用HindIII鉴定人乳腺癌BCAR4基因rs13334967多态性的方法与流程
本发明属于生物技术领域,涉及一种用hindiii鉴定人乳腺癌bcar4基因rs13334967多态性的方法。
背景技术:
单核苷酸多态性(singlenucleotidepolymorphism,snp),主要是指在基因组水平上由单个核苷酸的变异所引起的dna序列多态性,包括碱基的置换/插入和缺失等形式。snp是人类可遗传的变异中最常见的一种,现已广泛用于简单和复杂疾病的遗传连锁分析/关联分析及疾病易感基因的定位,指导易感基因克隆.较常见的单碱基多态性位点的检测方法包括聚合酶链式反应-限制性片段长度多态性技术(pcr-rflp),三引物等位基因扩增法(tp-pcr)和测序等方法。这些方法虽各有优点,但也各有其不足之处。
pcr-rflp是一种快速,简便,准确的检测snp基因型的经典方法。其原理是:限制性内切酶是一类识别dna特异位点(通常4-6bp),并在特异位点进行切割的酶类。酶切位点的特异性意味着对特定等位基因的完全消化会产生同样的片段序列。而碱基的置换,插入和缺失可以产生或消除一个特定酶切位点,从而改变酶切后产生片段的大小和数目。这些酶切片段带型的不同称为限制性片段长度多态性。如果snp产生和消除了某个限制性内切酶位点,则可以通过对pcr产物进行酶切、电泳加以检测。该方法的优点在于快速简单,终点判断准确。但其主要缺点在于酶切位点的选择,要求待测多态性位点和某一酶切位点相关。pcr-rflp酶的选择具有局限性,并不是所有的snp位点都可以用这种方法来鉴别,且部分内切酶价格昂贵,实验成本高。三引物等位基因扩增法(tp-pcr)是一种不需要限制性内切酶和连接酶的重组dna方法。反应体系中有2种模板和3种引物,从而在同一个反应体系中产生一个重组dna分子。这种方法的主要优点是快速,不依赖连接片段的限制酶位点。但其缺点是扩增效率低,扩增特异性差,无法保证pcr产物的特异性和稳定性,分型结果易造成误判。taqman荧光探针法是一种新型高效准确的基因分型方法,其优点是检测灵敏度高,分型准确。但该方法需要昂贵的仪器和较长的步骤,成本较高。
bcar4(breastcanceranti-estrogenresistance4)是一种长链非编码rna,其编码基因位于人类染色体16p13.13。bcar4最初在乳腺癌细胞中抵抗雌激素耐药基因的筛选中被发现表达异常。后来的研究显示:ccl21释放snip的抑制剂——p300依赖的乙酰化组蛋白,使bcar4结合到转录子snip1和pnuts上,激活非经典hedgehog/gli2转录途径来促进细胞侵袭。在小鼠模型中,bcar4的表达与晚期乳腺癌相关,并且bcar4的靶向治疗剂lnas能够明显抑制乳腺癌的转移。另有研究显示:高水平bcar4的mrna与乳腺癌病人较短的无转移生存率和总生存率相关。bcar是一种强大的转化基因,能够引起雌激素独立生长,抗雌激素抵抗,使肿瘤在体内形成。越来越多的研究发现lncrna的单核苷酸多态性(singlenucleotidepolymorphism,snp)与乳腺癌易感性相关。bcar4已被证实是和乳腺癌密切相关的lncrna,而基因突变有可能影响lncrna的调节作用,促使细胞发生癌变以及肿瘤发生、发展。
bcar4基因rs13334967位点多态性在人群中存在三种基因型:aa型(人基因组rs13334967多态位点的两个等位基因碱基均为a),at型(人基因组rs13334967多态位点的两个等位基因碱基分别为a和t)和tt型(人基因组rs13334967多态位点的两个等位基因碱基均为t)(美国国立卫生研究院国立医学图书馆生物信息技术中心,http://www.ncbi.nlm.nih.gov)。
理论上人乳腺癌bcar4基因多态rs13334967的鉴定应采用pcr-rflp方法。但是目前没有鉴定人乳腺癌bcar4rs13334967基因多态性使用的限制性内切酶,对snp实验造成了一定阻碍。
因此,本领域存在操作简单,成本低,使用范围广的新型检测snp方法的需求。
技术实现要素:
为了克服现有技术中存在的缺陷,本发明提供一种操作简单、成本低、适用范围广泛的用hindiii鉴定人乳腺癌bcar4基因rs13334967多态性的方法,在节约实验成本的前提下,通过创造酶切位点法(createdrestrictionsitepcr,crs-pcr),应用引物3’端错配技术,在pcr产物进行酶切电泳后进行基因型的鉴定,从而提供一种操作简单、成本低、适用范围广泛的检测人乳腺癌bcar4rs13334967多态性的方法及检测试剂盒。
其技术方案如下:
用hindiii鉴定人乳腺癌bcar4基因rs13334967多态性的方法,包括以下步骤:
(a)抽提样品的基因组dna;
(b)提供扩增人bcar4基因多态性rs13334967位点附近序列的正向引物和反向引物,以所述待测人基因组dna为模板,进行pcr扩增反应,获取扩增产物。
(c)使用限制性内切酶对所述扩增产物进行酶切,获取相应的酶切产物;
(d)将酶切产物采用3%的琼脂糖凝胶进行电泳,以判定bcar4基因多态性rs13334967位点的各基因型。
进一步,步骤(b)中所述正向引物和反向引物的设计与合成具体为:碱基序列信息从ncbi的dbsnp数据库获取,在chip网站上进行核对,确定bcar4多态位点碱基变异数据.
含bcar4rs13334967的部分基因序列如seq:id:no:1所示;基因序列的第507个碱基r代表了a/g多态,即为rs8012083的多态性位点。基因序列的第501个碱基w代表了a/t多态,即为rs13334967的多态性位点。根据rs13334967的具体序列和多态性碱基的位置采用primerpremier6.0设计出的上下游引物,正向引物序列如seq:id:no:2所示;反向引物序列如seq:id:no:3所示,其中正向引物序列中倒数第2位碱基为a(形成的扩增产物对应位置为错配碱基a),因此错配后pcr产物中多态附近序列由atgcta或atgctt更改为aagcta或aagctt(其中第2个碱基a为错配碱基,第6个碱基a/t为多态位点)。因此扩增产物中t等位基因片段可被识别a^agctt序列的限制性内切酶hindⅲ识别(即t等位基因片段可被内切酶切开。进而,可以根据片段切开情况来判断多态基因型。
按照正向引物序列和反向引物序列合成引物,引物的合成可以采用本领域通用的方法(如固相合成法),亦可委托生物公司合成并检测正向和反向引物。
进一步,步骤(b)中pcr扩增反应中制备pcr扩增体系具体为:2×taqpcrmix7.5μl,双蒸水5.9μl,上游引物0.3μl下游引物0.3μl以及模板dna1.0μl,充分混匀后即得15.0μlpcr扩增反应体系;按照第一阶段:94℃变性5min,第二阶段共包括35个循环三个步骤,首先94℃变性30s,62.1℃退火45s,最后72℃延伸45s,第三阶段:72℃延伸5min,最终4℃储存以备用,即得长为163bp的pcr扩增产物。扩增后的产物序列如seq:id:no:4所示。
进一步,步骤(c)中进行酶切的酶切体系具体为:pcr反应产物5μl,限制性内切酶0.5μl,buffer1.0μl以及无核酸酶纯水8.5μl共计15μl酶切体系,于水浴锅中37℃酶切1-16h,即得酶切产物。综合考虑经济实用性以及简单操作性,选择hindⅲ进行酶切鉴定。
进一步,步骤(d)琼脂糖凝胶电泳,确定基因多态性具体为:将酶切产物用3%的琼脂糖凝胶在4-10v/cm的条件下,电泳20-40min,至紫外灯下能分清明显的条带后并拍照鉴定。对于不同的基因型,rs13334967位点不同基因型展现出不同的条带,野生纯合基因型aa,长为163bp一条带;突变杂合基因型at,长为163bp,140bp及23bp三条带;突变杂合基因型tt,140bp,23bp两条带。
与现有技术相比,本发明的有益效果:
本发明采用创造酶切位点法(createdrestrictionsitepcr,crs-pcr),利用引物碱基错配技术设计检测bcar4多态性rs13334967。由于这种方法应用了引物3’端错配技术,pcr产物进行酶切电泳后即可进行基因型的鉴定工作,因此在实际运用中具有很大的灵活性,且检测方法简单易行,是一种进行单碱基突变位点基因型鉴定的较好方法。本发明建立的bcar4多态性rs13334967基因分型方法简单、快速、安全、准确、灵敏度高,值得在临床和研究中推广,并可借鉴用于其它多态性基因的分型。
附图说明
图1是bcar4rs13334967位点的电泳图谱;
图2是bcar4rs13334967位点aa基因型测序图谱(逆向;
图3是bcar4rs13334967位点at基因型测序图谱(逆向);
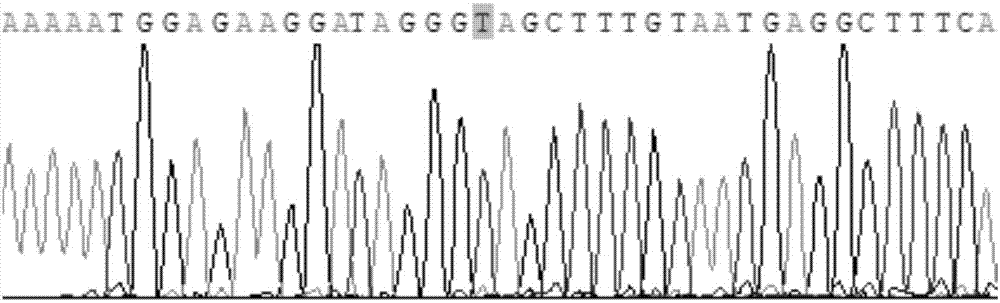
图4是bcar4rs13334967位点tt基因型测序图谱(逆向。
具体实施方式
下面结合附图和实施例进一步说明本发明的技术方案。
本发明的方法包括以下步骤:
a)抽提样品的基因组dna;
(b)提供扩增人乳腺癌bcar4基因多态性rs13334967位点附近序列的正向引物和反向引物,以所述待测人基因组dna为模板,进行pcr扩增反应,获取扩增产物。
(c)使用限制性内切酶对所述扩增产物进行酶切,获取相应的酶切产物;
(d)将酶切产物采用3%的琼脂糖凝胶进行电泳,以判定bcar4基因多态性rs13334967位点的各基因型。
其中,所述正向引物其3’末端倒数第1位碱基与多态rs13334967碱基相近,倒数第2位碱基为a(扩增产物的对应位置为错配碱基a),以便在扩增产物中与多态t等位基因形成aagctt结构(第2个碱基a为错配碱基,第6个碱基t为多态位点)。
本发明的方法可以通过引物上的错配碱基将snp附近的序列改变为可被预先确定的限制性内切酶识别的序列。因此,可以克服传统的pcr-rflp方法所存在的内切酶选择受限制,或者需要采用昂贵内切酶的缺陷。具体而言:由于该正向引物序列中倒数第2位碱基为a(扩增产物的对应位置为错配碱基a),因此错配后pcr产物中多态附近序列由atgcta或atgctt更改为aagcta或aagctt(其中第2个碱基a为错配碱基,第6个碱基a/t为多态位点)。因此扩增产物中t等位基因片段可被识别aagctt序列的限制性内切酶识别(即t等位基因片段可被内切酶切开);进而,可以根据片段切开情况来判断多态基因型。更具体地,经序列分析可知,基因原始序列中a/t多态没有内切酶可以识别切割。如通过碱基错配pcr将多态性位点前面第4位碱基t改变为a,则该多态为t等位基因经pcr扩增后包含aagctt序列可被识别aagctt序列的限制性内切酶hindⅲ识别,而a等位基因不能切开。
经序列分析发现,检测该a/t多态性,没有可以直接识别相应位点的内切酶。在采用创造酶切位点法(createdrestrictionsitepcr,crs-pcr)对正向引物序列中倒数第2位碱基由t改变为a后,pcr产物中多态附近序列生成aagctt序列,从而可被限制性内切酶hindⅲ识别。由于识别特定位点的限制性内切酶hindⅲ适用范围广,价钱较为经济,不仅解决了没有内切酶识别该snp的问题,而且检测snp的成本较为低廉。因此,综合考虑简单操作性与经济实用性,限制性内切酶hindⅲ是不二选择。
限制性内切酶hindⅲ的价格从neb公司(http://www.neb-china.com),限制性内切酶的选择从watcut限制性内切酶分析软件上获(http://watcut.uwaterloo.ca/template.php?act=snp_new),以此作为限制性内切酶识别位点和价格的参考。限制性内切酶hindⅲ识别序列为a^agctt,其价格为205元/5000u,价格低廉,经济实用。
在本发明的实施方案中,正向引物和反向引物的设计采用primer6.0软件进行,设计的原则综合考虑引物对pcr扩增的灵敏度、特异度以及扩增效率的影响。按照碱基互不配对的原则设计引物,引物长度一般在15~30碱基之间,过长或短均会造成特异性差,过长还会导致其延伸温度大于74℃,不适于taqdna聚合酶进行反应。引物gc含量在40%~60%之间,tm值最好接近72℃,gc含量过高或过低都不利于引发反应。碱基要随机分布,引物自身及引物之间不应存在互补序列,否则引物自身会折叠成发夹结构使引物本身复性。引物5′端和中间△g值应该相对较高,而3′端△g值较低。扩增产物的单链不能形成二级结构。引物应具有特异性,引物设计完成以后,应对其进行blast检测,以确保其与其它基因不具有互补性。
最终正向引物由选自下列的核苷酸序列构成:gagtgctgaaagcctcattacaaag。
反向引物由选自下列的核苷酸列:caggaaatagacagaagccacaaga。
正向引物和反向引物的设计主要考虑引物对pcr扩增的灵敏度、特异度和扩增效率的影响。通常按照碱基互补配对原则设计引物,引物与模板的序列要紧密互补。引物长度为15-30bp,过短或过长可导致特异性差,过长亦会导致其延伸温度大于74℃而不利于pcr反应。正向引物在其3’末端倒数第2位含有碱基a,以便在扩增产物中与多态t等位基因形成aagctt结构,从而可以被hindⅲ内切酶识别。扩增产物总长度为163bp.扩增产物如下:
下划线部分分别为正向引物和反向引物,第28位碱基w代表a/t多态即snp位点rs13334967。第24位碱基为错配后的碱基a(原为碱基t)。该长为163bp的扩增产物经限制性内切酶hindⅲ切后可产生163bp,140bp,23bp共三种片段类型。基因型判定结果:纯合野生基因型aa,长为163bp一条带;突变杂合基因型at,长为163bp,140bp及23bp三条带;突变杂合基因型tt,140bp,23bp两条带。
凝胶电泳分为琼脂糖凝胶电泳和聚丙烯酰胺凝胶电泳,琼脂糖(agarose)是一种线性多糖聚合物,系从红色海藻产物琼脂中提取而来的。当琼脂糖溶液加热到沸点后冷却凝固便会形成良好的电泳介质,其密度是由琼脂糖的浓度决定的,可用于dna片段的制备电泳。聚丙烯酰胺凝胶主要有两种方式:一是用于分离和纯化双链dna片段的非变性聚丙烯酰胺凝胶,二是用于分离及纯化单链dna片段的变性聚丙烯酰胺凝胶。但综合考虑实用性、便利性以及经济性,使用琼脂糖凝胶电泳不失为一种最佳选择。在本发明中,目的扩增产物使用限制性核酸内切酶hindⅲ3u水浴锅中酶切1-16h。酶切产物采用3%的琼脂糖进行琼脂糖凝胶电泳,在紫外灯照射下根据不同的条带判断野生纯合基因型,突变杂合基因型以及突变纯合基因型。
在本发明的具体实施方案中,本发明检测人乳腺癌bcar4基因多态rs13334967的具体步骤如下:
(l)提取待测人基因组dna模板,所述人基因组dna模板为人体任何部分取得的人基因组模板。
(2)pcr扩增基因组dna,对提取的模板基因组dna进行pcr扩增,获得含多态附近序列的pcr产物。
(3)对pcr扩增产物用限制性核酸内切酶hindⅲ进行酶切反应,得到酶切产物;
(4)酶切产物进行琼脂糖进行琼脂糖凝胶电泳,在紫外灯照射下根据不同的条带判断野生纯合基因型,突变杂合基因型以及突变纯合基因型。
下面对上述各个主要步骤进行详细描述:
(1)引物设计与合成
碱基序列信息从ncbi的dbsnp数据库获取,在chip网站上进行核对,确定bcar4多态位点碱基变异数据.
含bcar4rs13334967的部分基因序列如下:
基因序列的第501个碱基w代表了a/t多态,即为rs13334967的多态性位点。根据rs13334967的具体序列和多态性碱基的位置采用primerpremier6.0设计出的最有上下游引物如下;
正向引物序列:5’-gagtgctgaaagcctcattacaaag-3’
反向引物序列:5’-caggaaatagacagaagccacaaga-3’。
其中正向引物序列中倒数第2位碱基为a(形成的扩增产物对应位置为错配碱基a),因此错配后pcr产物中多态附近序列由atgcta或atgctt更改为aagcta或aagctt(其中第2个碱基a为错配碱基,第6个碱基a/t为多态位点)。因此扩增产物中t等位基因片段可被识别a^agctt序列的限制性内切酶hindⅲ识别(即t等位基因片段可被内切酶切开。进而,可以根据片段切开情况来判断多态基因型。
按照正向引物序列和反向引物序列合成引物,引物的合成可以采用本领域通用的方法(如固相合成法),亦可委托生物公司合成并检测正向和反向引物。
(2)制备pcr扩增体系:2×taqpcrmix7.5μl,双蒸水5.9μl,上游引物0.3μl下游引物0.3μl以及模板dna1.0μl,充分混匀后即得15.0μlpcr扩增反应体系;按照第一阶段:94℃变性5min,第二阶段共包括35个循环三个步骤,首先94℃变性30s,62.1℃退火45s,最后72℃延伸45s,第三阶段:72℃延伸5min,最终4℃储存以备用,即得长为163bp的pcr扩增产物。扩增后的产物序列为:
下划线部分分别为正向引物和反向引物,第28位碱基w代表a/t多态即snp位点rs13334967。第24位碱基为错配后的碱基a(原为碱基t)。
(3)酶切体系:pcr反应产物5μl,限制性内切酶0.5μl,buffer1.0μl以及无核酸酶纯水8.5μl共计15μl酶切体系,于水浴锅中37℃酶切1-16h,即得酶切产物。综合考虑经济实用性以及简单操作性,选择hindⅲ进行酶切鉴定。
(4)琼脂糖凝胶电泳,确定基因多态性:将酶切产物用3%的琼脂糖凝胶在4-10v/cm的条件下,电泳20-40min,至紫外灯下能分清明显的条带后并拍照鉴定。对于不同的基因型,rs13334967位点不同基因型展现出不同的条带(见图1),野生纯合基因型aa,长为163bp一条带;突变杂合基因型at,长为163bp,140bp及23bp三条带(23bp带不可见);突变杂合基因型tt,140bp,23bp两条带(23bp带不可见)。各基因型均经测序以进一步鉴定,测序结果(见图2-图4)显示与现有技术测得的结果完全相同。
实施例1.人外周血全血标本测定人乳腺癌bcar4rs13334967多态性
1材料与方法
1.1主要仪器与试剂
仪器:bcd-228ch冰箱(新飞电器),hh-2数显恒温水浴锅(华峰仪器),smartge凝胶成像仪(北京赛智创业),gt9612梯度pcr仪(百泰克生物),wd900sl23-2型号微波炉(格兰仕电器),dg-300c型电泳仪(鼎国昌盛生物)等。
试剂:nep004-1dna提取试剂盒(鼎国昌盛生物),50bpdnaladder(莱风生物),2×taqpcrmix(莱风生物),hindⅲ(thermo),琼脂糖(sigma)等。
1.2引物设计
在ncbi的dbsnp数据库中,查找bcar4rs13334967的核苷酸序列,在引物设计软件primerpremier6.0中,设置引物长度和扩增目的片段长度等参数进行引物设计,根据gc含量和退火温度等选择最优上下游引物,并将反向引物序列中倒数第2位碱基设为a(形成的扩增产物对应位置为错配碱基a),因此错配后pcr产物中多态附近序列由atgcta或atgctt更改为aagcta或aagctt(其中第2个碱基a为错配碱基,第6个碱基a/t为多态位点)。因此扩增产物中t等位基因片段可被识别a^agctt序列的限制性内切酶hindⅲ识别(即t等位基因片段可被内切酶切开)。再将此引物与pubmed中的blast序列比对功能(http://blast.ncbi.nlm.nih.gov/blast.cgi)进行引物比对,最终确定的上下游引物为
正向引物序列:5’-gagtgctgaaagcctcattacaaag-3’
反向引物序列:5’-caggaaatagacagaagccacaaga-3’。
1.3限制性内切酶的选择
根据dbsnp中rs13334967前后五个位点的碱基序列,用watcut在线限制性内切酶分析软件,在线搜索获得可识别突变位点的限制性内切酶的信息,综合考虑其酶切特异性及经济适用性选择最佳的限制性内切酶hindⅲ。
1.4从全血中提取待测样本的基因组dna
严格按照离心柱型dna提取试剂盒操作步骤提取基因组dna。
l)在1.5ml离心管中加入300μl血细胞后,再加入900μl细胞裂解液混和均匀,在冰上放置10min后,在离心机中12000rpm离心1min,弃上清,再次加入900μl细胞裂解液,用枪吹起沉淀并混匀后,重复上述步骤;
2)向沉淀物中加入600μlsolutionb溶液,用移液枪轻轻吹起沉淀后,加入10μl蛋白酶k混匀,在70℃水浴锅中放置10min后,12000rpm离心5min。
3)将离心管中的上清转入编过号的新的离心管中,再向新的离心管中加入500μl无水乙醇,期间可能会出现絮状沉淀物,该絮状物即为dna;
4)将混匀的液体转入离心柱中(若一次转不完,可分次转入),室温静置2min,再12000rpm离心1min,弃废液;
5)加入700μl已加入相应体积无水乙醇的solutionc漂洗液,室温静置2min,12000rpm离心1min,弃废液;
6)加入700μl已加入相应体积无水乙醇的solutiond漂洗液,室温静置2min,12000rpm离心1min,弃废液;
7)加入500μlsolutiond漂洗液,12000rpm离心1min,弃废液;
8)再次离心2min,将离心柱置于新的编过号的离心管中,敞口放入37℃恒温箱内10min直至无明显乙醇味;
9)在硅基质膜中央加入已经预热到65℃的solutione100μl,室温放置5min,12000rpm离心1min,重复上述步骤一次,终离心后的液体即为提取出的基因组dna;
10)吸取2μl提取出的dna用nanophotometerpearl微量分光光度计测定其浓度和纯度。2结果
2.1pcr扩增
(1)根据提取基因组dna的浓度,将研究对象的dna进行稀释,使终浓度为20μg/μl。
(2)pcr扩增体系为2×taqpcrmix7.5μl、上游引物和下游引物各0.3μl、模板dna1.0μl,最后用双蒸水补充总体积至15μl。
(3)3个位点的pcr反应条件:第一个阶段为预变性阶段,94℃/5min;第二个阶段包括三个步骤共35个循环,依次设置为94℃/35s、62.1℃退火时间45s、72℃/30s;第三个阶段72℃/5min。
2.2酶切反应
酶切体系为pcr扩增产物5μl、限制性内切酶0.5μl、buffer1.0μl,最后用双蒸水补充总体积至15μl。混匀后置于水浴锅中37℃水浴1-16小时。
2.3基因型的判定
表1rs13334967位点基因型判定
实施例2.人乳腺癌组织标本测定人乳腺癌bcar4rs13334967多态性
与实施例1的步骤基本相同,只是采用下面的方法从乳腺癌组织标本中提取dna作为待测dna。
切除乳腺癌组织,酚-氯仿法提取乳腺癌组织基因组dna作为待测dna。
1)将乳腺癌组织块解冻,用生理盐水洗去血污,剪取0.1g左右的组织碾磨,加入1ml的灭菌水,颠倒混匀,10000转离心10分钟,弃上清。管底应该有沉淀。重复两次
2)加入200ul的dna裂解液,5ul的蛋白酶k混匀,55度过夜消化。
3)消化完成后加入等体积的酚氯仿混合液(1:1),剧烈震荡,使其变成奶咖色。12000转离心10分钟。
4)取上清,注意不要洗到中间介质以及下层液体。加入等体积的氯仿,颠倒混匀。12000转离心10分钟。
5)取上清,注意不要洗到中间介质以及下层液体。加入10分之一的醋酸钠以及2.5倍的无水乙醇,颠倒混匀。
6)12000转离心10分钟。弃上清。
7)加入1ml70%乙醇,使其白色沉淀悬浮,颠倒混匀数次。12000转离心10分钟。
8)弃上清,然后室温开盖静置分钟使其乙醇挥发干净。
9)加入灭菌水溶解dna,一般加入50ul灭菌水溶解
10)-20℃保存,即得乳腺癌组织基因组dna
结果:将酶切产物用3%的琼脂糖凝胶电泳20-40min,至紫外灯下能分清明显的条带并拍照鉴定。对于不同的基因型,rs13334967位点不同基因型展现出不同的条带(见图1),野生纯合基因型aa,长为163bp一条带;突变杂合基因型at,长为163bp,140bp及23bp三条带;突变杂合基因型tt,140bp,23bp两条带。各基因型均经测序以进一步鉴定,测序结果(见图2-图4)显示与现有技术测得的结果完全相同。
从上述实施例可以看出,本发明针对使用crs-pcr鉴定bcar4rs13334967多态性,对传统pcr-rflp法进行改进,应用了引物3’端错配技术,克服了传统pcr-rflp法内切酶选择余地小,价格昂贵,实验成本高等缺点,本研究建立的bcar4rs13334967多态性的检测方法,内切酶价格十分便宜,同时pcr产物纯度较高,酶切结果易于分辨。该检测方法通过测序验证,其结果准确可靠。
snp检测技术已经很成熟,较常见的单碱基多态性位点的检测方法包括测序、三引物等位基因扩增法(tp-pcr)、聚合酶链式反应-限制性片段长度多态性技术(pcr-rflp)等方法。这些方法虽各有优点,但也各有其不足之处。测序可以直接测出dna序列中的所有突变位点的碱基替代情况,但该方法需要昂贵的仪器和较长的步骤,成本较高;tp-pcr方法准确性低,结果易造成误判;pcr-rflp方法比较成熟,但要求待测多态性位点和某一酶切位点相关,酶的选择具有局限性,往往成本较高。
因此,鉴于本发明所使用的限制性内切酶价格低廉,且引物合成以及各试剂均较为经济,加之crs-pcr操作简单,重复性好,因此是最经济有效的基因型检测方法。
本发明对传统pcr-rflp技术进行改进,采用创造酶切位点法(createdrestrictionsitepcr,crs-pcr),利用引物碱基错配技术设计检测bcar4多态性rs13334967。由于这种方法应用了引物3’端错配技术,pcr产物进行酶切电泳后即可进行基因型的鉴定工作,因此在实际运用中具有很大的灵活性,且检测方法简单易行,是一种进行单碱基突变位点基因型鉴定的较好方法。本项发明的技术关键点是设计出的bcar4多态性rs13334967的上下游引物,正向引物序列:5’-gagtgctgaaagcctcattacaaag-3’,反向引物序列:5’-caggaaatagacagaagccacaaga-3’。以及限制性内切酶hindⅲ的使用。总之,本发明建立的bcar4多态性rs13334967基因分型方法简单、快速、安全、准确、灵敏度高,值得在临床和研究中推广,并可借鉴用于其它多态性基因的分型。
以上所述仅为本发明最佳实施方式,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可显而易见地得到技术方案的简单变化或等效替换均落入本发明的保护范围内。
序列表
<110>郑州大学
<120>用hindiii鉴定人乳腺癌bcar4基因rs13334967多态性的方法
<160>4
<170>siposequencelisting1.0
<210>1
<211>1001
<212>dna
<213>人工序列(artificialsequence)
<400>1
agacaagaagagactataaattgctgacccacccacgctgagacccatgcgtatttgatt60
ttttttttttttttttttgagacggagtttcgctctttttgcccaggccggagtgcaatg120
gtgcaatctcggctcaccgcgacctctgcctcccgggttcaagcatttctcctggctcag180
cctcccaagtagctgggattacaggcgcctgccaccccgtctggcgaattttttgtattt240
ttagtagagacaggtttcaccacgttggcctggattgtctcgatctcctgacctcgtgat300
ccacccgcctcggcctcccaaagtgctgggattacaggcgtgagccactgcacccggccg360
catatttgatttctatgtttatctacaggtcaagtacagattgcccaagttcaagagaaa420
gacttgattgttcttcctctctttccttttcacatgcaacacgtggattgagtgagtgct480
gaaagcctcattacaaagctwccctatccttctccattttttcttttcacctctcccctc540
ctgcccactttttccctttaaatattgaagccctttaaatcctctttggaaaaagtgctg600
actccagatcctcttgtggcttctgtctatttcctgggcatgtcctcaatgttagctaaa660
ataaacctctacagtgattgagaccttctgagacttctggtttacaagtgaaatttcatg720
aagctatctcaagacatccaaaaagactgcttctgaaaatgaatcctcacgcttgggagg780
ccgaggcaggtggatcgcttgagcccaggagttcaagactagcctggggaacgtggcaaa840
accctgtctctacaaaaaataaataaataaattaaataaaatacaaaaaattagccacgc900
atggtggcacgtgcctgtagtaccagataccctgggagctgaggcttcagtaagccttga960
ttgtgccactgcactccagcctgggagatagaaaggcccca1001
<210>2
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>2
gagtgctgaaagcctcattacaaag25
<210>3
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>3
caggaaatagacagaagccacaaga25
<210>4
<211>162
<212>dna
<213>人工序列(artificialsequence)
<400>4
gagtgctgaaagcctcattacaagctwccctatccttctccattttttcttttcacctct60
cccctcctgcccactttttccctttaaatattgaagccctttaaatcctctttggaaaaa120
gtgctgactccagatcctcttgtggcttctgtctatttcctg162
- 还没有人留言评论。精彩留言会获得点赞!
- 一种基于片状锶铁氧体双重放大的乳腺癌易感基因免疫传感器的制备方法
- 基于pcr引物3’末端核苷酸双脱氧修饰的乳腺癌易感基因snp快速、灵敏检测方法
- 用于乳腺癌易感基因BRCA1和BRCA2的Long-range PCR检测的引物组、方法及试剂盒的制作方法
- 一种基于纳米银及纳米铜联合构建的双通道乳腺癌易感基因传感器的制备方法及应用
- 预测乳腺癌结果的nano46基因和方法
- Brca基因易感snp位点检测组合物的制作方法
- 3d热乳癌检测的制作方法
- 基于微流控芯片的基因治疗乳腺癌的药物的制备方法
- 一种检测原发性肝细胞癌的易感基因的荧光定量pcr基因分型方法及其试剂盒的制作方法
- Akr1b10基因蛋白作为乳腺癌诊断标志物和药物靶标的制作方法