丁腈橡胶选择性非均相溶液加氢催化剂的制备和使用方法与流程
本发明涉及一种可简单回收并重复使用的用于nbr选择性加氢的双功能化催化剂的制备和使用方法,属于聚合物加氢制备特种橡胶的领域。
背景技术:
nbr(丁腈橡胶)是由丁二烯和丙烯腈通过乳液共聚制取,其分子链中带有极性腈基,具有耐油性、耐热性、良好的加工性能和耐磨性等。但由于nbr分子链中含有大量碳-碳双键,导致耐热和耐氧性较差,使其应用范围受到限制。通过对nbr中的双键进行选择性加氢,可以在保留nbr原有性能的基础上,极大提高橡胶的耐热性、耐氧性、抗老化性,所得到的hnbr(氢化丁腈橡胶)在拉伸强度、伸长率、耐磨性和硬度等机械性质方面表现优异,被广泛应用于汽车工业、石油工业和航空航天等领域。
hnbr的制备方法主要有乙烯-丙烯腈共聚法、nbr乳液加氢法和溶液加氢法。其中溶液加氢法是现阶段工业化生产hnbr所采用的主要方法,又分为均相溶液加氢和非均相溶液加氢两种。均相溶液加氢是反应物nbr和催化剂在同一相(液相)中进行加氢反应(martinp,mcmanusnt,rempelgl,adetailedstudyofthehydrogenationofnitrile-butadienerubberandothersubstratescatalyzedbyru(ii)complexes[j],j.mol.catala.chem.,1997,126,115-131;周淑芹,一种新型催化剂制备氢化丁腈橡胶的研究[j],高分子材料科学与工程,2002,18,69-72;周淑芹,姚明,徐瑞清,铑钌双金属双配体催化剂制备氢化丁腈橡胶[j],北京化工大学学报,2002,29,17-19;weizl,wujl,panqm,rempelgl,directcatalytichydrogenationofanacrylonitrile-butadienerubberlatexusingwilkinson'scatalyst[j],macromol.rapidcomm.,2005,26,1768-1772;mahittikula,prasassarakichp,rempelgl,hydrogenationofnaturalrubberlatexinthepresenceofoshcl(co)(o2)(pcy3)2[j],j.appl.polym.sci.,2006,100,640-655)。由于活性组分以均相状态分散到胶液体系,能够与反应物充分接触,不存在扩散传质的影响,因此在较为温和的条件下即能使聚合物达到高的加氢程度,是目前工业化生产的主流方法之一,德国的lanxess公司即采用该方法生产hnbr。然而,均相溶液加氢法存在的一个共同缺点是催化剂与产品分离困难,虽然目前已开发了多种技术,如采用离子交换树脂对贵金属进行离子交换(李继霞,白文玉,羰基合成用废铑催化剂的再生与铑的回收[j],2008,29,53-55),利用氯化亚锡与铑催化剂的络合作用进行水相萃取回收贵金属铑(陈子尧,邢海琳,李超,张立群,岳冬梅,铑催化剂在nbr加氢领域的应用及回收方法,稀有金属材料与工程,2012,41,343-347),采用温度控制的相转移催化剂等(ningsm,yangsf,weixp,wangwm,zhanglq,yuedm,selectivehydrogenationofnitrile-butadienerubbercatalyzedbythermoregulatedphasetransferphosphinerhodiumcomplex[j],j.appl.polym.sci.,2012,123,1040-1046),但这些方法均难以完全脱除聚合物中残存的贵金属催化剂或可能产生凝胶影响产品质量,而且大规模工业化的成本极高。贵金属在加氢产物中的残留,一方面会造成生产成本增加和贵金属资源的浪费;另一方面会导致hnbr老化速率加快,影响聚合物的机械加工性能。
与均相加氢催化体系相比,非均相催化反应体系采用负载型贵金属催化剂,能够很好地解决催化剂与加氢产品分离的难题,不仅使得贵金属催化剂的重复利用成为可能,而且有效地避免了贵金属在聚合物中的残留。而目前负载型催化剂的制备还是依赖传统的浸渍法,因而导致活性组分团聚严重,颗粒尺寸较大,明显降低了活性中心数目,而且由于活性组分与载体间相互作用较弱,在强烈搅拌反应后容易造成贵金属从载体表面的脱离和流失,降低了催化剂的可利用度也影响了加氢产物的性能。近年来发现对载体表面进行功能化修饰,可成功的锚定活性组分。岳冬梅课题组将rh负载在氨基修饰过的sio2上,对nbr进行加氢反应,一次加氢度可达95%,通过icp测试产物hnbr中的rh含量,发现活性金属rh并未流失太多。但是回收出来的催化剂二次加氢反应活性较差,二次加氢度下降明显(caop,niy,zour,etal.enhancedcatalyticpropertiesofrhodiumnanoparticlesdepositedonchemicallymodifiedsio2forhydrogenationofnitrilebutadienerubber[j].rscadvances,2015,5,3417-3424)。此课题组又将rh负载的石墨烯或碳纳米管上,同样发现回收回来的催化剂二次加氢活性很低(caop,huangcy,zhanglq,yuedm,one-stepfabricationofrgo/hnbrcompositesviaselectivehydrogenationofnbrwithgraphene-basedcatalyst[j],rscadvances,2015,5,41098-41102;zour,lic,zhangl,etal.selectivehydrogenationofnitrilebutadienerubber(nbr)withrhodiumnanoparticlessupportedoncarbonnanotubesatroomtemperature[j].catalysiscommunications,2016,81,4-9)。这在一定程度上限制了非均相催化剂的工业化应用。因此,开发一种高性能的、可简单回收并能重复使用的nbr选择性加氢负载型催化剂,成为工业化生产高附加值hnbr的关键所在。
技术实现要素:
为解决上述技术问题,本发明的目的在于开发一种新型多功能化负载型贵金属加氢催化剂,以二氧化硅微球为载体,采用多种具有不同功能的硅烷偶联剂修饰载体,然后以嫁接法负载贵金属作为活性组分,制备出活性颗粒分散度高、载体表面具有氨基及疏水基官能团化的负载型催化剂。该催化剂的特点是:一方面,此制备方法得到的催化剂活性组分分散性好,颗粒尺寸可控,即使在高负载量下仍能保持良好的分散度和较小的颗粒尺寸(1-3nm),反应后不会发生团聚和流失,能够提高催化剂的可利用度并保证hnbr的产品性能;另一方面,此制备方法得到的催化剂表面具有疏水官能团,对反应物中的极性基团腈基(-cn)具有排斥作用,避免腈基吸附在活性位上进行加氢,而对nbr碳-碳双键进行选择性催化加氢,保证选择性100%,更重要的是,可以使产物在反应后快速脱离活性组分,有效避免产物对活性组分的覆盖而引起的催化剂失活,达到催化剂简单回收并重复使用的目的。
为达到上述目的,本发明提供的nbr选择性非均相溶液加氢催化剂用于制备hnbr的方法,其包括以下步骤:
(1)以二氧化硅微球为载体,以偶联剂嫁接法,在载体表面先后修饰具有氨基官能团和疏水性基团的不同功能的硅烷偶联剂n和c,负载活性组分m,得到负载量为0.5-6wt%的m/nc-sio2非均相加氢催化剂,其活性颗粒粒径为1-3nm;
(2)以有机溶剂100ml溶解1-8gnbr,nbr平均分子量1000-300000,取一定量的m/nc-sio2为非均相催化剂。反应条件:温度为30-80oc,氢气压力为0.5-6mpa,反应时间为0.5-12h;
(3)对得到的胶液进行离心分离,回收m/nc-sio2非均相加氢催化剂,然后将离心分离后的催化剂分散于有机溶剂中,搅拌数小时,离心分离用于下一次加氢反应。
在上述氢化丁腈橡胶的制备方法中,所述步骤(2)中溶解nbr的有机溶剂,包括丙酮、二氯甲烷、丁酮、二甲基甲酰胺、环己酮、乙酸乙酯和四氢呋喃等中的一种或几种的混合物。更优选的,所采用的有机溶剂为丙酮和/或四氢呋喃。
在上述氢化丁腈橡胶的制备方法中,该方法还包括以丙酮、二氯甲烷、丁酮、二甲基甲酰胺、环己酮、乙酸乙酯和四氢呋喃的单溶剂或混合溶剂对回收的非均相加氢催化剂进行洗涤以及重复用于丁腈橡胶的加氢处理的步骤。更优选的,所采用的有机溶剂为丙酮和/或二甲基甲酰胺。
在上述氢化丁腈橡胶的制备方法中,优选的,非均相加氢催化剂的用量占反应溶液质量的0.1-6wt%,更有选的,催化剂用量占反应溶液质量的0.2-3wt%。
在上述氢化丁腈橡胶的制备方法中,优选的,丁腈橡胶分子量为1000-300000,更优选的,分子量为1000-100000。
在上述氢化丁腈橡胶的制备方法中,优选的,所采用的活性组分m为pd、rh等。
在上述氢化丁腈橡胶的制备方法中,优选的,嫁接法采用带氨基的硅烷偶联剂n及带疏水基团的硅烷偶联剂c,采用的贵金属前驱体为mcl2或m(oac)2或mcl3;其中带氨基的硅烷偶联剂n,包括氨乙基氨丙基三甲氧基硅烷、氨基三亚甲基膦酸盐和3-氨丙基三乙氧基硅烷中的一种或两种;所述的带疏水基团的硅烷偶联剂c,包括三甲基氯硅烷、三乙基氯硅烷、乙基三甲氧基硅烷和氟硅烷中的一种或两种。更优选的,采用氨乙基氨丙基三甲氧基硅烷及甲基氯硅烷偶联剂作为连接剂,m(oac)2作为活性组分前驱体。
在上述氢化丁腈橡胶的制备方法中,m/nc-sio2非均相加氢催化剂中,活性组分m的负载量为0.5-6wt%,优选的,负载量为2-5wt%。
用于氢化丁腈橡胶催化剂的制备方法,其中,所述具有不同功能的硅烷偶联剂修饰载体是通过以下步骤制备的:
1)取1-5gsio2在100oc下烘干,冷却到室温,分散在15-80ml甲苯中;将0.5-11.0mmol带氨基的硅烷偶联剂n,溶解在5-40ml甲苯中,制备前体液ⅰ;将前体液ⅰ和sio2分散液混合,在室温下搅拌10min后,转移至50-100oc恒温水浴锅反应24h;待冷却至室温,离心分离,用甲苯、乙醇、丙酮溶剂依次洗涤以除去载体表面未反应的物质;洗涤后将样品分散在乙醇中,在60oc水浴锅中搅拌8h,冷却至室温,分离,在100oc烘箱内烘干,得氨基修饰的sio2;
2)取1-5g经过氨基修饰过的sio2在100oc下烘干,冷却到室温,分散在15-80ml甲苯中;将0.5-15.0mmol带疏水基团的硅烷偶联剂c,溶解在5-40ml甲苯中,制备前体液ⅱ;将前体液ⅱ和经过氨基修饰过的sio2分散液混合,在室温下搅拌10min后,转移至50-100oc恒温水浴锅反应24h;待冷却至室温,离心分离,用甲苯、乙醇、丙酮溶剂依次洗涤以除去载体表面未反应的物质;洗涤后将样品分散在乙醇中,在60oc水浴锅中搅拌8h,冷却至室温,分离,在100oc烘箱内烘干,制备得到表面经过双官能团修饰的载体nc-sio2。
本发明的显著优点:本发明所提供的上述高性能、可简单回收并能重复使用的nbr选择性加氢催化剂的制备方法,以二氧化硅为载体,采用多种具有不同功能的硅烷偶联剂分别修饰载体后负载贵金属,制备出活性组分分散度高、载体表面具有氨基及疏水基官能团化的负载型催化剂,疏水官能团对反应物中的极性基团腈基(-cn)具有排斥作用。将催化剂加入到nbr溶液中,在高压反应釜中对nbr碳-碳双键进行选择性催化加氢,在较低的温度、压力及短时间内即可得到加氢度高的hnbr,其反应条件为:氢气压力0.5-6mpa,加热温度30-80°c,反应时间0.5-12h,反应后加氢度大于90%,选择性100%,所得产品性能优异。反应后的催化剂通过离心分离即可回收,在有机溶剂中洗涤后重复使用数次,活性不会明显下降。该发明的特点在于利用两种硅烷偶联剂修饰二氧化硅载体表面将其功能化修饰,然后利用化学嫁接法制备负载型贵金属催化剂。一方面,此制备方法得到的催化剂活性组分分散性好,颗粒尺寸可控,即使在高负载量下仍能保持良好的分散度和较小的颗粒尺寸,反应后不会发生团聚和流失,能够提高催化剂的可利用度并保证hnbr的产品性能;另一方面,此制备方法得到的催化剂表面具有疏水官能团,对反应物中的极性基团腈基具有排斥作用,避免腈基吸附在活性位上进行加氢,保证选择性100%,更重要的是,可以使产物在反应后快速脱离活性组分,有效避免产物对活性组分的覆盖而引起的催化剂失活,达到催化剂简单回收并重复使用的目的。
附图说明
图1为实施例所制备不同加氢程度的hnbr与nbr红外光谱图;
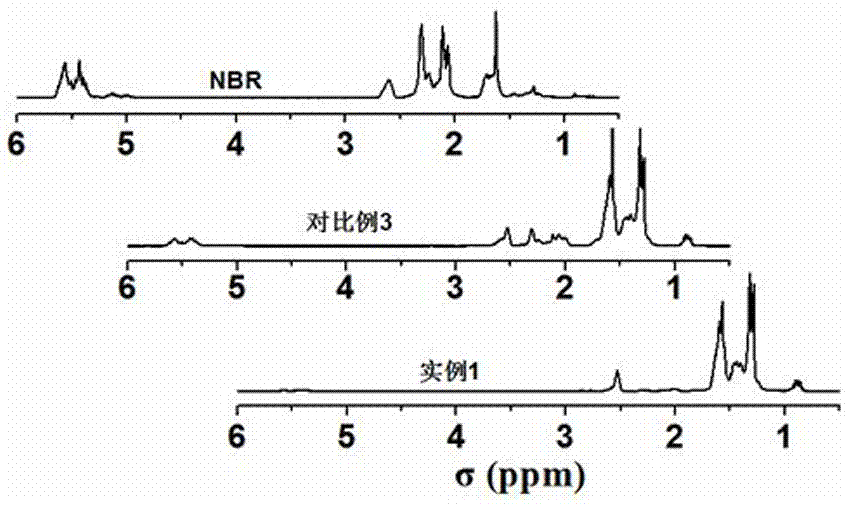
图2为实施例所制备不同加氢程度的hnbr与nbr的1hnmr谱图;
图3为实施例5所采用的pd/nc-sio2催化剂的tem图及贵金属晶粒粒径分布图;
图4为实施例8所采用的pd/nc-sio2催化剂用于nbr反应后的tem图及贵金属晶粒粒径分布图;
图5为实施例5-8和对比例1-3催化剂回收重复利用的加氢度对比。
具体实施方式
为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现对本发明的技术方案进行以下详细说明,但不能理解为对本发明的可实施范围的限定。
实施例中,sem图片由荷兰feiquanta200f场发射扫描电子显微镜获得;tem图片由日本jem2100lab6透射电子显微镜获得;红外谱图由美国magna-ir560e.s.p型ft-ir光谱仪获得;1hnmr谱图由日本jnm-la300ft-nmr核磁共振波谱仪获得。
实施例1
采用氨乙基氨丙基三甲氧基硅烷功能化修饰载体sio2,反应结束样品烘干后再采用三甲基氯硅烷功能化修饰氨基修饰过的载体,利用pd(oac)2溶液为贵金属前驱体溶液,制备负载量为3wt%的非均相催化剂pd/nc-sio2。将1g丁腈橡胶(nbr)溶解在100ml丙酮溶剂中,取0.2g非均相催化剂pd/nc-sio2加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度60℃,氢气压力2mpa,反应时间2h。反应结束后,非均相催化剂离心分离回收再利用,胶液蒸发有机溶剂得到产品hnbr,加氢度列于表1中。
实施例2
采用氨丙基三乙氧基硅烷功能化修饰载体sio2,反应结束样品烘干后再采用乙基三甲氧基硅烷功能化修饰氨基修饰过的载体,利用pd(oac)2溶液为贵金属前驱体溶液,制备负载量为3wt%的非均相催化剂pd/nc-sio2。将1g丁腈橡胶(nbr)溶解在100ml丙酮溶剂中,取0.2g非均相催化剂pd/f-sio2加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度60℃,氢气压力2mpa,反应时间2h。反应结束后,非均相催化剂离心分离回收再利用,胶液蒸发有机溶剂得到产品hnbr,加氢度列于表1中。
实施例3
采用氨乙基氨丙基三甲氧基硅烷功能化修饰载体sio2,反应结束样品烘干后再采用三甲基氯硅烷功能化修饰氨基修饰过的载体,利用pd(oac)2溶液为贵金属前驱体溶液,制备负载量为3wt%的非均相催化剂pd/nc-sio2。将1g丁腈橡胶(nbr)溶解在100ml丙酮溶剂中,取0.2g非均相催化剂pd/nc-sio2加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度40℃,氢气压力2mpa,反应时间2h。反应结束后,非均相催化剂离心分离回收再利用,胶液蒸发有机溶剂得到产品hnbr,加氢度列于表1中。
实施例4
采用氨乙基氨丙基三甲氧基硅烷功能化修饰载体sio2,反应结束样品烘干后再采用三甲基氯硅烷功能化修饰氨基修饰过的载体,利用pd(oac)2溶液为贵金属前驱体溶液,制备负载量为3wt%的非均相催化剂pd/nc-sio2。将1g丁腈橡胶(nbr)溶解在100ml丙酮溶剂中,取0.2g非均相催化剂pd/nc-sio2加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度40℃,氢气压力2mpa,反应时间0.5h。反应结束后,非均相催化剂离心分离回收再利用,胶液蒸发有机溶剂得到产品hnbr,加氢度列于表1中。
实施例5
采用氨乙基氨丙基三甲氧基硅烷功能化修饰载体sio2,反应结束样品烘干后再采用三甲基氯硅烷功能化修饰氨基修饰过的载体,利用pd(oac)2溶液为贵金属前驱体溶液,制备负载量为3wt%的非均相催化剂pd/nc-sio2。将1g丁腈橡胶(nbr)溶解在100ml丙酮溶剂中,取1g非均相催化剂pd/nc-sio2加入到胶液中,在高压加氢反应釜中进行催化加氢反应。反应条件为:温度60℃,氢气压力2mpa,反应时间2h。反应结束后,非均相催化剂离心分离回收再利用,胶液蒸发有机溶剂得到产品hnbr,加氢度列于表1中。
实施例6
将实施例5反应结束后的非均相催化剂离心分离回收,于有机溶剂丙酮中搅拌洗涤4h,离心分离。采用实施例5相同的反应条件,重新将其加入到反应体系中,考察其nbr加氢催化性能,加氢度列于表1中。
实施例7
将实施例6反应结束后的非均相催化剂离心分离回收,于有机溶剂丙酮中搅拌洗涤4h,离心分离。采用实施例5相同的反应条件,重新将其加入到反应体系中,考察其nbr加氢催化性能,加氢度列于表1中。
实施例8
将实施例7反应结束后的非均相催化剂离心分离回收,于有机溶剂丙酮中搅拌洗涤4h,离心分离。采用实施例5相同的反应条件,重新将其加入到反应体系中,考察其nbr加氢催化性能,加氢度列于表1中。
对比例1
本对比例提供一种仅采用氨乙基氨丙基三甲氧基硅烷功能化修饰过的二氧化硅微球作为载体制备的钯基催化剂,催化nbr加氢制备hnbr,以便与实施例5-8进行对比。其余步骤按照实施例5中的方法制备催化剂,nbr加氢实验条件不变。
对比例2
将对比例1反应结束后的非均相催化剂离心分离回收,于有机溶剂丙酮中搅拌洗涤4h,离心分离。采用对比例1相同的反应条件,重新将其加入到反应体系中,考察其nbr加氢催化性能,加氢度列于表1中。
对比例3
将对比例2反应结束后的非均相催化剂离心分离回收,于有机溶剂丙酮中搅拌洗涤4h,离心分离。采用对比例1相同的反应条件,重新将其加入到反应体系中,考察其nbr加氢催化性能,加氢度列于表1中。
实施例1-8及对比例1-3所制备的催化剂用于nbr加氢得到不同加氢度的hnbr,其加氢度及选择性列于表1,还对实施例1-8所制备的不同加氢度的hnbr进行了红外谱图检测,结果如图1所示。由图1可知,2230.0cm-1处的吸收峰为-cn,其强度在加氢后始终未减弱,而且在3500.0cm-1并未出现代表-nh2的吸收峰,说明反应前后-cn没有发生变化,即-cn没有被加氢,选择性为100%;在971.3cm-1及920.0cm-1处的1,4-双键基团和1,2-双键基团的吸收峰在加氢后有不同程度地减弱,甚至消失,说明双键基团被完全加氢。同时,加氢后在723.1cm-1出现新的峰,属于-[ch2]n-(其中n>4)结构,即为分子链中c=c加氢饱和后新出现的结构。利用红外谱图吸收峰的强度,得到加氢度,计算公式为:
图2为实施例1和对比例3所制备的hnbr与nbr的1hnmr谱图。c=c双键环境的质子吸收峰化学位移分别在1.64(-c-ch2-c-c-c=c-)、2.27和2.09(-c-ch2-c=c-)、5.09和4.97(-c-c=ch2)、5.53和5.37(-ch=ch-),由图2可知,实施例1得到的hnbr其上述质子吸收峰基本完全消失,并新增了代表饱和烷烃链段的质子吸收峰,化学位移分别在1.57、1.29、0.86;而对比例3得到的hnbr在5.53和5.37(-ch=ch-)以及2.27和2.09(-c-ch2-c=c-)处还存在有质子吸收峰,说明仍有较多的c=c键存在。
对实施例5所制备的pd/nc-sio2催化剂进行了tem表征(如图3所示),由图3可知,催化剂活性组分颗粒分布均匀,而且颗粒尺寸小,由粒径统计图可知,活性颗粒尺寸大约集中在1.7nm。图4是实施例8催化剂重复利用4次后的tem表征,说明利用本发明提供的方法得到的催化剂在反应后活性组分颗粒仍然分布均匀,颗粒尺寸小,由粒径统计图可知,活性颗粒尺寸大约集中在2.1nm。
表1实施例和对比例所制备的hnbr加氢度及选择性对比数据
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
- 还没有人留言评论。精彩留言会获得点赞!