一种大黄酸酰胺类衍生物纳米混悬剂及其制备方法和应用与流程
本发明属于药物制剂领域,具体地涉及一种大黄酸酰胺类衍生物纳米混悬剂及其制备方法和应用。
背景技术:
大黄来源于蓼科多年生草本植物掌叶大黄(Rheum palmatum L.)、唐古特大黄(R.tanguticum Maxim.ex Balf.)或药用大黄(Rheum officinale Baill.)的干燥根及根茎。始载于《神农本草经》,其性味苦寒,归脾、胃、大肠、肝、心经,具有逐癖痛经,泄热通肠,凉血解毒的功效。临床常用于胆囊炎、肝炎、糖尿病、肾病、慢性肾功能衰竭以及便秘等疾病的治疗。大黄的主要药理成分是蒽醌衍生物(含量约占3%~5%)和少量二蒽酮类衍生物。其中,游离型蒽醌为大黄的主要抗菌成分,结合型蒽醌则为大黄的主要泻下成分。现代研究表明,游离型蒽醌类化合物具有抗肿瘤活性。
大黄酸(Rhein,RH)作为中药大黄、中药芦荟等的主要有效成分,属单蒽核类1,8-二羟基蒽醌衍生物,大量的药理学研究结果表明,大黄酸在降糖调脂、抗肿瘤、抗病毒、抗纤维化、抗菌、保肝等多方面具有广泛的药理活性,特别是在治疗骨关节炎、糖尿病、肾病及协同抗肿瘤方面有突出的治疗作用,是近年来研究热点之一。但是由于其水溶性低,生物利用度差,极大地限制了大黄酸的开发和临床应用。现有技术中均只是对大黄酸进行了结构改造,而没有相关制剂的深入研究。
纳米混悬剂是20世纪末发展起来的一种新型纳米给药系统,由于不使用添加剂,纳米混悬剂可以避免由添加剂在体内消除和聚集引起的毒性问题。与传统小分子抗肿瘤药物相比,纳米混悬剂具有如下突出优点:1)可使药物具有控释效果,能够延长药物在体内的半衰期;2)可使药物具有靶向输送的效果,纳米混悬剂通过其高通透性和滞留效应(EPR),能够将药物靶向输送至肿瘤部位,从而降低其对正常组织的毒副作用,提高药物的生物利用度;3)可通过化学键键和、表面吸附或者物理增溶等方式提高疏水性抗癌药物的溶解性,有效改善其给药方式;4)可以通过胞吞方式进入肿瘤细胞,并解决有效克服上皮和内皮运输障碍的问题;5)可同时输送多种药物,实现联合给药,增强药物抗肿瘤等作用;6)可通过包载或与显像剂连接,使药物输送可视化,便于实时跟踪研究药物在体内的输送和释放过程。
因此,为了进一步提高原化合物的生物利用度,同时具有缓释作用,开发大黄酸酰胺类衍生物的新剂型,具有重要的作用。
技术实现要素:
本发明的目的之一是,提供一种更加简单、快速、高产率的大黄酸酰胺类衍生物混悬剂的制备方法。
本发明的目的之二是,提供大黄酸酰胺类衍生物纳米混悬剂在临床上的应用。
本发明采用的技术方案是:一种大黄酸酰胺类衍生物纳米混悬剂,制备方法包括如下步骤:
1)将大黄酸酰胺类衍生物溶于有机溶剂中,作为油相;将稳定剂溶于水中,作为水相;
2)将水相置于冰水浴中,搅拌下,逐滴加入油相,分散完全后继续搅拌30-40min,旋蒸除去有机溶剂,在20000rpm条件下,用高速剪切机连续剪切,静置,消泡后,得到粗混悬剂;
3)将粗混悬剂经超声法和/或高压均质法处理,得大黄酸酰胺类衍生物纳米混悬剂。
上述的一种大黄酸酰胺类衍生物纳米混悬剂,所述的大黄酸酰胺类衍生物具有通式(Ⅰ)的结构,
其中,R为L-丙氨酸、L-缬氨酸、L-亮氨酸、L-异亮氨酸、L-脯氨酸、L-苯丙氨酸、L-色氨酸、L-蛋氨酸、L-甘氨酸、L-丝氨酸、L-苏氨酸、L-半胱氨酸、L-酪氨酸、L-天冬酰胺、L-谷氨酰胺、L-赖氨酸、L-组氨酸、L-天冬氨酸、L-谷氨酸。优选的,R为L-异亮氨酸或L-苯丙氨酸。
上述的一种大黄酸酰胺类衍生物纳米混悬剂,按重量比,大黄酸酰胺类衍生物:稳定剂=1:0.3-1。优选的,大黄酸酰胺类衍生物:稳定剂=1:1。
上述的一种大黄酸酰胺类衍生物纳米混悬剂,所述的有机溶剂选自甲醇、无水乙醇、乙酸乙酯、氯仿、丙酮、DMSO或DMF中的一种或二种以上的混合。优选的,有机溶剂是甲醇,其用量控制在,2~8mL/50毫克大黄酸酰胺类衍生物,更优选的,甲醇用量为4mL。
上述的一种大黄酸酰胺类衍生物纳米混悬剂,所述的稳定剂选自泊洛沙姆188(F68)、卵磷脂(LC)、聚维酮(PVP)或羟丙基纤维素(HPC)中的一种或二种以上的混合。优选的,稳定剂是,按重量比,泊洛沙姆188:卵磷脂=1:1的混合。
上述的一种大黄酸酰胺类衍生物纳米混悬剂,将油相匀速逐滴加入水相中,匀速是指0.5~2.0mL/min,优选1.0mL/min。
上述的冻干保护剂选自甘露醇、乳糖、蔗糖、海藻糖、麦芽糖中的一种或几种的组合,优选甘露醇。其用量控制在1~10%,优选5%。
上述的一种大黄酸酰胺类衍生物纳米混悬剂,所述的超声法是:将粗混悬剂转移至超声波细胞粉碎机中,于超声功率135~360W下,探头超声时间控制在2~8min。优选的,超声功率为315W,探头超声时间控制在4min
上述的一种大黄酸酰胺类衍生物纳米混悬剂,所述的高压均质法是:将粗混悬剂转移至高压微射流机中,4660psi均质3次后,于6990~16310psi压力下均质5~25次。优选的,4660psi均质3次后,于11650psi压力下均质20次。
本发明在对粗混悬剂进行处理时,可以单一的选择超声法或高压均质法,也可以两者同时使用。
为了方便长期储存及动物试验,上述的一种大黄酸酰胺类衍生物纳米混悬剂,包括冻干步骤,所述的冻干步骤是:于大黄酸酰胺类衍生物纳米混悬剂中,加入冻干保护剂,完全溶解后,在-80℃条件下预冻48h后取出,放入冷冻干燥机中冻干,得纳米混悬剂冻干粉。此冻干粉可进一步加工制备成胶囊剂、片剂、注射剂等。
上述的一种大黄酸酰胺类衍生物纳米混悬剂,所述的冻干保护剂选自甘露醇、乳糖、蔗糖、海藻糖、麦芽糖中的一种或二种以上的组合。
本发明提供的大黄酸酰胺类衍生物纳米混悬剂在制备治疗抗肿瘤药物中的应用。
本发明提供的大黄酸酰胺类衍生物的合成方法如下:
1)大黄酰氨基酸甲酯的合成:称取大黄酸(RH)于茄形瓶中,加入过量羧基保护的氨基酸甲酯盐酸盐(或二乙酯盐酸盐),RH与羧基保护的氨基酸甲酯盐酸盐(或二乙酯盐酸盐)摩尔比控制在1:1~1:5之间,置于磁力搅拌上,继续加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC·HCl),1-羟基苯并三氮唑(HOBt),沿壁加入三乙胺,加入二氯甲烷。室温搅拌反应,TLC(氯仿做展开剂)监测反应进程。6h反应完毕,将上述反应液倒入分液漏斗中,缓慢加入18%盐酸溶液萃取三次,收集二氯甲烷层液体,二氯甲烷相用饱和NaHCO3水溶液反复萃取至上层无色,二氯甲烷相无水硫酸钠干燥,过滤,滤液减压蒸发浓缩,即得大黄酰氨基酸甲酯。
2)大黄酸酰胺类衍生物的合成:向上述大黄酰氨基酸甲酯中加入适量1M NaOH溶液,超声溶解后,45℃搅拌反应,TLC(氯仿做展开剂)监测反应,1h水解完全,过滤,收集滤液。将反应液置于0℃冰水浴中,搅拌下逐滴加入10%盐酸溶液,调节pH 1~2,静置过夜,析出固体,抽滤,收集滤饼,干燥,即得大黄酸酰胺类衍生物。
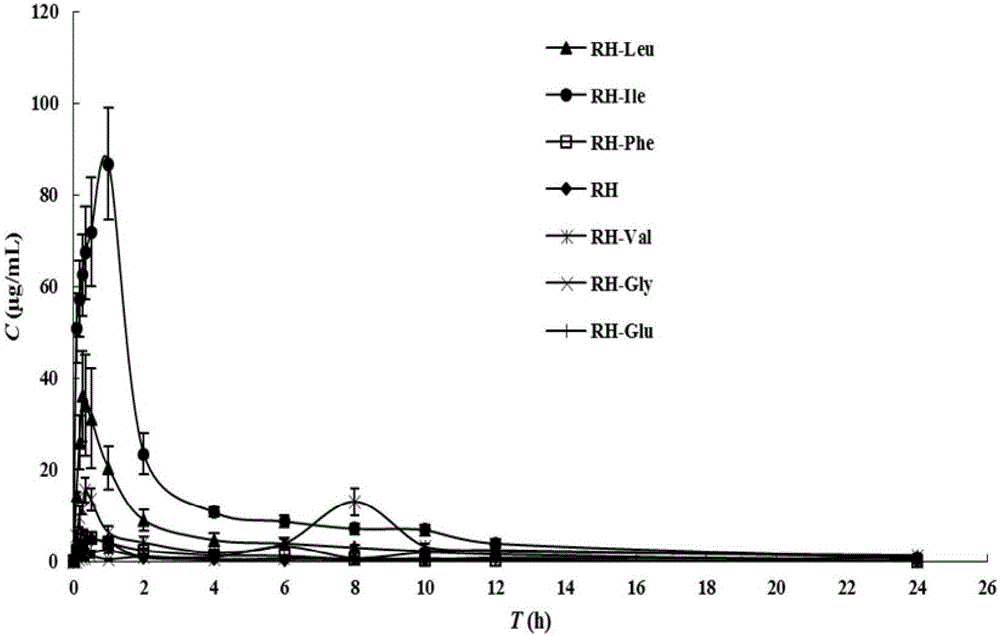
本发明采用高效液相(HPLC)法测定了各化合物在不同有机溶剂和不同pH缓冲液中的溶解度(见表1、表2),以及不同pH缓冲液中的油水分配系数(见表3),结果表明有部分化合物的水溶性及脂溶性较RH均有明显提高,油水分配系数也有所提高;采用MTT法(见表4)初步测定各化合物体外活性,结果证明部分化合物对肝癌HepG2细胞有明显的抑制增殖作用;体内药代动力学研究(见图1)结果表明部分衍生物的血药浓度较同摩尔量的RH有明显提高。综合比较各化合物以上指标,优选出两种最优化合物大黄酰异亮氨酸(Rhein-isoleucine,RH-Ile)和大黄酰苯丙氨酸(Rhein-phenylalanine,RH-Phe),进行深入研究。
表1部分化合物在不同溶剂中的平衡溶解度(Mean±SD,n=3)
表2部分化合物在不同pH缓冲液中的平衡溶解度(Mean±SD,n=3)
表3部分化合物在不同溶剂中的表观油水分配系数(Mean±SD,n=3)
表4部分化合物对肝癌HepG2细胞的增殖抑制作用
注:“—”表示低浓度促进,高浓度抑制,有明显地促进增殖作用。
本发明的优点是:本发明,制备了一系列大黄酸酰胺类衍生物纳米混悬剂,制备方法简单可行,产物产率和纯度较高。本发明,对制备方法进行了选择比较,并对影响制备工艺的重要因素进行了考察,以粒径、Zeta电位和多分散系数(PDI)为指标确定最优的处方工艺,通过比较制剂和原化合物的药动学参数,结果表明,制备成制剂后,RH-Ile和RH-Phe的生物利用度分别提高了2倍和3.5倍左右,半衰期分别延长了3.45h和6.37h,具有缓释效果。本发明首次将大黄酸的结构改造产物制备成了纳米混悬剂,并探讨了制备成制剂后的体内药代动力学,为大黄酸类结构改造产物的临床应用奠定了基础。
附图说明
图1:大鼠口服给予RH及大黄酸酰胺类衍生物后平均血药浓度-时间曲线(n=6)。
图2:粒径及Zeta电位图。
其中,A:RH-Ile-NS粒径图;B:RH-Ile-NS的Zeta电位图;C:RH-Ile-NS冻干粉粒径图;D:RH-Ile-NS冻干粉Zeta电位图。
图3:粒径及Zeta电位图。
其中,A:RH-Phe-NS粒径图;B:RH-Phe-NS的Zeta电位图;C:RH-Phe-NS冻干粉粒径图;D:RH-Phe-NS冻干粉Zeta电位图。
图4:纳米混悬剂冻干粉TEM图。
其中,A:RH-Ile-NS;B:RH-Phe-NS。
图5:差示量热扫描图谱结果。
图6:大鼠口服给予RH-Ile-NS后平均血药浓度-时间曲线(n=6)。
图7:大鼠口服给予RH-Phe-NS后平均血药浓度-时间曲线(n=6)。
具体实施方式
大黄酰苯丙氨酸(Rhein-phenylalanine,RH-Phe)的制备:称取RH 1.76mmol于100mL茄形瓶中,加入3.52mmol L-苯丙氨酸甲酯盐酸盐,置于磁力搅拌上,继续加入EDC·HCl 2.50mmol,HOBt 1.85mmol,沿壁加入三乙胺1mL,加入二氯甲烷10~20mL。室温搅拌反应,TLC(氯仿做展开剂)监测反应进程。6h反应完毕,将上述反应液倒入分液漏斗中,缓慢加入18%盐酸溶液萃取三次,收集二氯甲烷层液体,二氯甲烷相用饱和NaHCO3水溶液反复萃取至上层无色,二氯甲烷相无水硫酸钠干燥,过滤,滤液减压蒸发浓缩,即得大黄酰苯丙氨酸甲酯。向上述大黄酰苯丙氨酸甲酯中加入适量1M NaOH溶液,超声溶解后,45℃搅拌反应,TLC(氯仿做展开剂)监测反应,1h水解完全,过滤,收集滤液。将反应液置于0℃冰水浴中,搅拌下逐滴加入10%盐酸溶液,调节pH 1~2,静置过夜,析出固体,抽滤,收集滤饼,干燥,即得大黄酰苯丙氨酸。
RH-Phe,C24H17NO7,黄棕色粉末状固体,产率76%。1H-NMR(400MHz,DMSO-d6/TMS):δ11.98(2H,d,J=4.9Hz,1-OH和8-OH),9.20(1H,d,J=8.2Hz,2’-H),8.06(1H,s,4-H),7.77-7.67(3H,m,5,6,7-H),7.36-7.27(5H,m,7’,8’,9’,10’,11’-H),7.18(1H,d,J=7.2Hz,2-H),4.71-4.64(1H,s,3’-H),3.26-3.11(2H,m,5’-H)。IR(KBr):3304.06(缔合OH),1728.22(s,COOH),1676.14(酰胺羰基),1635.64(醌羰基),1610.56,1564.27,1475.54,1452.40,1373.32,1298.09,1261.45,1201.65,1157.29,748.38,717.52,704.02(苯环)cm-1。MS(ESI)m/z(%):432.1[M+H]+,454.0[M+Na]+,表明化合物分子量为431,与RH-Phe分子量相符。
大黄酰异亮氨酸(Rhein-isoleucine,RH-Ile)的制备:称取RH 1.76mmol于100mL茄形瓶中,加入3.52mmol L-异亮氨酸甲酯盐酸盐,置于磁力搅拌上,继续加入EDC·HCl2.50mmol,HOBt 1.85mmol,沿壁加入三乙胺1mL,加入二氯甲烷10~20mL。室温搅拌反应,TLC(氯仿做展开剂)监测反应进程。6h反应完毕,将上述反应液倒入分液漏斗中,缓慢加入18%盐酸溶液萃取三次,收集二氯甲烷层液体,二氯甲烷相用饱和NaHCO3水溶液反复萃取至上层无色,二氯甲烷相无水硫酸钠干燥,过滤,滤液减压蒸发浓缩,即得大黄酰异亮氨酸甲酯。向上述大黄酰异亮氨酸甲酯中加入适量1M NaOH溶液,超声溶解后,45℃搅拌反应,TLC(氯仿做展开剂)监测反应,1h水解完全,过滤,收集滤液。将反应液置于0℃冰水浴中,搅拌下逐滴加入10%盐酸溶液,调节pH 1~2,静置过夜,析出固体,抽滤,收集滤饼,干燥,即得大黄酰异亮氨酸。
RH-Ile,C21H19NO7,黄色粉末状固体,产率78%。1H-NMR(400MHz,DMSO-d6/TMS):δ12.72(1H,s,4’-COOH),11.94(2H,s,1-OH和8-OH),9.00(1H,d,J=7.9Hz,2’-H),8.16(1H,s,4-H),7.85-7.77(3H,m,5,6,7-H),7.43(1H,d,J=9.4Hz,2-H),4.36(1H,t,J=7.5Hz,3’-H),1.99(1H,s,5’-H),1.58-1.24(2H,m,7’-H),1.03-0.79(6H,m,6’-H和8’-H)。IR(KBr):3421.72(宽,w),3288.63(缔合OH),1718.58(s,COOH),1674.21(酰胺羰基),1645.28(醌羰基),1629.85,1533.41,1477.47,1454.33,1379.10,1276.88,1201.65,1157.29,752.24,704.02(苯环)cm-1。MS(ESI)m/z(%):398.1[M+H]+,420.1[M+Na]+,表明化合物分子量为397,与RH-Ile分子量相符。
实施例1超声法和高压均质法对纳米混悬剂的影响
(一)大黄酰异亮氨酸纳米混悬剂(RH-Ile-NS)
1)沉淀-超声法(Precipitation-ultrasonic method,PU method):称取RH-Ile 50mg,用甲醇4mL溶解作为油相备用。称取泊洛沙姆188(Poloxamer,F68)25mg,卵磷脂(Lecithin,LC)25mg于100mL烧杯中,加入纯净水40mL,超声溶解作为水相。将水相置于冰水浴中,边搅拌边匀速(1.0mL/min)逐滴加入油相,分散完全后继续搅拌30min,旋蒸除去甲醇。在20000rpm条件下,用高速剪切机剪切3min,静置10min,消泡后,转移至超声波细胞粉碎机中,在超声功率315W条件下,探头超声4min(脉冲开2s停2s),即得。
2)沉淀-高压均质法(High pressure homogenization method,HPH method):称取RH-Ile50mg,用甲醇4mL溶解作为油相备用。称取F68 25mg,LC 25mg于100mL烧杯中,加入纯净水40mL,超声溶解作为水相。将水相置于冰水浴中,边搅拌边匀速(1.0mL/min)逐滴加入油相,分散完全后继续搅拌30min,旋蒸除去甲醇。在20000rpm条件下,用高速剪切机剪切3min,静置10min,消泡后,转移至高压微射流机中,4660psi均质3次,11650psi均质20次,即得。
(二)大黄酰苯丙氨酸纳米混悬剂(RH-Phe-NS)
1)沉淀-超声法(Precipitation-ultrasonic method,PU method):方法同大黄酰异亮氨酸纳米混悬剂的沉淀-超声法。
2)沉淀-高压均质法(High pressure homogenization method,HPH method):方法同大黄酰异亮氨酸纳米混悬剂的沉淀-高压均质法。
(三)测定粒径和Zeta电位
采用激光粒度仪测定RH-Ile-NS和RH-Phe-NS的粒径和Zeta电位分布,结果如表5。
表5超声法和高压均质法对粒径、Zeta电位和PDI的影响(Mean±SD,n=3)
由表5可见,沉淀-高压均质法制备的纳米混悬剂粒径更小且较稳定、均一,因此优选高压均质法。
实施例2均质压力对纳米混悬剂的影响
分别称取RH-Ile和RH-Phe 50mg,用甲醇4mL溶解作为油相备用。称取F68 25mg,LC 25mg于100mL烧杯中,加入纯净水40mL,超声溶解作为水相。将水相置于冰水浴中,边搅拌边匀速逐滴加入油相,分散完全后继续搅拌30min,旋蒸除去甲醇。在20000rpm条件下,用高速剪切机剪切3min,静置10min,消泡后,转移至高压微射流机中,4660psi均质3次后,分别于6990psi、9320psi、11650psi、13980psi、16310psi条件下均质20次,分别得到RH-Ile-NS和RH-Phe-NS。测定粒径和Zeta电位,结果如表6和表7。
表6均质压力对RH-Ile-NS粒径、Zeta电位和PDI的影响(Mean±SD,n=3)
表7均质压力对RH-Phe-NS粒径、Zeta电位和PDI的影响(Mean±SD,n=3)
由表6和表7可见,对于RH-Ile-NS,随着均质压力的增大,Zeta电位几乎没有变化,说明Zeta电位与均质压力无关,而粒径和PDI则表现出递减趋势,在13980psi时粒径最小,当压力继续增加时,粒径反而变大。当PDI<0.3时,认为粒径相对比较均一,当均质压力≥11650psi时,PDI均符合要求,其中,均质压力在16310psi时,PDI最小,而研究发现均质压力在11650psi和13980psi时粒径相差不大,考虑到仪器的损耗,优选,均质压力11650psi。对于RH-Phe-NS,随着均质压力增大,Zeta电位同样没有明显变化,粒径表现出递减趋势,均质压力16310psi时粒径最小,但研究发现PDI只有在均质压力11650psi时符合要求,这可能是由于均质压力过大时,会导致体系中小粒径部分变得更小,因此PDI变大,体系粒径差异较大,综合以上原因,最终选择均质压力11650psi。
实施例3均质次数对混悬剂的影响
分别称取RH-Ile和RH-Phe 50mg,用甲醇4mL溶解作为油相备用。另称取F68 25mg,LC 25mg于100mL烧杯中,加入纯净水40mL,超声溶解作为水相。将水相置于冰水浴中,边搅拌边匀速逐滴加入油相,分散完全后继续搅拌30min,旋蒸除去甲醇。在20000rpm条件下,用高速剪切机剪切3min,静置10min消泡后,转移至高压微射流中,4660psi均质3次后,于11650psi压力下分别均质5次、10次、15次、20次、25次,分别得到RH-Ile-NS和RH-Phe-NS。测定粒径和Zeta电位,结果如表8和表9。
表8均质次数对RH-Ile-NS粒径、Zeta电位和PDI的影响(Mean±SD,n=3)
表9均质次数对RH-Phe-NS粒径、Zeta电位和PDI的影响(Mean±SD,n=3)
由表8和表9可见,对于RH-Ile-NS,随着均质次数的增加,制剂的粒径表现出先减小后增大的趋势,在均质20次时粒径最小,Zeta电位同样没有明显变化,PDI则呈现出递减趋势,均质25次时PDI最小,但在均质次数≥20次时PDI均小于0.3,综合考虑,优选均质20次。对于RH-Phe-NS,随着均质次数增加,粒径同样呈现出先减小后增大的趋势,在20次时粒径最小,Zeta电位没有明显变化,但总体比RH-Ile-NS的Zeta电位更小,表明RH-Phe-NS的体系可能更稳定。PDI随着均质次数增加先减小后增大,在均质20次时达到最小值,综合考虑,优选选择均质20次。
实施例4稳定剂对混悬剂的影响
分别称取RH-Ile和RH-Phe 50mg,用甲醇4mL溶解作为油相备用。按表10和表11称取稳定剂50mg(两种稳定剂则各25mg)于100mL烧杯中,加入纯净水40mL,超声溶解作为水相。将水相置于冰水浴中,边搅拌边匀速逐滴加入油相,分散完全后继续搅拌30min,旋蒸除去甲醇。在20000rpm条件下,用高速剪切机剪切3min,静置10min消泡后,转移至高压微射流机中,4660psi均质3次后,于11650psi压力下分别均质20次,即得RH-Ile-NS和RH-Phe-NS。测定粒径和Zeta电位,结果如表10和表11。
表10 RH-Ile-NS处方考察(Mean±SD,n=3)
表11 RH-Phe-NS处方考察(Mean±SD,n=3)
由表10和表11可见,采用两种稳定剂混合制得的纳米混悬剂粒径相对更小,更稳定一些。对于RH-Ile-NS,同时选用F68和LC作为稳定剂时,得到的纳米混悬剂粒径最小,Zeta电位相对稳定,PDI<0.3,符合要求;对于RH-Phe-NS,粒径整体相对于RH-Ile-NS更小,同时选用F68和LC时粒径同样表现出最小,Zeta电位绝对值相比于RH-Ile-NS更大,表示体系更稳定些,PDI同样符合要求,因此选用F68和LC作为稳定剂。
实施例5 F68和LC的比例对混悬剂的影响
分别称取RH-Ile和RH-Phe 50mg,用甲醇4mL溶解作为油相备用。按表12和表13称取F68和LC于100mL烧杯中,加入纯净水40mL,超声溶解作为水相。将水相置于冰水浴中,边搅拌边匀速逐滴加入油相,分散完全后继续搅拌30min,旋蒸除去甲醇。在20000rpm条件下,用高速剪切机剪切3min,静置10min消泡后,转移至高压微射流中,4660psi均质3次后,于11650psi压力下分别均质20次,即得RH-Ile-NS和RH-Phe-NS。测定粒径和Zeta电位,结果如12和表13。
表12 RH-Ile-NS处方考察(Mean±SD,n=3)
表13 RH-Phe-NS处方考察(Mean±SD,n=3)
由表12和表13可见,分别考察了稳定剂F68和LC在15~30mg范围内的用量及二者的比例,结果发现,LC的用量比F68的用量对粒径的影响更大一些,这可能是因为LC的稳定作用更强,可以使化合物粒子分散地更均匀稳定一些。研究表明,当Zeta电位的绝对值大于20mV时,纳米混悬剂具有较好的稳定性。当F68和LC都为25mg时,粒径最小,Zeta电位符合要求,且PDI也均小于0.3,因此最终选择F68 25mg和LC 25mg作为稳定剂。
实施例6大黄酰异亮氨酸纳米混悬剂冻干粉和大黄酰苯丙氨酸纳米混悬剂冻干粉的制备
称取适量甘露醇于西林瓶中,分别加入2mL RH-Ile-NS和RH-Phe-NS,轻晃至甘露醇完全溶解后,放入超低温冰箱,在-80℃条件下预冻48h后取出,放入冷冻干燥机中冻干,通过考察冻干粉的外观、再分散性以及粒径确定最终处方,冻干粉复溶后粒径变化情况见表14。
表14冻干保护剂对粒径的影响(Mean±SD,n=3)
表14表明,不添加冻干保护剂直接冻干后,制剂紧贴在西林瓶壁上,加入纯净水超声复溶,不能够完全分散,粒径也最大;添加2.5%甘露醇冻干后,样品外观有萎缩,部分结块,加入纯净水超声复溶,能够分散均匀,但粒径变化较大;加入5%甘露醇冻干后,样品质地疏松,颜色均一,加入纯净水后超声片刻即可分散均匀,测定复溶后粒径变化较小,稳定性也较好,因此选择加入5%甘露醇作为冻干保护剂制备冻干粉。
采用激光粒度仪测定RH-Ile-NS和RH-Phe-NS的冻干前后粒径及Zeta电位分布,测定结果如图2和图3,结果显示,冻干后二者粒径均增加20~30nm,Zeta电位则无明显变化。
采用透射电镜(TEM)测定RH-Ile-NS冻干粉和RH-Phe-NS冻干粉复溶后粒径,结果如图4,二者粒径均在200nm左右,相差不大,粒子呈球形,彼此之间无黏连,制剂稳定性良好。
采用差示量热扫描仪(DSC)分别测定F68、LC、甘露醇、RH-Ile、RH-Phe、RH-Ile-NS冻干粉、RH-Phe-NS冻干粉以及二者各自的物理混合物,设定温度以10℃/min的速度从25℃升到300℃,测定结果见图5。结果显示,辅料LC不出峰,F68在55.67℃处有吸热峰,冻干保护剂甘露醇在169.5℃处出现吸热峰,RH-Ile的吸热峰在201.33℃,同样在其物理混合物和RH-Ile-NS中均能够看到RH-Ile的吸热峰,以及甘露醇和F68的吸热峰。在RH-Phe中存在一个184℃的放热峰和232.33℃的吸热峰,在其物理混合物和制剂中也同样能够看到,说明RH-Ile和RH-Phe制备成纳米混悬剂后晶型没有发生改变,均为结晶形。
实施例7 RH-Ile-NS和RH-Phe-NS的体内药动学情况
选取SD大鼠为模型动物,分别灌胃给药,分析药时曲线(见图6、图7)和药动学参数,与RH-Ile混悬液和RH-Phe混悬液的大鼠体内药动学参数(见表15)进行比较,从而研究制备成纳米混悬剂后对原料药的口服吸收的促进作用。结果表明,将RH-Ile和RH-Phe制备成纳米混悬剂之后,达到了预期的进一步提高生物利用度的目的,并具有缓释效果,增强了药效。
表15大鼠口服给予大黄酸酰胺类衍生物和制剂后的药动学参数(Mean±SD,n=6)
Note:*P<0.05 compared with rhein-isoleucine(rhein-phenylalanine)suspensions
**P<0.01compared with rhein-isoleucine(rhein-phenylalanine)suspensions。
- 还没有人留言评论。精彩留言会获得点赞!