgactcactatagcgagcaattaagcgactcag41
<210>28
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>28
cgccagggttttcccagtcacgac24
<210>29
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>29
agcggataacaatttcacacagga24
<210>30
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>30
cgtccmarrggawactgatc20
<210>31
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>31
gcmcagggwcataayaatgg20
<210>32
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>32
aagggmgtaaccgaaawcggt21
<210>33
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>33
gtacctkcwggatcagccat20
<210>34
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>34
gctgcataagcactagca18
<210>35
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>35
tgcaggtgtggataatagag20
<210>36
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>36
tgcttgtgcgagtcacat18
<210>37
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>37
atcatgctggcagctcta18
<210>38
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>38
taacgtctgcagttaaggtt20
<210>39
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>39
cactattttggaggactgga20
<210>40
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>40
catgtctgctatactgctt19
<210>41
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>41
ttgaggaatatgatttgcag20
<210>42
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>42
gatgatctgcaacaagacat20
<210>43
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>43
aagaacacgtagagaaaccc20
<210>44
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>44
cgttggagtcgttcctgtc19
<210>45
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>45
cgacgcagagaaacacaag19
- 还没有人留言评论。精彩留言会获得点赞!