MDM2抑制剂和其组合的制作方法
本公开涉及药物组合,包括(a)mdm2抑制剂和(b)(i)mek抑制剂和/或(b)(ii)bcl2抑制剂,特别用于治疗癌症。本公开还涉及这类组合在制备治疗癌症药物中的应用;在需要的对象中治疗癌症的方法,包括向所述对象给予联合治疗有效量的所述组合;含这类组合和其商业包装的药物组合物。
发明背景
癌症靶向疗法的出现增加了多种恶性肿瘤患者寿命且有助于在药物抗性机制研究中理解肿瘤的复杂性。对靶向药剂的一般不完全和/或短暂临床反应的情况由多种因素引起,所述因素能大致分成2类:阻碍最优药物给药并因而限制靶标交会的毒性(brana和siu2012,chapman,solit等.2014),和癌症适应并维持其增殖潜能抵御扰动的能力(druker2008,chandarlapaty2012,doebele,pilling等.2012,duncan,whittle等.2012,katayama,shaw等.2012,lito,rosen等.2013,sullivan和flaherty2013,solit和rosen2014)。药物组合能如下应对这两类因素:改善总体功效和同时靶向肿瘤顽强性(robustness)及复杂性以抵消抗性(robert,karaszewska等.2015,turner,ro等.2015)。尚不清楚需要多少药物且需要联合靶向哪些过程以克服癌症。但几乎可以肯定,需要抑制不同通路或驱动因子,最可能需要2种或更多药物(bozic,reiter等.2013)。这由以下的成功支持:联合常规化疗剂以治疗癌症(devita1975),联合疗法用于传染病如hiv(porter,babiker等.2003),以及理论方法显示如何能通过提升干扰级来挑战生物顽强性(lehar,krueger等.2008)。
尽管就特定类型癌症患者而言有许多治疗选择,仍需要有效和安全的联合疗法,其能给药用于有效长期癌症治疗。
技术实现要素:
本公开的一个目的是提供药物以改善癌症治疗,特别是通过抑制细胞生长(增殖)和诱导细胞凋亡来改善癌症治疗。本公开的一个目的是发现新型联合疗法,其选择性协同抑制增殖和/或诱导细胞凋亡。
抑制剂诸如mdm2抑制剂、mek抑制剂和bcl2抑制剂作为单一疗法在体外和体内临床前试验中展现抗增殖(细胞抑制)和促凋亡(细胞毒性)活性。
意外发现包含以下的药物组合
(a)mdm2抑制剂,选自
(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮或其药学上可接受盐,和(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮或其药学上可接受盐;和
(b)
(i)mek抑制剂,选自曲美替尼,6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺,(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺,pd0325901,pd-184352,rdea119,xl518,as-701255,as-701173,as703026,rdea436,e6201,ro4987655,rg7167和rg7420或其药学上可接受盐;
和/或
(ii)bcl2抑制剂,选自abt-737,abt-263(navitoclax)和abt-199或其药学上可接受盐,
具有有益的协同相互作用、提高的抗癌活性、改善的抗增殖效果和改善的促凋亡效果,这些组合在细胞生长抑制和通过凋亡诱导细胞死亡中展现协同效应。
此外,发现以下组合
(a)mdm2抑制剂,选自
(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮或其药学上可接受盐,和(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮或其药学上可接受盐;和
(b)
(i)mek抑制剂,选自曲美替尼,6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺,(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺,pd0325901,pd-184352,rdea119,xl518,as-701255,as-701173,as703026,rdea436,e6201,ro4987655,rg7167和rg7420或其药学上可接受盐;
和/或
(ii)bcl2抑制剂,选自abt-737,abt-263(navitoclax)和abt-199或其药学上可接受盐,
可有利地包括选自egfr抑制剂、pi3k抑制剂和braf抑制剂的其它抑制剂。另外,cdk4/6抑制剂或护理标准如紫杉醇能加至mdm2抑制剂(“mdm2i”)与曲美替尼的组合,其能引起进一步的协同效应或强诱导细胞凋亡。mdm2抑制剂与bcl2抑制剂的组合能补充有braf抑制剂(如达拉菲尼)和cmet抑制剂(如pf-04217903)以形成四重组合。发现后一种组合为弱协同,但强烈诱导细胞凋亡。
另一方面,本公开涉及药物组合物,包括本公开的药物组合和至少一种药学上可接受载体。
一方面,本公开涉及本公开药物组合或药物组合物,其用作药物。
一方面,本公开涉及本公开药物组合或药物组合物,其用于治疗癌症。
另一方面,本公开提供本公开药物组合在制备治疗癌症药物中的应用。
另一方面,本公开涉及在需要的对象中治疗癌症的方法,包括向对象给予治疗有效量的本公开药物组合或本公开药物组合物。
特别地,本公开分别单独或联合提供下列方面、有利特征和特定实施方式,如以下权利要求所列。
附图简要说明
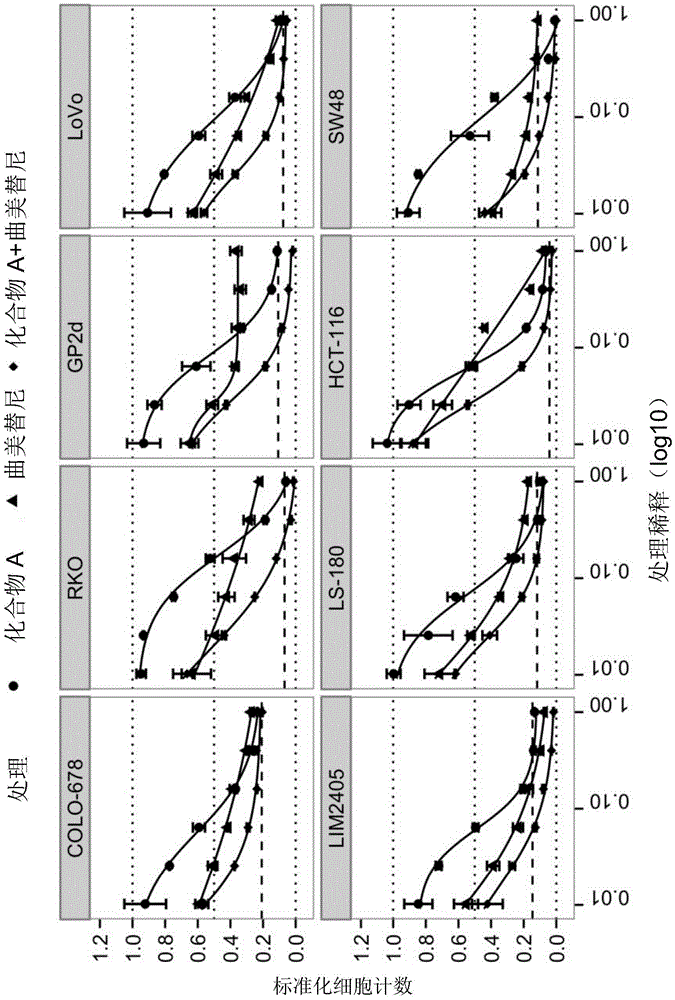
图18个tp53野生型结直肠癌细胞系的剂量-反应曲线,使用mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)(圆形)和mek抑制剂曲美替尼(三角形)以及其组合(菱形)。x轴表示log10(处理稀释);y轴表示治疗后相对于dmso的细胞计数。组合产生自单一药剂的固定比例(1:1)组合。强虚线表示治疗开始前的细胞数(‘基线’)。
图28个tp53野生型结直肠癌细胞系的剂量-反应曲线,使用mdm2抑制剂(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮(化合物b)(圆形)和mek抑制剂曲美替尼(三角形)以及其组合(菱形)。x轴表示log10(处理稀释);y轴表示治疗后相对于dmso的细胞计数。组合产生自单一药剂的固定比例(1:1)组合。强虚线表示治疗开始前的细胞数(‘基线’)。
图38个tp53野生型结直肠癌细胞系中mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)或mdm2抑制剂(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮(化合物b)(y轴)与mek抑制剂曲美替尼(x轴)的组合在75%抑制水平的等效应图分析。对角曲线上的点表示累加效应,其右边的点表示拮抗作用,其左边的点表示协同作用。空心圆显示联合指数最低(协同作用最强)的组合(见表2的值)。
图4显示mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼和其组合在5个tp53野生型结直肠癌细胞系中并在24小时、48小时和72小时后(不同色度的灰色)的最大半胱天冬酶3/7诱导。x轴表示处理;y轴表示各处理所见的最大半胱天冬酶3/7诱导(%细胞)。
图5mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)与mek抑制剂曲美替尼的单一药剂和组合的长期集落形成试验。“化合物a(l)”:0.33μm;“化合物a(h)”:1μm;“曲美替尼(l)”用于lim2405和sw48以外的所有细胞系:4nm;“曲美替尼(h)”用于lim2405和sw48以外的所有细胞系:12nm;“曲美替尼(l)”用于lim2405和sw48:1nm,“曲美替尼(h)”用于lim2405和sw48:3nm。(a)结晶紫染色后的代表性细胞图像。(b)定量来自(a)的结晶紫信号。柱显示均值±标准偏差,n=3个重复。显著性检验参见表3。rfu=相对荧光单位。
图6mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼和其组合在24小时处理后的facs分析。“化合物a(l)”:0.33μm;“化合物a(h)”:1μm;“曲美替尼(l)”用于lim2405和sw48以外的所有细胞系:4nm;“曲美替尼(h)”用于lim2405和sw48以外的所有细胞系:12nm;“曲美替尼(l)”用于lim2405和sw48:1nm,“曲美替尼(h)”用于lim2405和sw48:3nm。堆叠柱表示处于各细胞周期时相:subg1、g1、s和g2的细胞群百分数。
图7mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼和其组合在24小时处理后的蛋白质印迹分析。“化合物a(l)”:0.33μm;“化合物a(h)”:1μm;“曲美替尼(l)”用于lim2405和sw48以外的所有细胞系:4nm;“曲美替尼(h)”用于lim2405和sw48以外的所有细胞系:12nm;“曲美替尼(l)”用于lim2405和sw48:1nm,“曲美替尼(h)”用于lim2405和sw48:3nm。
图8mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼和其组合在10小时处理后的5个靶基因qrt-pcr分析。“化合物a(l)”:0.33μm;“化合物a(h)”:1μm;“曲美替尼(l)”用于lim2405和sw48以外的所有细胞系:4nm;“曲美替尼(h)”用于lim2405和sw48以外的所有细胞系:12nm;“曲美替尼(l)”用于lim2405和sw48:1nm,“曲美替尼(h)”用于lim2405和sw48:3nm。柱显示相较dmso处理的log2尺度差异表达,误差线显示n=2次重复的标准偏差。
图9mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)(圆形)、mek抑制剂曲美替尼(化合物b,三角形)、bcl-2/-xl抑制剂navitoclax(abt-263)(化合物c,菱形)以及其组合a+b(圆形,点虚线)、a+c(三角形)、b+c(菱形)和a+b+c(圆形,实线)在5个tp53野生型结直肠癌细胞系中的剂量-反应曲线。x轴表示log10(治疗处理);y轴表示处理后相对于dmso的细胞计数。强虚线表示处理开始前的细胞数(‘基线’)。
图10mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼(b)、bcl-2/-xl抑制剂navitoclax(abt-263)(c)及其组合a+b、a+c、b+c和a+b+c在5个tp53野生型结直肠癌细胞系中且在24小时、48小时和72小时后(不同色度的灰色)的最大半胱天冬酶3/7诱导。x轴表示处理;y轴表示各处理所见的最大半胱天冬酶3/7诱导(%细胞)。
图11mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)(圆形)、mek抑制剂曲美替尼(化合物b,三角形)、egfr抑制剂埃罗替尼(化合物c,菱形)以及其组合a+b(圆形,点虚线)、a+c(三角形)、b+c(菱形)和a+b+c(圆形,实线)在5个tp53野生型结直肠癌细胞系中的剂量-反应曲线。x轴表示log10(处理稀释);y轴表示处理后相对于dmso的细胞计数。强虚线表示处理开始前的细胞数(‘基线’)。
图12mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼(化合物b)、egfr抑制剂埃罗替尼(化合物c)及其组合a+b、a+c、b+c和a+b+c在5个tp53野生型结直肠癌细胞系中且在24小时、48小时和72小时后(不同色度的灰色)的最大半胱天冬酶3/7诱导。x轴表示处理;y轴表示各处理所见的最大半胱天冬酶3/7诱导(%细胞)。
图13pik3ca抑制剂(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)(化合物a)(圆形)、mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物b)(三角形)、bcl-2/-xl抑制剂navitoclax(abt-263)(化合物c)(菱形)以及其组合a+b(圆形,点虚线)、a+c(三角形)、b+c(菱形)和a+b+c(圆形,实线)在5个tp53野生型结直肠癌细胞系中的剂量-反应曲线。x轴表示log10(处理稀释);y轴表示处理后相对于dmso的细胞计数。强虚线表示治疗开始前的细胞数(‘基线’)。
图14pik3ca抑制剂即(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)(化合物a)、mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物b)、bcl-2/-xl抑制剂navitoclax(abt-263)(化合物c)、a+b、a+c、b+c和a+b+c在5个tp53野生型结直肠癌细胞系中且在24小时、48小时和72小时后(不同色度的灰色)的最大半胱天冬酶3/7诱导。x轴表示处理;y轴表示各处理所见的最大半胱天冬酶3/7诱导(%细胞)。
图15mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)(圆形)、bcl-2/-xl抑制剂navitoclax(abt-263)(三角形)以及化合物a与abt-263组合(菱形)在5个tp53野生型结直肠癌细胞系中的剂量-反应曲线。x轴表示log10(处理稀释);y轴表示处理后相对于dmso的细胞计数。强虚线表示处理开始前的细胞数(‘基线’)。
图16化合物a和abt-263及mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)与bcl-2/-xl抑制剂navitoclax(abt-263)组合在5个tp53野生型结直肠癌细胞系中且在24小时、48小时和72小时后(不同色度的灰色)的最大半胱天冬酶3/7诱导。x轴表示处理;y轴表示各处理所见的最大半胱天冬酶3/7诱导(%细胞)。
图17kras突变hct-116异种移植用mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼(化合物b)和bcl-2/-xl抑制剂abt-263(化合物c)或其组合处理。特别地,所述异种移植用载剂(g1)、abt-263(g2,每日100mg/kg)、化合物a(g3,每周三次100mg/kg)、曲美替尼(g4,每日0.3mg/kg)、化合物a与曲美替尼组合(g5)、或所有三种药剂的组合(g6)处理。在第9天,向g3-g5加入abt-263。显示相对于初始肿瘤体积的平均肿瘤体积变化百分比。误差线代表sem。
图18瀑布图显示9天治疗(a)和19天治疗(b)(依序加入abt-263后10天)后,g3-g6组中个体肿瘤的肿瘤体积变化百分比(相对于初始体积)(如实施例10和图17所述)。
发明详述
一方面,本公开涉及药物组合,包括:
(a)mdm2抑制剂,选自
(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮或其药学上可接受盐,和(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮或其药学上可接受盐;和
(b)
(i)mek抑制剂,选自曲美替尼,6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺,(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺,pd0325901,pd-184352,rdea119,xl518,as-701255,as-701173,as703026,rdea436,e6201,ro4987655,rg7167和rg7420或其药学上可接受盐;
和/或
(ii)bcl2抑制剂,选自abt-737,abt-263(navitoclax)和abt-199或其药学上可接受盐。
已确定所述组合能用于有效治疗癌症。具体地,已确定由于在抑制细胞生长和/或诱导细胞死亡中的协同效应,该组合能用于有效治疗癌症。因此,本公开的组合特别是三重和进一步的组合,可使“细胞抑制”反应转变为“细胞毒性”反应,从而实现癌症消退。
术语“一个”和“一种”和“所述”以及描述本公开上下文中的类似提法(特别是下列权利要求的上下文中)应解释为涵盖单数和复数,除非本文另有说明或与上下文明显矛盾。复数用于化合物、患者、癌症等时,也指单一化合物、患者等。
本文所用的术语“协同效应”指2种或3种治疗剂如式(i)化合物例如化合物a和本公开的至少一种mek抑制剂化合物例如化合物a与本公开的至少一种bcl2抑制剂化合物作用以产生效果,例如减缓增生性疾病特别是癌症或其症状的进展,该效果大于所给予各药物自身所得效果的简单叠加。协同效应能够算出,例如用合适方法,如sigmoid-emax方程(holford,n.h.g.和scheiner,l.b.,clin.pharmacokinet.6:429-453(1981))、loewe叠加性方程(loewe,s.和muischnek,h.,arch.exp.patholpharmacol.114:313-326(1926))和中效方程(chou,t.c.和talalay,p.,adv.enzymeregul.22:27-55(1984))。上面所提各方程能应用于实验数据以产生对应图,从而协助评价药物组合效果。与上面所提方程相关的对应图分别是浓度-效应曲线、等效应图曲线和联合指数曲线。
具体地,已证明相较各单一药剂,在tp53野生型结直肠癌中联合抑制mdm2和mek可提供改善(实施例1,图1和2,表2)和更持久的反应(实施例1,图5,表3)。同样,相较单一药剂,在tp53野生型结直肠癌中联合抑制mdm2和bcl2显示更强的细胞凋亡诱导(实施例5,图16)。更进一步,mdm2抑制剂、mek抑制剂和bcl2抑制剂的三重组合在2/5所测试tp53野生型结直肠癌细胞模型中相较药物对引起协同抑制(实施例2,表5),三重组合在4个这些细胞系中相较成对组合显示更强的细胞凋亡(实施例2,图10)。因此,本公开的组合提供相较各单一药剂能改善反应并能引起临床更持久反应的有效治疗选项。
本文所用的术语“mdm2抑制剂”或“hdm2抑制剂”或“mdm2抑制剂”指抑制hdm2/p53(mdm2/p53)相互作用关联的任何化合物。hdm2(鼠双微体2的人同源物)是p53的负调节物。mdm2抑制剂用于人或兽用的药物组合物,用于需要抑制mdm2/p53关联的情形,例如在治疗肿瘤和/或癌细胞生长中。具体地,mdm2抑制剂用于治疗人类癌症,因为这些癌症的发展可能至少部分依赖于压过p53的“看门人”功能,例如mdm2过表达。
根据本公开,mdm2抑制剂是选自下组的化合物:
(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮或其药学上可接受盐,和
(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮或其药学上可接受盐。
mdm2抑制剂可以是(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮或其药学上可接受盐。mdm2抑制剂(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮属于新类型的咪唑并吡咯烷酮化合物,并显示有效抑制mdm2/p53相互作用(此术语特别包括hdm2/p53相互作用)。具体地,此化合物通过结合mdm2而用作mdm2与p53相互作用的抑制剂。mdm2抑制剂(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮是本公开所述最优选的mdm2i抑制剂,是式i的化合物并描述于wo2013/111105的实施例102,所述专利通过引用全文纳入本文:
(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮的结晶形式描述于wo2013/111105的ex6、ex7和ex8。本公开涵盖(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮化合物的琥珀酸共晶。所述化合物也能采用乙醇溶剂合物的形式。
mdm2抑制剂还可以是(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮或其药学上可接受盐。mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮是式ii的化合物并描述于wo2011/076786的实施例106,所述专利通过引用全文纳入本文:
在一个实施方式中,(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮的药学上可接受盐是硫酸氢盐。(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮的硫酸氢盐结晶形式描述于wo2012/066095。
本文定义的术语“mek抑制剂”指靶向、减少或抑制map激酶即mek的激酶活性的化合物。mek抑制剂的靶标包括但不限于erk。mek抑制剂的间接靶标包括但不限于细胞周期素d1。
本公开的药物组合能包括选自下组的至少一种mek抑制剂:曲美替尼,6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺,(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺,pd0325901,pd-184352,rdea119,xl518,as-701255,as-701173,as703026,rdea436,e6201,ro4987655,rg7167和rg7420或其药学上可接受盐。
优选地,mek抑制剂是曲美替尼(n-(3-{3-环丙基-5-[(2-氟-4-碘苯基)氨基]-6,8-二甲基-2,4,7-三氧代-3,4,6,7-四氢吡啶并[4,3-d]嘧啶-1(2h)-基}苯基)乙酰胺,也称为jpt-74057或gsk1120212。曲美替尼(gsk1120212)描述于pct公开号wo05/121142,所述专利通过引用全文纳入本文。所述化合物作为
根据本公开,本公开组合的另一合适mek抑制剂是式(iii)化合物6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺
mek抑制剂化合物6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺描述于pct申请号wo03/077914,其制备方法描述于例如其中的实施例18。
本公开组合的额外合适mek抑制剂(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺是式(iv)化合物
mek抑制剂化合物(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺描述于pct申请号wo2007/044084的实施例25-bb,其制备方法描述于已在其中描述。
6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺的特别优选盐是盐酸或硫酸盐。适合本公开的6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺和(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺的额外药学上可接受盐包括pct申请号wo03/077914和pct申请号wo2007/044084公开的盐,其都通过引用在此纳入本申请。
可用于本公开组合的额外mek抑制剂包括但不限于pd0325901(辉瑞(pfizer))(参见pct公开号wo02/06213),pd-184352(辉瑞),rdea119(ardea生物科技(ardeabiosciences)),xl518(exelexis),as-701255(默克雪兰诺(merckserono)),as-701173(默克雪兰诺),as703026(默克雪兰诺),rdea436(ardea生物科技,e6201(卫材(eisai))(参见goto等,journalofpharmacologyandexperimentaltherapeutics,3331(2):485-495(2009)),ro4987655(豪夫迈·罗氏公司(hoffmann-laroche)),rg7167和/或rg7420。
本文定义的术语“bcl2抑制剂”或“bcl2抑制剂”或“bcl-2抑制剂”或“bcl-2抑制剂”指靶向、减少或抑制抗凋亡b细胞淋巴瘤-2(bcl-2)家族蛋白(bcl-2,bcl-xl,bcl-w,mcl-1,bfl1/a-1和/或bcl-b)的化合物。
在一个实施方式中,本公开的药物组合包括选自下组的至少一种bcl2抑制剂化合物:abt-737、abt-263(navitoclax)和abt-199。
本公开的特别优选bcl2抑制剂是navitoclax(abt-263)或其药学上可接受盐。navitoclax是bcl-2和相关细胞凋亡抑制因子bcl-xl的选择性高亲和小分子抑制剂(参见c,shoemakerar,adickesj,andersonmg,chenj,jins等.《abt-263:有效和可口服生物利用的bcl-2家族抑制剂》(abt-263:apotentandorallybioavailablebcl-2familyinhibitor).cancerres2008;68:3421–8)。
根据本公开,药物组合可包括mdm2抑制剂和mek抑制剂;或其可包括mdm2抑制剂和bcl2抑制剂。根据本公开,含mdm2抑制剂和mek抑制剂或mdm2和bcl2抑制剂的药物组合还可有利地包括另一抑制剂,其甚至进一步提高组合的抗肿瘤活性。因此,mdm2抑制剂、mek抑制剂和bcl2抑制剂的三重组合在2/5所测试tp53野生型结直肠癌细胞模型中相较药物对引起协同抑制(实施例2,表5),三重组合在4个这些细胞系中相较成对组合显示更强的细胞凋亡(实施例2,图10)。
类似地,本公开的药物组合包括(a)mdm2抑制剂和(b)(i)mek抑制剂和/或(ii)bcl2,还可有利地包括egfr抑制剂。
本文定义的术语“egfr抑制剂”指靶向、减少或抑制受体酪氨酸激酶表皮生长因子家族(egfr、erbb2、erbb3、erbb4作为同或异二聚体)活性或者结合egf或egf相关配体的化合物。
用于本公开组合的egfr抑制剂选自埃罗替尼、吉非替尼、拉帕替尼、卡奈替尼、培利替尼、来那替尼、(r,e)-n-(7-氯-1-(1-(4-(二甲氨基)丁-2-烯酰基)氮杂环庚-3-基)-1h-苯并[d]咪唑-2-基)-2-甲基异烟酰胺、帕尼单抗、马妥珠单抗、帕妥珠单抗、尼妥珠单抗、扎鲁木单抗、埃克替尼、阿法替尼和西妥昔单抗及其药学上可接受盐。
优选地,egfr抑制剂是埃罗替尼或其药学上可接受盐。
在一个实施方式中,含mdm2抑制剂和mek抑制剂的药物组合还可有利地包括egfr抑制剂。意外发现此三重组合相较成对组合显示更强的细胞凋亡(实施例3,图12)。
在一个优选实施方式中,所述药物组合包括mdm2抑制剂,选自(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮或其药学上可接受盐,和(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮或其药学上可接受盐;mek抑制剂曲美替尼或其药学上可接受盐,以及egfr抑制剂埃罗替尼或其药学上可接受盐。
根据本公开,本公开的药物组合包括(a)mdm2抑制剂和(b)(i)mek抑制剂和/或(ii)bcl2,还可有利地包括pi3k抑制剂。
本文定义的术语“磷脂酰肌醇3-激酶抑制剂”或“pi3k抑制剂”指靶向、减少或抑制pi3-激酶的化合物。pi3-激酶活性显示响应一些激素和生长因子刺激而增加,这些因子包括胰岛素、血小板衍生生长因子、胰岛素样生长因子、表皮生长因子、集落刺激因子和肝细胞生长因子,且参与涉及细胞生长和转化的过程。
适合本公开的磷脂酰肌醇3-激酶(pi3k)抑制剂选自2-甲基-2-[4-(3-甲基-2-氧代-8-喹啉-3-基-2,3-二氢-咪唑并[4,5-c]喹啉-1-基)-苯基]-丙腈或其药学上可接受盐,5-(2,6-二吗啉-4-基-嘧啶-4-基)-4-三氟甲基-吡啶-2-基胺或其药学上可接受盐;和(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)或其药学上可接受盐。
wo2006/122806描述咪唑喹啉衍生物,其被描述成抑制pi3k活性。化合物2-甲基-2-[4-(3-甲基-2-氧代-8-喹啉-3-基-2,3-二氢-咪唑并[4,5-c]喹啉-1-基)-苯基]-丙腈具有式(v)的化学结构
所述化合物、其用作pi3k抑制剂的效用以及合成2-甲基-2-[4-(3-甲基-2-氧代-8-喹啉-3-基-2,3-二氢-咪唑并[4,5-c]喹啉-1-基)-苯基]-单甲苯磺酸盐描述于wo2006/122806,所述专利通过引用全文纳入本文,例如分别在实施例7和实施例152-3中。化合物2-甲基-2-[4-(3-甲基-2-氧代-8-喹啉-3-基-2,3-二氢-咪唑并[4,5-c]喹啉-1-基)-苯基]-丙腈可采用游离碱或其任何药学上可接受盐形式存在。优选地,2-甲基-2-[4-(3-甲基-2-氧代-8-喹啉-3-基-2,3-二氢-咪唑并[4,5-c]喹啉-1-基)-苯基]-丙腈以其单甲苯磺酸盐形式存在。
wo07/084786描述特定嘧啶衍生物,发现其抑制pi3k活性。化合物5-(2,6-二吗啉-4-基-嘧啶-4-基)-4-三氟甲基-吡啶-2-基胺具有式(vi)的化学结构
所述化合物、其盐、其用作pi3k抑制剂的效用以及合成化合物5-(2,6-二吗啉-4-基-嘧啶-4-基)-4-三氟甲基-吡啶-2-基胺描述于wo2007/084786,所述专利通过引用全文纳入本文,例如在实施例10中。化合物5-(2,6-二吗啉-4-基-嘧啶-4-基)-4-三氟甲基-吡啶-2-基胺可采用游离碱或其任何药学上可接受盐形式存在。优选地,5-(2,6-二吗啉-4-基-嘧啶-4-基)-4-三氟甲基-吡啶-2-基胺采用其盐酸盐形式存在。
wo2010/029082描述特定2-甲酰胺环氨基尿素衍生物,发现其对α-同种型pi3k有高选择性并能加入本公开组合。化合物(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)具有式(vii)的化学结构
所述化合物、其盐、其用作α-同种型选择性pi3k抑制剂的效用以及合成化合物(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)描述于wo2010/029082,所述专利通过引用全文纳入本文,例如在实施例15中。化合物(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)可采用游离碱或其任何药学上可接受盐形式存在。优选地,(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)采用其游离碱形式存在。
优选地,用于本公开组合的pi3k抑制剂化合物是(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)或其任何药学上可接受盐。
在一个实施方式中,含mdm2抑制剂和bcl2抑制剂的药物组合还可有利地包括pi3k抑制剂。意外发现此三重组合在2/5所测试细胞模型中相较药物对引起协同抑制(实施例4,表9)且相较成对组合显示更强的细胞凋亡(实施例4,图14)。
在一个优选实施方式中,所述药物组合包括mdm2抑制剂,选自(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮或其药学上可接受盐,和(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮或其药学上可接受盐;bcl2抑制剂navitoclax或其药学上可接受盐以及pi3k抑制剂(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)或其任何药学上可接受盐。
此外,根据本公开,本公开的药物组合包括(a)mdm2抑制剂和(b)(i)mek抑制剂和/或(ii)bcl2,还可有利地包括braf抑制剂。
此外,本公开的药物组合可有利地包括(a)mdm2抑制剂、(b)mek抑制剂、(c)bcl2抑制剂和(d)braf抑制剂。
本文定义的术语“braf抑制剂”指靶向、减少或抑制丝氨酸/苏氨酸蛋白激酶b-raf活性的化合物。
如前述权利要求中任一项所述的药物组合,其中braf抑制剂选自raf265、达拉菲尼、(s)-甲基-1-(4-(3-(5-氯-2-氟-3-(甲磺酰氨基)苯基)-1-异丙基-1h-吡唑-4-基)嘧啶-2-基氨基)丙烷-2-基氨基甲酸酯、甲基n-[(2s)-1-({4-[3-(5-氯-2-氟-3-甲磺酰氨基苯基)-1-(丙烷-2-基)-1h-吡唑-4-基]嘧啶-2-基}氨基)丙烷-2-基]氨基甲酸酯和威罗菲尼、或其药学上可接受盐。
根据本公开,braf抑制剂优选是达拉菲尼或其药学上可接受盐。在一个实施方式中,所述加入组合的braf抑制剂是raf265。
本公开组合特别是mdm2抑制剂和mek抑制剂(如曲美替尼)的组合还能包括cdk4/6抑制剂。本文定义的“周期素依赖性激酶4/6(cdk4/6)抑制剂”指与周期素-cdk复合体相互作用以阻断激酶活性的小分子。周期素依赖性激酶(cdk)是调节哺乳动物细胞周期起始、进展和完成的蛋白激酶大家族。优选地,cdk4/6抑制剂是7-环戊基-n,n-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7h-吡咯并[2,3-d]嘧啶-6-甲酰胺或其药学上可接受盐。
术语“药学上可接受盐”指保留化合物生物有效性和特性的盐,且其通常在生物学或其它方面不是不合需要的。所述化合物可通过存在氨基而能形成酸加成盐。
除非另有规定或由文本明确说明,提及用于本公开药物组合的治疗剂包括化合物的游离碱,以及化合物的所有药学上可接受盐。
本文定义的术语“组合”或“药物组合”指采用一个剂量单位形式的固定组合,或指联合给药的成套药盒或非固定组合,其中治疗剂可共同给药,同时独立给药或在一定时间间隔内分开地给药,这些时间间隔优选允许组合伴侣显示协作如协同效应。因此,本公开药物组合的那些单一化合物能同时或依序给药。
此外,本公开药物组合可采用固定组合或非固定组合形式。
术语“固定组合”指治疗剂如组合的这些单一化合物采用单一实体或剂型形式。
术语“非固定组合”指治疗剂如组合的这些单一化合物作为分开实体或剂型给予患者,同时或没有特定时间限制地依序给予,其中优选这类给药在需要的对象如哺乳动物或人体内提供治疗有效量的2种治疗剂。
药物组合还能包括至少一种药学上可接受载体。因此,本公开涉及含本公开药物组合和至少一种药学上可接受载体的药物组合物。
本文所用的术语“载体”或“药学上可接受载体”包括任何和所有溶剂、分散介质、包衣、表面活性剂、抗氧化剂、防腐剂(如抗菌药、抗真菌剂)、等渗剂、吸收延迟剂、盐、防腐剂、药物稳定剂、粘合剂、赋形剂、崩解剂、润滑剂、甜味剂、增味剂、染料等和其组合,如本领域技术人员已知(参见例如《雷明顿药物科学》,第18版,麦克出版公司,1990,第1289-1329页)。除非任何常规载体与活性成分不相容,否则考虑其用于治疗或药物组合物。
本文所用的短语“药学上可接受”指在合理医学判断范围内,适于接触人和动物组织的那些化合物、材料、组合物和/或剂型,而没有过度毒性、刺激、过敏反应和其它问题或并发症,并具有合理的效益/风险比。
一般,本文定义的术语“药物组合物”指混合物或溶液包含至少一种待给予对象如哺乳动物或人的治疗剂。本药物组合能配制于合适药物组合物以用于肠部或胃肠外给药,例如采用单位剂型的那些,如包糖衣片剂、片剂、胶囊或栓剂或安瓶。除非另有说明,这些以本身已知的方式制备,例如通过本领域技术人员显而易见的多种常规混合、粉碎、直接压片、制粒、包糖衣、溶解、冻干工艺或制造技术。应理解各剂型单独剂量所含组合伴侣的单位内容本身不需构成有效量,因为必要的有效量可通过给予多个剂量单位来实现。所述药物组合物可包含约0.1%-约99.9%,优选约1%-约60%的治疗剂。本领域普通技术人员可通过常规实验根据所需特定剂型特性来选择一种或多种上述载体,而没有任何过度负担。所用各载体的量可在本领域常规范围内变化。下列参考文献公开了用于配制口服剂型的技术和赋形剂。参见《药用赋形剂手册》(thehandbookofpharmaceuticalexcipients),第4版,rowe等编,美国药学协会(americanpharmaceuticalsassociation)(2003);和《雷明顿:药学科学与实践》(remington:thescienceandpracticeofpharmacy),第20版,gennaro编,利平科特威廉姆斯.威尔金斯(lippincottwilliams&wilkins)(2003)。这些可任选的额外常规载体可如下纳入口服剂型:将一种或多种常规载体在造粒前或期间纳入初始混合物或者在口服剂型中联合一种或多种常规载体与含药剂组合或药剂组合中单独药剂的颗粒。在后一个实施方式中,组合的混合物可进一步混合,如通过v型混合器,随后压成或模塑成片剂例如单层片剂,胶囊包封,或填充入小袋。显然,本公开的药物组合能用于制造药物。
本公开涉及特定能用作药物的药物组合或药物组合物。
特别地,本公开的组合或组合物能应用于治疗癌症。
本公开还涉及本公开组合或药物组合物在制备治疗癌症药物中的应用,和在需要的对象中治疗癌症的方法,包括向对象给予治疗有效量的本公开药物组合或本公开药物组合物。
本文所用的术语“治疗”(或“处理”)包括减缓、减轻或缓解对象中至少一种症状,提高无进展生存、总生存、延长反应持续时间或延迟疾病发展的治疗。例如,治疗可以是减少一种或多种疾病症状或者完全消除疾病,如癌症。在本公开意义内,术语“治疗”也指在患者如哺乳动物(尤其患者是人)中阻滞、延迟疾病发生(即疾病临床表征前的阶段)和/或降低疾病发展或恶化的风险。本文所用的术语“治疗”指抑制肿瘤生长,包括直接抑制原发性肿瘤生长和/或系统抑制转移性癌细胞。
“对象”、“个体”或“患者”在本文中可互换使用,指脊椎动物,优选哺乳动物,更优选人。哺乳动物包括但不限于小鼠、类人猿、人、耕畜、运动型动物和宠物。
术语“治疗有效量”的本公开化合物(如化学实体或生物制剂)指一定量的本公开化合物,其会引起对象生物或医学反应例如降低或抑制酶或蛋白活性,或改善症状、缓解病症、减缓或延迟疾病发展或预防疾病等。在一个实施方式中,体内治疗有效量的范围取决于给药途径,为约0.1-500mg/kg或约1-100mg/kg。
用于治疗癌症的各组合伴侣的最优剂量能用已知方法就各个体凭经验确定,并取决于多种因素,包括但不限于:疾病进展程度;个体的年龄、体重、总体健康、性别和饮食;给药时间和途径;和个体服用的其它药物。最优剂量可用常规测试和本领域熟知的程序确立。各组合伴侣可联合载体材料以生成单一剂型的量会根据所治疗个体和特定给药模式而变化。在一些实施方式中,含本文所述药剂组合的单位剂型所包含组合中各药剂的量是该药剂单独给予时通常施用的量。
剂量频率可根据所用化合物和待治疗或预防的特定病症而变化。一般,优选足以提供有效治疗的最小剂量。一般可监测患者的疗效,使用为本领域普通技术人员熟悉的适合治疗或预防病症的试验。
(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮的治疗量或剂量在口服时范围可以是每3周100-1500mg,特别是每3周100-800mg或每日50-600mg。(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮的治疗量或剂量的可以是400mg,更优选300mg,用于每28日周期前21天的每日给药。或者,(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮的总治疗量或总剂量的是每周期560mg(40mg每日一次(qd)服用2周/停用2周,或80mg每日一次(qd)服用1周/停用3周)。静脉内剂量需要相应降低。
(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮的治疗量或剂量在口服时是500-2000mg,特别是500-1200mg。在一个优选实施方式中,(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮的治疗量或剂量是500mg,更优选800mg。静脉内剂量需要相应降低。
mek抑制剂曲美替尼的建议剂量是每日2mg。管理不良反应可能需要剂量降低,多至每日1mg。
mek抑制剂化合物6-(4-溴-2-氟苯基氨基)-7-氟-3-甲基-3h-苯并咪唑-5-羧酸(2-羟乙氧基)-酰胺可以单一或分开剂量每日给予合适对象,有效剂量范围是每日约0.001-100mg/kg体重,优选约1-约35mg/kg/日,采用单一或分开剂量。对于70kg人,这相当于约0.05-7g/日的优选剂量范围,优选约0.05-约2.5g/日。
mek抑制剂化合物(s)-5-氟-2-(2-氟-4-(甲硫基)苯基氨基)-n-(2-羟丙氧基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺可以单一或分开剂量每日给予合适对象,有效剂量范围是每日约0.001-约100mg/kg体重,优选约1mg/kg/日-约35mg/kg/日,采用单一或分开剂量。对于70kg人,这相当于约0.07-2.45g/日的优选剂量范围,优选约0.05-约1.0g/日。
bcl-2抑制剂navitoclax的有效剂量范围可以是每日约100mg-约500mg。剂量可减少或采用150mg7日引入剂量。引入剂量后,能每日给予325mg剂量或多至425mg剂量。
egfr抑制剂埃罗替尼的建议剂量是每日100mg或150mg。
pi3k抑制剂(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)一般口服给药,成人剂量范围是每日约30mg-450mg,例如每日100-400mg。日剂量能以每日一次(qd)或每日两次(bid)安排给药。(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)可以单一或分开剂量每日给予合适对象,有效剂量范围是每日约0.05-约50mg/kg体重,优选约0.1-25mg/kg/日,更优选约0.5-10mg/kg/日,采用单一或分开剂量。对于70kg人,这相当于约35-700mg每日的优选剂量范围。更优选地,剂量范围是约35–400mg/日。
pi3k抑制剂化合物2-甲基-2-[4-(3-甲基-2-氧代-8-喹啉-3-基-2,3-二氢-咪唑并[4,5-c]喹啉-1-基)-苯基]-丙腈一般口服给药,成人剂量范围是每日约100mg-1200mg,或约200mg-1000mg,或约300mg-800mg,或约400mg-600mg。日剂量能以每日一次(qd)或每日两次(bid)安排给药。
pi3k抑制剂化合物5-(2,6-二吗啉-4-基-嘧啶-4-基)-4-三氟甲基-吡啶-2-基胺一般口服给药,成人剂量范围是每日约30mg-300mg,或约60mg-120mg,或约100mg。日剂量能以每日一次(qd)或每日两次(bid)安排给药。
braf抑制剂达拉菲尼的建议剂量是150mg每日口服2次,作为单一药剂或联合曲美替尼2mg每日口服1次。
应理解各治疗剂可方便地给予,例如在一个单独剂量单位中或分成多个剂量单位。应理解各治疗剂可方便地以每日1次的剂量到多至每日4次的剂量给药。
本文所用的术语“癌症”指宽范围的肿瘤,尤其是实体瘤。这类肿瘤的示例包括但不限于选自以下的良性或恶性肿瘤:肺(包括小细胞肺癌和非小细胞肺癌)、支气管、前列腺、乳腺(包括散发性乳腺癌和考登病患者)、胰腺、胃肠道、结肠、直肠、结肠癌、结直肠癌、甲状腺、肝、胆道、肝内胆管、肝细胞、肾上腺、胃、胃部、胶质瘤、成胶质细胞瘤、子宫内膜、肾、肾盂、膀胱、子宫、宫颈、阴道、卵巢、多发性骨髓瘤、食管、颈或头、脑、口腔和咽、喉、小肠、黑素瘤、结肠绒毛腺瘤、肉瘤、瘤形成、上皮特征性肿瘤、乳腺癌、基底细胞癌、鳞状细胞癌、光化性角化病、红细胞增多、原发性血小板增多症、白血病(包括急性骨髓性白血病、慢性髓细胞性白血病、淋巴细胞性白血病和骨髓性白血病)、淋巴瘤(包括非霍奇金淋巴瘤和霍奇金淋巴瘤)、髓样化生性骨髓纤维化、瓦尔登斯特伦氏病和barret腺癌。
优选地,所述癌是结直肠癌、黑素瘤、脂肪肉瘤、成胶质细胞瘤、成神经细胞瘤、淋巴瘤或白血病。在一个优选实施方式中,所述癌是结直肠癌。本文所用的术语“结直肠癌”指在结肠或直肠中的癌,也称为结肠癌、直肠癌或肠癌。在一个实施方式中,本公开涉及转移性结直肠癌。
预期所述组合在功能性p53或p53野生型癌症中实现优良效果。tp53基因是人类癌症中最频繁突变的基因之一。因此,在近50%人类癌症中,肿瘤抑制因子p53因突变或缺失而功能受损。在剩余的人类癌症中,p53保留野生型状态,但其功能受其主要细胞抑制因子鼠双微体2(mdm2,mdm2;hdm2(鼠双微体2的人同源物))抑制。mdm2是p53肿瘤抑制因子的负调节物。mdm2蛋白作为e3泛素连接酶和p53转录活化抑制剂起作用,e3泛素连接酶引起p53蛋白酶体降解。通常,发现mdm2在p53野生型肿瘤中扩增。由于mdm2与p53之间的相互作用是抑制癌症(其保留野生型p53)中p53功能的主要机制,含mdm2抑制剂的本公开组合对于治疗功能性p53或p53野生型癌症特别有用。
另外,预期所述组合的功效在癌症中增加,所述癌症表征为kras突变和/或braf突变和/或mek1突变和/或pik3ca突变和/或pik3ca过表达中的一个或多个。
携带kras或braf突变的结直肠癌患者共同构成50%-60%的所报告结直肠癌病例(fearon2011),一般与不良预后相关(arrington,heinrich等.2012,safaeeardekani,jafarnejad等.2012)。本公开组合特别用于治疗包括一个或多个kras突变或者一个或多个braf突变的癌症。。
braf突变示例包括但不限于:v600e,r461i,i462s,g463e,g463v,g465a,g465e,g465v,g468a,g468e,n580s,e585k,d593v,f594l,g595r,l596v,t598i,v599d,v599e,v599k,v599r,v600k,a727v。这些突变大部分聚集在2个区域:n叶的甘氨酸富集p环以及激活区段和侧翼区。v600e突变在多种癌症中检测到,这归因于在核苷酸1799用腺嘌呤取代胸腺嘧啶。这引起密码子600处缬氨酸(v)被谷氨酸(e)取代(现在称为v600e)。
例如,mek1突变可以是mek1s72g突变。
pik3ca突变和/或pik3ca过表达的示例包括但不限于:扩增α同种型pi3k,pik3ca体细胞突变,pten的胚系突变或体细胞突变,用于上调p85-p110复合体的p85α突变和易位,或者扩增或表达β同种型pi3k。
本公开的药物组合特别用于治疗癌症,尤其是结直肠癌,其中所述癌症抗egfr抑制剂治疗,或正发展出对egfr抑制剂治疗的抗性,或有高风险发展出对egfr抑制剂治疗的抗性,尤其是其中egfr抑制剂选自埃罗替尼、吉非替尼和阿法替尼。
本公开的药物组合也适合治疗不良预后患者,特别是有癌症尤其是结直肠癌的那些不良预后患者,所述癌变得抗egfr抑制剂治疗,如初始响应egfr抑制剂治疗而随后复发的癌症患者。另一示例中,所述患者未接受采用fgfr抑制剂的治疗。此癌症在用一种或多种egfr抑制剂的先前治疗中可能产生获得性耐药性。例如,egfr靶向疗法可包括用如下的治疗:吉非替尼,埃罗替尼,拉帕替尼,xl-647,hki-272(neratinib),bibw2992(afatinib),ekb-569(pelitinib),av-412,卡奈替尼,pf00299804,bms690514,hm781-36b,wz4002,ap-26113,西妥昔单抗,帕尼单抗,马妥珠单抗,曲妥珠单抗,帕妥珠单抗,或其药学上可接受盐。具体地,egfr靶向治疗可包括用吉非替尼、埃罗替尼和拉帕替尼的治疗。获得性耐药性的机制包括但不限于:egfr基因本身发展出第二突变,如t790m,egfr扩增;和/或fgfr失调、fgfr突变、fgfr配体突变、fgfr扩增或fgfr配体扩增。
以下实施例阐明上述公开;然而其不意在以任何方式限制本公开范围。相关领域技术人员已知的其它测试模型也能确定所要求权利的公开的有益效果。
实施例
“化合物a”、“化合物b”等在本文指特定化合物。提及各化合物就所有实施例或组合而言可能不一样。相反,所述化合物在各实施例中重新指示。
实施例1:联合mdm2抑制剂和mek抑制剂对增殖的体外效果。
此研究设计成探索在tp53野生型结直肠癌细胞系中联合mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)或mdm2抑制剂(6s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢吡啶-3-基)-6-(4-氯苯基)-2-(2,4-二甲氧基嘧啶-5-基)-1-(丙烷-2-基)-5,6-二氢吡咯并[3,4-d]咪唑-4(1h)-酮(化合物b)与mek抑制剂曲美替尼(化合物c)对增殖的体外效果。
方法
化合物a、b和c以20mm浓度溶于100%dmso(西格玛(sigma),产品目录号d2650),-20℃保存待用。
从商业公司atcc、ecacc、dsmz和cellbank澳大利亚(cellbankaustralia)获得用于此研究的结直肠癌细胞系,培养并处理(表1)。所有细胞系培养基补充有10%fbs(海克隆(hyclone),产品目录号sh30071.03)。lim2405的培养基额外补充有0.6ug/ml胰岛素(西格玛,产品目录号i9278)、1ug/ml氢化可的松(西格玛,产品目录号h0135)和10μm1-硫代甘油(西格玛,产品目录号m6145)。
表1.细胞系信息。
细胞系在37℃和5%co2培养箱中培养,在t-75培养瓶中扩增。所有情况下,细胞从冷冻储液中解冻,用1:3稀释通过≥1次传代来扩增,用vicell计数器(贝克曼库尔特(beckman-coulter))计数并评价活力,然后接种。为拆分并扩增细胞系,细胞用0.25%胰蛋白酶-edta(gibco,产品目录号25200)从培养瓶中移出。确定所有细胞系没有支原体污染,这通过idexxradil(美国密苏里州哥伦比亚)进行的pcr检测方法确定并通过检测snp组来正确鉴定。
为测试化合物a或化合物b与化合物c组合对细胞增殖的效果,细胞接种入透明底黑色384孔微量板(matrix/赛默科技(thermoscientific),产品目录号4332)中的50μl培养基/每孔,细胞密度为500-1250细胞/孔(表1),并允许在37度,5%co2孵育24小时。24小时后,各细胞系制备一块384孔板以通过显微镜检查(见下)进行细胞计数,而不接受处理(=‘基线)。其它细胞板用hpd300数字分配器(帝肯(tecan))处理,产生6点剂量反应曲线,采用2.5x稀释步骤。
化合物a在51nm-5um的终浓度范围中使用,化合物b在10nm-1um的终浓度范围中使用,化合物c在1nm-100nm的终浓度范围中使用。
对于化合物a或化合物b与化合物c的组合,单一药剂以6个单药剂剂量组合,产生6x6=36种组合处理的剂量矩阵。另外,转移阴性对照(dmso=‘载剂’)和阳性对照(星孢菌素=杀死细胞,7点1:2稀释系列用于16nm-1um剂量范围)作为处理对照。细胞根据其倍增时间处理72小时-96小时(表1)。处理结束时,制备细胞以通过显微镜检查进行细胞计数。细胞在溶于pbs(波士顿生物产品公司(bostonbioproducts),产品目录号bm-220)的4%pfa(ems(electronmicroscopysciences),产品目录号15714)、0.12%tx-100(ems,产品目录号22140)中固定并透化45分钟。细胞用pbs洗3次后,其dna用终浓度为4μg/ml的hoechst33342(赛默飞世尔,产品目录号h3570)染色30分钟。细胞用pbs洗3次,然后板用以铝密封件(安捷伦科技(agilenttechnologies),产品目录号06644-001)使用plateloc(安捷伦科技)热密封,4℃保存直至成像。通过荧光显微镜在单一图像中捕获每孔所有细胞/处理,使用装有4x物镜和dapi激发/发射滤光片的incell分析仪2000(通用电气医疗集团(gehealthcare))。
为测试所述组合对诱导细胞凋亡的效果,半胱天冬酶3/7试验用就上述增殖试验设置的类似实验步骤进行,并且仅检测化合物a与化合物c的组合。化合物排列在在药物靠模板(葛莱娜(greiner),产品目录号788876)中并以2000x浓度3倍连续稀释(7级)。细胞如下处理:用ats声学液体分配器(ecd生物系统公司((ecdbiosystems)))从药物靠模板转移25nl2000x化合物到50ul细胞,产生最终1x浓度。
化合物a在13nm-10μm的终浓度范围中使用,化合物c在0.4nm-0.3um的终浓度范围中使用。另外,转移阴性对照(dmso=‘载剂’)和阳性对照(星孢菌素=杀死细胞,7点1:2稀释系列给出16nm-1μm剂量范围)作为处理对照。
化合物添加后,用hpd300数字分配器将50nl2mmcellevent半胱天冬酶-3/7绿色检测试剂(赛默飞世尔(thermofisher),产品目录号c10423)加入三个重复之一。半胱天冬酶3/7诱导作为处理诱导的细胞凋亡替代物测量。细胞根据其倍增时间处理72小时-96小时(表1),每24小时通过显微镜检查测量半胱天冬酶3/7活化,使用装有4x物镜和fitc激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。处理结束时,制备细胞以通过显微镜进行细胞计数和成像,如上就细胞增殖试验所述。
适应前述方法(horn,sandmann等)和使用r中的bioconductor包ebimage(pau,fuchs等.2010)后,分析图像。2个通道即dapi(用于hoechst/dna)和fitc(用于半胱天冬酶3/7)中的对象通过自适应阈值化单独分段并计数。比较阴性对照(dmso)和阳性对照(星孢菌素)后,手动确定各细胞系的半胱天冬酶3/7阳性对象阈值。通过分析dna通道中的17个额外实体/核特征(形状和强度特征),鉴定碎片/片段化核。为此,手动比较每个细胞系的阳性对照(星孢菌素)与阴性对照(dmso)之间的额外特征分布。能在条件之间区分的特征(如比较dmso与星孢菌素的特征测量分布的变化)用于确定‘碎片’群与‘活’核群。从原始核计数中减去碎片计数。所得核数目用作细胞增殖量度(‘细胞计数’)。
化合物对细胞增殖的作用计算自与阴性对照(dmso)的细胞计数相比较的处理细胞计数,图1和2中表示为y轴上的‘标准化细胞计数’(=‘xnorm’)。组合的协同作用通过等效应图分析(greco,bravo等.1995)和计算联合指数(chou,talalay1984)(表2)来评价,其测试loewe模型(loewe1928)下的协同作用。就75%等效应水平(相较组合处理,在单一药剂处理下的75%抑制)完成ci分析。‘最佳ci’(图3的红点和表2)是此组合在特定细胞系中观察到的最低联合指数。
联合指数(ci)是组合效果指示物:
ci<1:协同作用
ci=1:累加效应
ci>1:拮抗作用
例如,联合指数0.5表示,与达到相同效果(这里是75%抑制)所需的单独单一药剂剂量相比,联合时仅需要一半的各药剂。
ic75是相较dmso产生75%细胞计数的化合物浓度。ic75计算(见表2)通过对数据进行3参数逻辑回归来完成。
表2.各化合物的单一药剂ic50值以及化合物a和曲美替尼与化合物b和曲美替尼组合的最佳联合指数(最佳ci)。
化合物对细胞凋亡的作用如下确定:相对于原始细胞计数(减去碎片前),计算各处理和时间点有活化半胱天冬酶3/7的细胞百分数(图4的y轴)。未经实验测量的各时间点细胞计数通过回归分析由第0天和处理结束时细胞计数的对数变换(假设指数细胞生长)作线性模型拟合获得。
对于集落形成试验(图5),细胞接种于12孔组织培养处理板(costar,产品目录号3513)的1ml培养基:colo-6786000细胞/孔,sw485000细胞/孔,gp2d2000细胞/孔,lovo2500细胞/孔,ls-1802500细胞/孔,lim24052500细胞/孔,rko1000细胞/孔以及hct-1161000细胞/孔。细胞生长72小时,然后添加化合物,每48小时更新处理(新鲜培养基中)多至14天,使用hpd300数字分配器(帝肯)。处理结束时,细胞在pbs中洗一次,室温固定并染色30分钟,使用含4%pfa(ems,产品目录号15714)和2mg/ml结晶紫(emd,产品目录号192-12)的溶液,用水洗3次。板干燥过夜,用odyssee成像仪(licor)扫描。imagestudio软件(licor)用于定量图5的结晶紫信号。显著性检验参见表3。
表3.相较对应剂量的单独化合物a或曲美替尼,化合物a与曲美替尼组合的集落形成试验(图4)的差异显著性(单尾t检验)。*p<0.05,**p<0.01,***p<0.001.
.
对于细胞周期分析(图6),细胞接种于10cm组织培养处理皿(康宁(corning),产品目录号430167)的10ml培养基:colo-678350万细胞,sw48275万细胞,gp2d250万细胞,lovo250万细胞,ls-180250万细胞,lim2405150万细胞,rko150万细胞及hct-116200万细胞。药物处理在24小时后手工完成。处理后24小时如下收集样品:通过胰蛋白酶化来收集上清和收获细胞,使用0.25%胰蛋白酶-edta(gibco,产品目录号25200)。细胞沉淀,用pbs洗一次,再次沉淀,然后在1ml冰冷70%乙醇(逐滴加至沉淀,同时涡旋)中于4℃固定30分钟。接着,细胞在较高速度(850xg)沉淀5分钟,且沉淀在磷酸盐柠檬酸盐缓冲液(0.2mna2hpo4,0.1m柠檬酸,调至ph7.8)中洗2次并以同一速度离心(持续5分钟)。沉淀最终散入0.5mlpi/rnase染色缓冲液(bdpharmingen,产品目录号550825)。室温孵育15分钟后,在bdfacscantoii系统(分析10,000事件/条件)上分析细胞周期。用flowjo软件分析数据。
对于蛋白质印迹(图7),细胞接种于10cm组织培养处理皿(康宁(corning),产品目录号430167)的10ml培养基:colo-678800万细胞,sw48400万细胞,gp2d300万细胞,lovo400万细胞,ls-180450万细胞,lim2405200万细胞,rko350万细胞及hct-116350万细胞。细胞生长24小时,然后添加化合物。24小时处理后将细胞通过刮擦收集在冰冷pbs中,在含磷酸酶抑制剂(罗氏(roche),产品目录号04906837001)和蛋白酶抑制剂(罗氏,产品目录号04693116001)的裂解缓冲液(细胞信号转导公司(cellsignaling),产品目录号9803)中裂解30分钟。裂解物用bca蛋白试验试剂盒(赛默飞世尔(thermofisher),产品目录号23225)定量,浓度标准化,加入上样缓冲液(赛默飞世尔,产品目录号np0007),在预制4-15%聚丙烯酰胺梯度凝胶(伯乐(biorad),产品目录号5671084)上加样25-50ug蛋白,在300v运行电泳系统(伯乐)30-35分钟。蛋白转移至硝酸纤维素膜上,使用iblot转移系统(英杰(invitrogen))和iblotgel转移试剂盒(赛默飞世尔,产品目录号ib301001)。图5所示蛋白用以下一抗检测:p53(圣克鲁斯生物技术(santacruzbiotechnology),sc-126,1:500),mdm2(calbiochem,#op46,1:500),erk(cst(cellsignalingtechnology),#4695,1:1000),perk(cst#4370,1:1000),p21(cst,#2947,1:1000),p27(圣克鲁斯生物技术,sc-528,1:500),细胞周期素d1(圣克鲁斯生物技术,sc-718,1:500),bim(cst,#2819,1:500),cparp(cst,#9541,1:500),puma(cst,#4976,1:500)和β肌动蛋白(ambion,am4302,1:10000)。使用以下二抗:hrp山羊抗兔(伯乐,170-5046,1:10000),hrp山羊抗小鼠(伯乐,170-5047,1:10000)和
对于qrt-pcr分析(图8),细胞接种于6孔组织培养处理板(康宁,产品目录号3516):colo-67875万细胞,sw4850万细胞,gp2d40万细胞,lovo40万细胞,ls-18040万细胞,lim240525万细胞,rko25万细胞及hct-11630万细胞。药物处理在24小时后手工完成。处理后10小时收集样品.移出上清,细胞用pbs洗一次,然后rneasy迷你试剂盒(凯杰(qiagen),产品目录号74104)用于rna提取。rna用nanodrop1000定量,1μg总rna用于cdna合成,采用iscriptcdna合成试剂盒(伯乐,产品目录号170-8891)。taqmanqrt-pcr在384孔板中于abiviia7(应用生物系统公司(appliedbiosystems))上进行。每反应用6μl2xpcr反应混合物(赛默飞世尔,产品目录号4352042),0.6μlβ肌动蛋白引物/探针组(20x),0.6μl靶引物/探针组(20x)和4.8μlcdna(1:4稀释来自cdna合成的产物)。运行以下程序:50℃2分钟,95℃10分钟,40轮的95℃10秒和60℃1分钟。使用以下探针/引物组(赛默飞世尔,taqman基因表达试验):cdkn1a/p21(hs00355782_m1),bax(hs00180269_m1),bbc3/puma(hs00248075_m1),bmf(hs00372937_m1),noxa1(hs00736699_m1)。
结果
此报告中,在总共8个tp53野生型结直肠癌细胞中单独和联合评价2种mdm2抑制剂(化合物a和化合物b)以及mek抑制剂曲美替尼(化合物c)的功效。5个细胞系是kras突变(gp2d,ls-180,hct-116,lovo,colo-678),2个细胞系是braf突变(rko,lim2405),1个细胞系是mek1突变(sw48)。4个细胞系也是pik3ca突变(gp2d,rko,ls-180,hct-116)(表1)。作为单一药剂的化合物a以亚微摩尔到微摩尔ic75值抑制所有细胞系生长,,化合物b有亚微摩尔ic75值(除了colo-678),化合物c有纳摩尔ic75值(除了colo-678,rko和gp2d)(图1和2,表2)。组合处理在所测试全部细胞模型中引起协同抑制(根据loewe模型),如联合指数(ci)所示(图3和表2)。进一步机制研究聚焦于化合物a与曲美替尼组合。所述组合显示细胞凋亡弱诱导(通过测量半胱天冬酶3/7诱导来评价)(图4)。所述组合在集落形成试验中对集落过度生长的预防显著优于各单一药剂,还显示该组合的长期功效(图5和表3)。facs分析在24小时处理后进行,显示mdm2抑制使在细胞周期s期的细胞枯竭并使细胞停滞在g1和/或g2期(图6)。对mek抑制的反应更依赖细胞系,但在大部分模型中,其产生增多的g1群。所述组合主要显示s期枯竭且在5/8模型中也显示增加的sub-g1群,提示细胞死亡。为鉴定联合处理后在细胞周期阻滞和细胞死亡中起作用的因子,进行western(24小时处理后)和qpcr(10小时处理后)分析细胞周期及细胞凋亡的调节因子(图7和8)。western和qpcr结果表明,mdm2抑制后的g1阻滞归因于p21(cdkn1a)诱导。mek抑制在一些测试模型中诱导p27蛋白(cdkn1b)(参见图7的westerns),其能潜在加强组合中的细胞周期阻滞。mdm2抑制可转录诱导促凋亡因子puma和bax表达(图7),puma诱导也在westerns上得到确认(图8)。mek抑制显示提高水平的促凋亡蛋白bim(图7),和转录诱导的促凋亡因子bmf和noxa1表达(图8)。总体上,药物组合中这些基因和蛋白的诱导能解释colo-678以外所有模型的组合中所见的parp切割(cparp)增加(图7)。cparp是诱导细胞凋亡的指示物。
结论
总之,数据表明,联合抑制mdm2和mek能调节细胞周期阻滞蛋白互补组(p21和p27诱导)以诱导g1和/或g2细胞周期阻滞,和调节促凋亡蛋白(如诱导bax、bim和puma)以通过细胞凋亡诱导细胞死亡。在tp53野生型结直肠癌中联合抑制mdm2和mek可提供相较各单一药剂能够改善反应的有效治疗模式,并产生临床更持久的反应。
实施例2:联合mdm2抑制剂和mek抑制剂与bcl2抑制剂对增殖的体外效果。
此研究设计成探索在tp53野生型结直肠癌细胞系中联合mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)和mek抑制剂曲美替尼(化合物b)与bcl-2/-xl抑制剂navitoclax(abt-26)(化合物c)对增殖的体外效果。
方法
化合物a、b和c以20mm浓度溶于100%dmso(西格玛,产品目录号d2650),-20℃保存待用。化合物排列在药物靠模板(葛莱娜,产品目录号788876)中并以2000x浓度3倍连续稀释(7级)。
从商业公司atcc和ecacc获得用于此研究的结直肠癌细胞系,培养并处理(表4)。所有细胞系培养基补充有10%fbs(海克隆,产品目录号sh30071.03)。
表4.细胞系信息。
细胞系在37℃和5%co2培养箱中培养,在t-75培养瓶中扩增。所有情况下,细胞从冷冻储液中解冻,用1:3稀释通过≥1次传代来扩增,用vicell计数器(贝克曼库尔特)计数并评价活力,然后接种。为拆分并扩增细胞系,细胞用0.25%胰蛋白酶-edta(gibco,产品目录号25200)从培养瓶中移出。确定所有细胞系没有支原体污染,这通过idexxradil(美国密苏里州哥伦比亚)进行的pcr检测方法确定并通过检测snp组来正确鉴定。
为测试化合物a、化合物b与化合物c组合对细胞增殖的效果,细胞接种于有透明底的黑色384孔微量板(matrix/赛默科技,产品目录号4332)中的50μl培养基/每孔,细胞密度为500-1250细胞/孔(表4),并允许在37度,5%co2孵育24小时。24小时后,各细胞系制备一块384孔板以通过显微镜检查(见下)进行细胞计数,而不接受处理(=‘基线)。其它细胞板如下处理:用ats声学液体分配器(ecd生物系统公司)从药物靠模板转移25nl2000x化合物,产生最终1x浓度。化合物a在13nm-10μm的终浓度范围中使用,化合物b在13nm-10μm的终浓度范围中使用,化合物c在0.4nm-0.3μm的终浓度范围中使用(7级1:3稀释步骤)。为评价三重组合的效果,在同一实验中测试所有个体化合物(a,b,c)、所有3个成对组合(a+b,a+c,b+c)和三重组合(a+b+c)。成对组合和三重组合在各稀释度以1:1(用于药物对)和1:1:1(用于三联药物)固定比例测试,各处理产生7种组合条件处理。另外,阴性对照(dmso=‘载剂’)和阳性对照(星孢菌素=杀死细胞,7点1:2稀释系列用于16nm-1μm剂量范围)作为处理对照转移,在所测试细胞系中无功效的化合物与化合物a和化合物b联用作为组合对照(不超过更有效单一药剂功效的组合=‘非相互作用’组合)。化合物添加后,用hpd300数字分配器(帝肯)将50nl2mmcellevent半胱天冬酶-3/7绿色检测试剂(赛默飞世尔,产品目录号c10423)加入三个重复之一。半胱天冬酶3/7诱导作为处理诱导的细胞凋亡替代物测量。细胞根据其倍增时间处理72小时-96小时(表4),每24小时通过显微镜检查测量半胱天冬酶3/7活化,使用装有4x物镜和fitc激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。处理结束时,制备细胞以通过显微镜检查进行细胞计数。细胞在溶于pbs(波士顿生物产品公司,产品目录号bm-220)的4%pfa(ems,产品目录号15714)、0.12%tx-100(ems,产品目录号22140)中固定并透化45分钟。细胞用pbs洗3次后,其dna用终浓度为4μg/ml为hoechst33342(赛默飞世尔,产品目录号h3570)染色30分钟。细胞用pbs洗3次,然后板用铝密封件(安捷伦科技,产品目录号06644-001)使用plateloc(安捷伦科技)热密封,4℃保存直至成像。通过荧光显微镜在单一图像中捕获每孔所有细胞/处理,使用装有4x物镜和dapi激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。
适应前述方法(horn,sandmann等.2011)和使用r中的bioconductor包ebimage(pau,fuchs等.2010)后,分析图像。2个通道即dapi(用于hoechst/dna)和fitc(用于半胱天冬酶3/7)中的对象通过自适应阈值化单独分段并计数。比较阴性对照(dmso)和阳性对照(星孢菌素)后,手动确定每细胞系的半胱天冬酶3/7阳性对象阈值。通过分析dna通道中的17个额外对象/核特征(形状和强度特征),鉴定碎片/片段化核。为此,手动比较每个细胞系的阳性对照(星孢菌素)与阴性对照(dmso)之间的额外特征分布。能在条件之间区分的特征(如比较dmso与星孢菌素的特征测量分布的变化)用于确定‘碎片’群与‘活’核群。从原始核计数中减去碎片计数。所得核数目用作细胞增殖量度(‘细胞计数’)。
化合物对细胞增殖的作用计算自与阴性对照(dmso)的细胞计数相比较的处理的细胞计数,图9中表示为y轴上的‘标准化细胞计数’(=‘xnorm’)。协同组合用最高单一药剂模型(hsa)作为零假设(berenbaum1989)鉴定。超过hsa模型则预测受抑制靶标之间的功能联系(lehar,zimmermann等.2007,lehar,krueger等.2009)。模型输入是各药物剂量的抑制值:
i=1-xnorm
i:抑制
xnorm:标准化细胞计数(3个重复的中值)
在组合处理的各剂量点,计算组合抑制与2种单一药剂中更强者抑制之间的差异(=模型残差)。类似地,为评价三重组合在各剂量点的协同作用,计算三联药物抑制与最强药物对抑制之间的差异。为有利于高抑制下的组合效果,残差用同一剂量点观察到的抑制加权。药物组合的总体组合得分c是所有浓度中加权残差的总和:
c=σconc(i数据*(i数据–i模型))
i数据:测量的抑制
i模型:根据hsa零假设的抑制
稳健组合z得分(zc)计算为处理组合得分c与非相互作用组合绝对中位差(mad)之比:
zc=c/mad(c零)
c零:非相互作用组合的组合得分
zc是组合强度指示物:
zc≥3:协同作用
3>zc≥2:弱协同作用
zc<2:无协同作用
ic50是相对于dmso产生50%细胞计数的化合物浓度。完成ic50计算(见表5),使用r中的drc包(ritz和streibig2005年)并拟合四参数log-逻辑函数至数据。
化合物对细胞凋亡的作用如下确定:相对于原始细胞计数(减去碎片前),计算各处理和时间点有活化半胱天冬酶3/7的细胞百分数(图10的y轴)。未经实验测量的各时间点细胞计数通过回归分析由第0天和处理结束时细胞计数的对数变换(假设指数细胞生长)作线性模型拟合获得。
表5.各化合物的单一药剂ic50值以及化合物a、化合物b与化合物c组合的协同作用z得分的测量。
结果
此报告中,在总共5个tp53野生型结直肠癌细胞中单独和联合评价mdm2抑制剂(化合物a)、mek抑制剂(曲美替尼,化合物b)与bcl-2/-xl抑制剂(abt-263,化合物c)的功效。4个细胞系是kras突变(gp2d,ls-180,hct-116,lovo),1个细胞系是braf突变(rko)(表4)。作为单一药剂的化合物a以亚微摩尔到微摩尔ic50值抑制细胞系生长,。作为单一药剂的化合物b在除了1个(gp2d)细胞系以外的所有细胞系中以纳摩尔ic50值抑制生长,,而化合物c没有单一药剂功效(图9和表5)。三重组合(a+b+c)相比药物对在2/5所测试细胞模型中引起协同抑制(根据hsa模型)(表5)。在4个细胞系(gp2d,hct-116,rko,lovo)中,三重组合相较成对组合显示更强的细胞凋亡(通过测量半胱天冬酶3/7诱导来评价)(图10)。
结论
总之,在tp53野生型crc中联合抑制mdm2、mek和bcl-2/-xl可提供相较各单一药剂能够改善反应的有效治疗模式,并产生临床更持久的反应。
另外,将pi3k抑制剂(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)加入(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼(化合物b)与bcl-2/-xl抑制剂navitoclax(abt-263)(化合物c)的组合,以形成四重组合并在kras突变细胞系中一起测试,发现弱协同(ls-180,组合z得分为2.63)和强烈诱导细胞凋亡(最大为61%)。
实施例3:联合mdm2抑制剂和mek抑制剂与egfr抑制剂对增殖的体外效果。
此研究设计成探索在tp53野生型结直肠癌细胞系中联合mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)和mek抑制剂曲美替尼(化合物b)与egfr抑制剂埃罗替尼(化合物c)对增殖的体外效果。
方法
化合物a、b和c以20mm浓度溶于100%dmso(西格玛,产品目录号d2650),-20℃保存待用。化合物排列在药物靠模板(葛莱娜,产品目录号788876)中并以2000x浓度3倍连续稀释(7级)。
从商业公司atcc和ecacc获得用于此研究的结直肠癌细胞系,培养并处理(表6)。所有细胞系培养基补充有10%fbs(海克隆,产品目录号sh30071.03)。
表6.细胞系信息
细胞系在37℃和5%co2培养箱中培养,在t-75培养瓶中扩增。所有情况下,细胞从冷冻储液中解冻,用1:3稀释通过≥1次传代来扩增,用vicell计数器(贝克曼库尔特)计数并评价活力,然后接种。为拆分并扩增细胞系,细胞用0.25%胰蛋白酶-edta(gibco,产品目录号25200)从培养瓶中移出。确定所有细胞系没有支原体污染,这通过idexxradil(美国密苏里州哥伦比亚)进行的pcr检测方法确定并通过检测snp组来正确鉴定。
为测试化合物a、化合物b与化合物c组合对细胞增殖的效果,细胞接种于有透明底的黑色384孔微量板(matrix/赛默科技,产品目录号4332)中的50μl培养基/每孔,细胞密度为500-1250细胞/孔(表6),并允许在37度,5%co2孵育24小时。24小时后,各细胞制备一块384孔板/以通过显微镜检查(见下)进行细胞计数,而不接受处理(=‘基线)。其它细胞板如下处理:用ats声学液体分配器(ecd生物系统公司)从药物靠模板转移25nl2000x化合物,产生最终1x浓度。化合物a在13nm-10μm的终浓度范围中使用,化合物b在13nm-10μm的终浓度范围中使用,化合物c在13nm-10μm的终浓度范围中使用(7个1:3稀释步骤)。为评价三重组合的效果,在同一实验中测试所有个体化合物(a,b,c)、所有3个成对组合(a+b,a+c,b+c)和三重组合(a+b+c)。成对组合和三重组合在各稀释度以1:1(用于药物对)和1:1:1(用于三联药物)固定比例测试,各处理产生7种组合条件。另外,转移阴性对照(dmso=‘载剂’)和阳性对照(星孢菌素=杀死细胞,7点1:2稀释系列用于16nm-1μm剂量范围)作为处理对照,在所测试细胞系中无功效的化合物与化合物a和化合物b联用作为组合对照(不超过更有效单一药剂功效的组合=‘非相互作用’组合)。化合物添加后,用hpd300数字分配器(帝肯)将50nl2mmcellevent半胱天冬酶-3/7绿色检测试剂(赛默飞世尔,产品目录号c10423)加入三个重复之一。半胱天冬酶3/7诱导作为处理诱导的细胞凋亡替代物测量。细胞根据其倍增时间处理72小时-96小时(表6),每24小时通过显微镜检查测量半胱天冬酶3/7活化,使用装有4x物镜和fitc激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。处理结束时,制备细胞以通过显微镜检查进行细胞计数。细胞在溶于pbs(波士顿生物产品公司,产品目录号bm-220)的4%pfa(ems,产品目录号15714)、0.12%tx-100(ems,产品目录号22140)中固定并透化45分钟。细胞用pbs洗3次后,其dna用终浓度为4μg/ml的hoechst33342(赛默飞世尔,产品目录号h3570)染色30分钟。细胞用pbs洗3次,然后板用铝密封件(安捷伦科技,产品目录号06644-001)使用plateloc(安捷伦科技)热密封,4℃保存直至成像。通过荧光显微镜在单一图像中捕获每孔所有细胞/处理,使用装有4x物镜和dapi激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。
适应前述方法(horn,sandmann等.2011)和使用r中的bioconductor包ebimage(pau,fuchs等.2010)后,分析图像。2个通道中即dapi(用于hoechst/dna)和fitc(用于半胱天冬酶3/7)的对象通过自适应阈值化单独分段并计数。比较阴性对照(dmso)和阳性对照(星孢菌素)后,手动确定各细胞系的半胱天冬酶3/7阳性对象阈值。通过分析dna通道中的17个额外对象/核特征(形状和强度特征),鉴定碎片/片段化核。为此,手动比较各细胞系的阳性对照(星孢菌素)与阴性对照(dmso)之间的额外特征分布。能在条件之间区分的特征(如比较dmso与星孢菌素的特征测量分布的变化)用于确定‘碎片’群与‘活’核群。从原始核计数中减去碎片计数。所得核数目用作细胞增殖量度(‘细胞计数’)。
化合物对细胞增殖的作用计算自与阴性对照(dmso)的细胞计数相比较的处理的细胞计数,图11中表示为y轴上的‘标准化细胞计数’(=‘xnorm’)。协同组合用最高单一药剂模型(hsa)作为零假设(berenbaum1989)鉴定。超过hsa模型则预测受抑制靶标之间的功能联系(lehar,zimmermann等.2007,lehar,krueger等.2009)。模型输入是各药物剂量的抑制值:
i=1-xnorm
i:抑制
xnorm:标准化细胞计数(3个重复的中值)
在组合处理的各剂量点,计算组合抑制与2种单一药剂中更强者抑制之间的差异(=模型残差)。类似地,为评价三重组合在各剂量点的协同作用,计算三联药物抑制与最强药物对抑制之间的差异。为有利于高抑制下的组合效果,残差用同一剂量点观察到的抑制加权。药物组合的总体组合得分c是所有浓度中加权残差的总和:
c=σconc(i数据*(i数据–i模型))
i数据:测量的抑制
i模型:根据hsa零假设的抑制
稳健组合z得分(zc)计算为处理组合得分c与非相互作用组合绝对中位差(mad)之比:
zc=c/mad(c零)
c零:非相互作用组合的组合得分
zc是组合强度指示物:
zc≥3:协同作用
3>zc≥2:弱协同作用
zc<2:无协同作用
ic50是相对于dmso产生50%细胞计数的化合物浓度。完成ic50计算(见表7),使用r中的drc包(ritz和streibig2005年)并拟合四参数log-逻辑函数至数据。
化合物对细胞凋亡的作用如下确定:相对于原始细胞计数(减去碎片前),计算各处理和时间点有活化半胱天冬酶3/7的细胞百分数(图12的y轴)。未经实验测量的各时间点细胞计数通过回归分析由第0天和处理结束时细胞计数的对数变换(假设指数细胞生长)作线性模型拟合获得。
表7.各化合物的单一药剂ic50值以及化合物a、化合物b与化合物c组合的协同作用z得分的测量。
结果
此报告中,在总共5个tp53野生型结直肠癌细胞中单独和联合评价mdm2抑制剂(化合物a)、mek抑制剂(曲美替尼,化合物b)与egfr抑制剂(埃罗替尼,化合物c)的功效。4个细胞系是kras突变(gp2d,ls-180,hct-116,lovo),1个细胞系是braf突变(rko)(表6)。作为单一药剂的化合物a以亚微摩尔到微摩尔ic50值抑制细胞系生长。作为单一药剂的化合物b在除了1个(gp2d)细胞系以外的所有细胞系中以纳摩尔ic50值抑制生长,而化合物c在4/5细胞系没有单一药剂功效且在kras突变lovo中有微摩尔ic50(图11和表7)。三重组合(a+b+c)相比药物对在kras突变模型lovo中引起协同抑制(根据hsa模型)(表7)。此细胞系中,三重组合相较成对组合显示更强的细胞凋亡(通过测量半胱天冬酶3/7诱导来评价)(图12)。
结论
在tp53野生型crc中联合抑制mdm2、mek和egfr可提供相较各单一药剂能够改善反应的有效治疗模式,并产生临床更持久的反应。
实施例4:联合pi3k抑制剂和mdm2抑制剂与bcl2抑制剂对增殖的体外效果。
此研究设计成探索在tp53野生型结直肠癌细胞系中联合pik3ca抑制剂(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)(化合物a)和mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物b)与bcl-2/-xl抑制剂navitoclax(abt-263)(化合物c)对增殖的体外效果。
方法
化合物a、b和c以20mm浓度溶于100%dmso(西格玛,产品目录号d2650),-20℃保存待用。化合物排列在药物靠模板(葛莱娜,产品目录号788876)中并以2000x浓度3倍连续稀释(7级)。
从商业公司atcc和ecacc获得用于此研究的结直肠癌细胞系,培养并处理(表8)。所有细胞系培养基补充有10%fbs(海克隆,产品目录号sh30071.03)。
表8.细胞系信息
细胞系在37℃和5%co2培养箱中培养,在t-75培养瓶中扩增。所有情况下,细胞从冷冻储液中解冻,用1:3稀释通过≥1次传代来扩增,用vicell计数器(贝克曼库尔特)计数并评价活力,然后接种。为拆分并扩增细胞系,细胞用0.25%胰蛋白酶-edta(gibco,产品目录号25200)从培养瓶中移出。确定所有细胞系没有支原体污染,这通过idexxradil(美国密苏里州哥伦比亚)进行的pcr检测方法确定并通过检测snp组来正确鉴定。
为测试化合物a、化合物b与化合物c组合对细胞增殖的效果,细胞接种于有透明底的黑色384孔微量板(matrix/赛默科技,产品目录号4332)中的50μl培养基/每孔,细胞密度为500-1250细胞/孔(表8),并允许在37度,5%co2孵育24小时。24小时后,各细胞系制备一块384孔板以通过显微镜检查(见下)进行细胞计数,而不接受处理(=‘基线)。其它细胞板如下处理:用ats声学液体分配器(ecd生物系统公司)从药物靠模板转移25nl2000x化合物,产生最终1x浓度。化合物a在13nm-10μm的终浓度范围中使用,化合物b在13nm-10μm的终浓度范围中使用,化合物c在13nm-10μm的终浓度范围中使用(7个1:3稀释步骤)。为评价三重组合的效果,在同一实验中测试所有个体化合物(a,b,c)、所有3个成对组合(a+b,a+c,b+c)和三重组合(a+b+c)。成对组合和三重组合在各稀释度以1:1(用于药物对)和1:1:1(用于三联药物)固定比例测试,各处理产生7种组合条件。另外,转移阴性对照(dmso=‘载剂’)和阳性对照(星孢菌素=杀死细胞,7点1:2稀释系列用于16nm-1μm剂量范围)作为处理对照,在所测试细胞系中无功效的化合物与化合物a和化合物b联用作为组合对照(不超过更有效单一药剂功效的组合=‘非相互作用’组合)。化合物添加后,用hpd300数字分配器(帝肯)将50nl2mmcellevent半胱天冬酶-3/7绿色检测试剂(赛默飞世尔,产品目录号c10423)加入三个重复之一。半胱天冬酶3/7诱导作为处理诱导的细胞凋亡替代物测量。细胞根据其倍增时间处理72小时-96小时(表8),每24小时通过显微镜检查测量半胱天冬酶3/7活化,使用装有4x物镜和fitc激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。处理结束时,制备细胞以通过显微镜检查进行细胞计数。细胞在溶于pbs(波士顿生物产品公司,产品目录号bm-220)的4%pfa(ems,产品目录号15714)、0.12%tx-100(ems,产品目录号22140)中固定并透化45分钟。细胞用pbs洗3次后,其dna用终浓度为4μg/ml的hoechst33342(赛默飞世尔,产品目录号h3570)染色30分钟。细胞用pbs洗3次,然后板用铝密封件(安捷伦科技,产品目录号06644-001)使用plateloc(安捷伦科技)热密封,4℃保存直至成像。通过荧光显微镜在单一图像中捕获每孔所有细胞/处理,使用装有4x物镜和dapi激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。
适应前述方法(horn,sandmann等.2011)和使用r中的bioconductor包ebimage(pau,fuchs等.2010)后,分析图像。2个通道中即dapi(用于hoechst/dna)和fitc(用于半胱天冬酶3/7)的对象通过自适应阈值化单独分段并计数。比较阴性对照(dmso)和阳性对照(星孢菌素)后,手动确定各细胞系的半胱天冬酶3/7阳性对象阈值。通过分析dna通道中的17个额外对象/核特征(形状和强度特征),鉴定碎片/片段化核。为此,手动比较各细胞系阳性对照(星孢菌素)与阴性对照(dmso)之间的额外特征分布。能在条件之间区分的特征(如比较dmso与星孢菌素的特征测量分布的变化)用于确定‘碎片’群与‘活’核群。从原始核计数中减去碎片计数。所得核数目用作细胞增殖量度(‘细胞计数’)。
化合物对细胞增殖的作用计算自与阴性对照(dmso)的细胞计数相比较的处理的细胞计数,图13中表示为y轴上的‘标准化细胞计数’(=‘xnorm’)。协同组合用最高单一药剂模型(hsa)作为零假设(berenbaum1989)鉴定。超过hsa模型则预测受抑制靶标之间的功能联系(lehar,zimmermann等.2007,lehar,krueger等.2009)。模型输入是各药物剂量的抑制值:
i=1-xnorm
i:抑制
xnorm:标准化细胞计数(3个重复的中值)
在组合处理的各剂量点,计算组合抑制与2种单一药剂中更强者抑制之间的差异(=模型残差)。类似地,为评价三重组合在各剂量点的协同作用,计算三联药物抑制与最强药物对抑制之间的差异。为有利于高抑制下的组合效果,残差用同一剂量点观察到的抑制加权。药物组合的总体组合得分c是所有浓度中加权残差的总和:
c=σconc(i数据*(i数据–i模型))
i数据:测量的抑制
i模型:根据hsa零假设的抑制
稳健组合z得分(zc)计算为处理组合得分c与非相互作用组合绝对中位差(mad)之比:
zc=c/mad(c零)
c零:非相互作用组合的组合得分
zc是组合强度指示物:
zc≥3:协同作用
3>zc≥2:弱协同作用
zc<2:无协同作用
ic50是相对于dmso产生50%细胞计数的化合物浓度。完成ic50计算(见表9),使用r中的drc包(ritz和streibig2005年)并拟合四参数log-逻辑函数至数据。
化合物对细胞凋亡的作用如下确定:相对于原始细胞计数(减去碎片前),计算各处理和时间点有活化半胱天冬酶3/7的细胞百分数(图14的y轴)。未经实验测量的各时间点细胞计数通过回归分析由第0天和处理结束时细胞计数的对数变换(假设指数细胞生长)作线性模型拟合获得。
表9.各化合物的单一药剂ic50值以及化合物a、化合物b与化合物c组合的协同作用z得分的测量。
结果
此报告中,在总共5个tp53野生型结直肠癌细胞中单独和联合评价pik3ca抑制剂(byl719,化合物a)、mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物b)与bcl-2/-xl抑制剂(abt-263,化合物c)的功效。4个细胞系是kras突变(gp2d,ls-180,hct-116,lovo),1个细胞系是braf突变(rko)。作为单一药剂的化合物a以微摩尔ic50值抑制2个细胞系生长,且在3个其它细胞系中仅于最高剂量(10um)具有活性(图13和表9)。作为单一药剂的化合物b以亚微摩尔到微摩尔ic50值抑制细胞系生长,而化合物c没有单一药剂功效(图13和表9)。三重组合(a+b+c)相比药物对在2/5所测试细胞模型中引起协同抑制(根据hsa模型)。在4个细胞系(hct-116,lovo,rko,ls-180)中,三重组合相较成对组合显示更强的细胞凋亡(通过测量半胱天冬酶3/7诱导来评价)(图14)。
结论
总之,在tp53野生型crc中联合抑制pik3ca、mdm2和bcl-2/-xl可提供相较各单一药剂能够改善反应的有效治疗模式,并产生临床更持久的反应。
实施例5:联合mdm2抑制剂和bcl2抑制剂对增殖的体外效果
此研究设计成探索在tp53野生型结直肠癌细胞系中联合mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)与bcl-2/-xl抑制剂navitoclax(abt-263)(化合物b)对增殖的体外效果。
方法
化合物a和b以20mm浓度溶于100%dmso(西格玛,产品目录号d2650),-20℃保存待用。化合物排列在药物靠模板(葛莱娜,产品目录号788876)中并以2000x浓度3倍连续稀释(7级)。
从商业公司atcc和ecacc获得用于此研究的结直肠癌细胞系,培养并处理(表10)。所有细胞系培养基补充有10%fbs(海克隆,产品目录号sh30071.03)。
表10.细胞系信息
细胞系在37℃和5%co2培养箱中培养,在t-75培养瓶中扩增。所有情况下,细胞从冷冻储液中解冻,用1:3稀释通过≥1次传代来扩增,用vicell计数器(贝克曼库尔特)计数并评价活力,然后接种。为拆分并扩增细胞系,细胞用0.25%胰蛋白酶-edta(gibco,产品目录号25200)从培养瓶中移出。确定所有细胞系没有支原体污染,这通过idexxradil(美国密苏里州哥伦比亚)进行的pcr检测方法确定并通过检测snp组来正确鉴定。
为测试化合物a与化合物b组合对细胞增殖的效果,细胞接种于有透明底的黑色384孔微量板(matrix/赛默科技,产品目录号4332)中的50μl培养基/每孔,细胞密度为500-1250细胞/孔(表10),并允许在37度,5%co2孵育24小时。24小时后,各细胞系制备一块384孔板以通过显微镜检查(见下)进行细胞计数,而不接受处理(=‘基线)。其它细胞板如下处理:用ats声学液体分配器(ecd生物系统公司)从药物靠模板转移25nl2000x化合物,产生最终1x浓度。化合物a在13nm-10μm的终浓度范围中使用,化合物b在13nm-10μm的终浓度范围中使用(7个1:3稀释步骤)。对于化合物a与化合物b的组合,单一药剂在各稀释度以1:1固定比例合并,产生7种组合处理。另外,转移阴性对照(dmso=‘载剂’)和阳性对照(星孢菌素=杀死细胞,7点1:2稀释系列用于16nm-1μm剂量范围)作为处理对照,在所测试细胞系中无功效的化合物与化合物a和化合物b联用作为组合对照(不超过更有效单一药剂功效的组合=‘非相互作用’组合)。化合物添加后,用hpd300数字分配器(帝肯)将50nl2mmcellevent半胱天冬酶-3/7绿色检测试剂(赛默飞世尔,产品目录号c10423)加入三个重复之一。半胱天冬酶3/7诱导作为处理诱导的细胞凋亡替代物测量。细胞根据其倍增时间处理72小时-96小时(表10),每24小时通过显微镜检查测量半胱天冬酶3/7活化,使用装有4x物镜和fitc激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。处理结束时,制备细胞以通过显微镜检查进行细胞计数。细胞在溶于pbs(波士顿生物产品公司,产品目录号bm-220)的4%pfa(ems,产品目录号15714)、0.12%tx-100(ems,产品目录号22140)中固定并透化45分钟。细胞用pbs洗3次后,其dna用终浓度为4μg/ml的hoechst33342(赛默飞世尔,产品目录号h3570)染色30分钟。细胞用pbs洗3次,然后板用铝密封件(安捷伦科技,产品目录号06644-001)使用plateloc(安捷伦科技)热密封,4℃保存直至成像。通过荧光显微镜在单一图像中捕获每孔所有细胞/处理,使用装有4x物镜和dapi激发/发射滤光片的incell分析仪2000(通用电气医疗集团)。
适应前述方法(horn,sandmann等.2011)和使用r中的bioconductor包ebimage(pau,fuchs等.2010)后,分析图像。2个通道即dapi(用于hoechst/dna)和fitc(用于半胱天冬酶3/7)中的对象通过自适应阈值化单独分段并计数。比较阴性对照(dmso)和阳性对照(星孢菌素)后,手动确定各细胞系的半胱天冬酶3/7阳性对象阈值。通过分析dna通道中的17个额外对象/核特征(形状和强度特征),鉴定碎片/片段化核。为此,手动比较各细胞系阳性对照(星孢菌素)与阴性对照(dmso)之间的额外特征分布。能在条件之间区分的特征(如比较dmso与星孢菌素的特征测量分布的变化)用于确定‘碎片’群与‘活’核群。从原始核计数中减去碎片计数。所得核数目用作细胞增殖量度(‘细胞计数’)。
化合物对细胞增殖的作用计算自与阴性对照(dmso)的细胞计数相比较的处理的细胞计数,图15中表示为y轴上的‘标准化细胞计数’(=‘xnorm’)。协同组合用最高单一药剂模型(hsa)作为零假设(berenbaum1989)鉴定。超过hsa模型则预测受抑制靶标之间的功能联系(lehar,zimmermann等.2007,lehar,krueger等.2009)。模型输入是各药物剂量的抑制值:
i=1-xnorm
i:抑制
xnorm:标准化细胞计数(3个重复的中值)
在组合处理的各剂量点,计算组合抑制与2种单一药剂中更强者抑制之间的差异(=模型残差)。为有利于高抑制下的组合效果,残差用同一剂量点观察到的抑制加权。药物组合的总体组合得分c是所有浓度中加权残差的总和:
c=σconc(i数据*(i数据–i模型))
i数据:测量的抑制
i模型:根据hsa零假设的抑制
稳健组合z得分(zc)计算为处理组合得分c与非相互作用组合绝对中位差(mad)之比:
zc=c/mad(c零)
c零:非相互作用组合的组合得分
zc是组合强度指示物:
zc≥3:协同作用
3>zc≥2:弱协同作用
zc<2:无协同作用
ic50是相对于dmso产生50%细胞计数的化合物浓度。完成ic50计算(见表11),使用r中的drc包(ritz和streibig2005年)并拟合四参数log-逻辑函数至数据。
化合物对细胞凋亡的作用如下确定:相对于原始细胞计数(减去碎片前),计算各处理和时间点有活化半胱天冬酶3/7的细胞百分数(图16的y轴)。未经实验测量的各时间点细胞计数通过回归分析由第0天和处理结束时细胞计数的对数变换(假设指数细胞生长)作线性模型拟合获得。
表11.各化合物的单一药剂ic50值以及化合物a与化合物b组合的协同作用z得分的测量。
结果
此报告中,在总共5个tp53野生型结直肠癌细胞中单独和联合评价mdm2(化合物a)与bcl-2/-xl抑制剂(化合物b)的功效。4个细胞系是kras突变(gp2d,ls-180,hct-116,lovo),1个细胞系是braf突变(rko),4个细胞系也是pik3ca突变(gp2d,rko,ls-180,hct-116)(表10)。作为单一药剂的化合物a以亚微摩尔到微摩尔ic50值抑制所有细胞系生长(图15和表11)。化合物b没有单一药剂功效(图15和表11)。所述组合处理在3/5细胞模型中引起协同抑制(根据hsa模型),在另1个模型中引起弱协同抑制(表11)。所述组合相较单一药剂还显示更强的细胞凋亡诱导(通过测量半胱天冬酶3/7诱导来评价)(图16),在gp2d、hct-116和lovo中发现最强诱导。
结论
在tp53野生型、kras和braf突变结直肠癌中联合抑制mdm2和bcl-2/-xl可提供相较各单一药剂能够改善反应的有效治疗模式,并产生临床更持久的反应。
实施例6:联合mdm2抑制剂、mek抑制剂和bcl2抑制剂对增殖的体外效果
与前面实施例所述类似,在4个kras突变模型(hct-116,gp2d,lovo和ls-180)中测试(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮、曲美替尼和raf265的三重组合。所述组合在2/4模型(hct-116、gp2d,组合z得分分别是9.2和5.5)中协同,在1/4(lovo,组合z得分为2.8)中弱协同。在ls-180模型中没有观察到协同作用(组合z得分为0.2)。此外,在gp2d和lovo模型中,三重组合相较药物对显示更强的细胞凋亡诱导(通过测量半胱天冬酶3/7活化来评价)(gp2d中最高为67%细胞凋亡,lovo中为83%)。hct-116和ls-180中观察到的三重组合所引起细胞凋亡水平分别是49%和15%。
实施例7:联合mdm2抑制剂、mek抑制剂和braf抑制剂对增殖的体外效果
与前面实施例所述类似,在1个braf突变模型(rko)中测试(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮、曲美替尼和达拉菲尼的三重组合。我们发现三重组合具有协同性(组合z得分为3.5),且相较药物对显示更强(但总体弱,最高为11%)的细胞凋亡诱导(通过测量半胱天冬酶3/7活化来评价)。
实施例8:联合mdm2抑制剂、braf抑制剂和bcl2抑制剂以及可选的pi3k抑制剂或cmet抑制剂对增殖的体外效果
与前面实施例所述类似,在4个kras突变模型(hct-116,gp2d,lovo和ls-180)(rko)中测试(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮、raf265和navitoclax(abt-263)的三重组合。所述组合在1/4模型(gp2d,组合z得分为5)协同,且在所有4个测试模型(gp2d和lovo中最高为100%细胞凋亡,hct-116中为64%,ls-180中为32%)中相较药物对显示更强的细胞凋亡诱导(通过测量半胱天冬酶3/7活化来评价)。在hct-116、ls-180和lovo中没有观察到协同作用(组合z得分分别是1.3,1.2和0.5)。
同一类中,在1个braf突变模型(rko)中测试(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮、达拉菲尼和navitoclax(abt-263)的三重组合。我们发现三重组合具有协同性(组合z得分为3)且显示强烈诱导细胞凋亡(最高为100%),而所有药物对仅显示弱到没有细胞凋亡(通过测量半胱天冬酶3/7活化来评价)。
此外,测试2个四重组合,所述组合包括mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮、braf抑制剂达拉菲尼和bcl抑制剂navitoclax(abt-263)。第一个四重组合联同pi3k抑制剂(s)-吡咯烷-1,2-二羧酸2-酰胺1-({4-甲基-5-[2-(2,2,2-三氟-1,1-二甲基-乙基)-吡啶-4-基]-噻唑-2-基}-酰胺)且发现在1个braf突变细胞系(rko,组合z得分为2)中弱协同,并强烈诱导细胞凋亡(最高为79%)。第二个四重组合包括cmet抑制剂pf-04217903(辉瑞)。一旦测试,发现在1个braf突变细胞系(rko,组合z得分为4.4)中相较三重组合为弱协同,并强烈诱导细胞凋亡(最高为77%)。
实施例9:联合mdm2抑制剂、mek抑制剂和cd4/6抑制剂或紫杉醇对增殖的体外效果
将cd4/6抑制剂(特别是7-环戊基-n,n-二甲基-2-((5-(哌嗪-1-基)吡啶-2-基)氨基)-7h-吡咯并[2,3-d]嘧啶-6-甲酰胺)加入(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮和曲美替尼的组合,在2/5所测试模型中引起协同效应(hct-116,lovo;z得分分别为5.1,3.3),在2/5模型中有弱协同作用(ls-180,rko;z得分分别为2,2.8)。在gp2d(z得分为1.7)中没有观察到协同作用。没有观到到诱导细胞凋亡。
将紫杉醇(标准护理)加入组合,在1/5所测试模型中引起协同效应(gp2d,z得分为4.3),在3/5所测试模型中有弱协同效应(hct-116,lovo,ls-180;z得分分别为2.4,2和2.4)。在rko(z得分为1.1)中没有观察到协同作用。此外,组合强烈诱导细胞凋亡(gp2d:100%,hct-116:59%,lovo:61%,ls-180:20%,和rko65%)。
实施例10:在tp53野生型结直肠癌模型中联合mdm2抑制剂与mek抑制剂和bcl-2/-xl抑制剂对增殖的体外效果
产生荷瘤小鼠和治疗
所有动物实验严格遵守瑞士动物保护法完成。雌性crl:nu(ncr)-foxn1nu小鼠购自查尔斯河实验室国际公司(charlesriverlaboratoriesinternationalinc)(德国),保持在病原体受控环境中。hct-116皮下肿瘤(kras突变、pik3ca突变、p53野生型)如下诱发:在100μlpbs中浓缩300万细胞并将其注入裸小鼠右侧。小鼠随机分组,当肿瘤尺寸达到50-250mm3时,开始治疗。各组纳入8只小鼠。肿瘤尺寸每周监测3次,体积用下式计算:(mm3)=长度×宽度2×0.5.
粉末形式的mdm2抑制剂(s)-1-(4-氯-苯基)-7-异丙氧基-6-甲氧基-2-(4-{甲基-[4-(4-甲基-3-氧代-哌嗪-1-基)-反式-环己基甲基]-氨基}-苯基)-1,4-二氢-2h-异喹啉-3-酮(化合物a)、mek抑制剂曲美替尼(化合物b)和bcl-2/-xl抑制剂abt-263(化合物c)在+4℃保存。化合物a溶于0.5%羟丙甲纤维素,化合物b溶于含0.5%吐温(tween)-80%的1%羧甲基纤维素(ph7.6-8.0)的蒸馏水中,化合物c溶于微乳液预浓缩5。所有药物用5-10ml/kg口服给药。化合物a以100mg/kg一周给药3次(3qw)。化合物b和化合物c分别以0.3和100mg/kg每日给药(q24h)。组合给药方案和剂量与单一试剂相同。
检测6个治疗组(g1-g6):
-g1:载剂(dmso)
-g2:化合物c
-g3:化合物a=>9天治疗后,加入化合物c
-g4:化合物b=>9天治疗后,加入化合物c
-g5:化合物a+化合物b=>9天治疗后,加入化合物c
-g6:化合物a+化合物b+化合物c
统计分析
对于各时间点的各肿瘤,尺寸根据治疗开始前的尺寸标准化以获得“%肿瘤体积变化”(图17-18,y轴)。对于图17,计算各时间点的每组所有肿瘤平均尺寸,尺寸误差使用均值标准误差(sem)。对于图18,用单尾t检验计算p值。
结果
在hct-116细胞异种移植模型中,化合物a和曲美替尼的联合治疗(g5)相较各单一药剂治疗(g3-g4显示进行性疾病)显著更佳(稳定疾病),化合物a、曲美替尼和abt-263的三重组合(g6)引起显著肿瘤消退且相较g5有明显更佳反应(图17和图18a)。图17显示汇总的存活曲线,图18显示治疗开始后第9天和第19天的瀑布图。9天后依序加入abt-263到单一药剂化合物a不产生额外益处,而其加入单一药剂曲美替尼时终止肿瘤发展。abt-263在加入化合物a与曲美替尼的组合时引起显著肿瘤消退,第19天,同步和依序治疗的反应无法区分(图18b)。
参考文献
arrington,a.k.,e.l.heinrich,w.lee,m.duldulao,s.patel,j.sanchez,j.garcia-aguilar和j.kim(2012)."kras突变在结直肠癌中的预后和预测作用(prognosticandpredictiverolesofkrasmutationincolorectalcancer.)"intjmolsci13(10):12153-12168.
berenbaum,m.c.(1989)."协同作用是什么?(whatissynergy?)"pharmacolrev41(2):93-141.
bozic,i.,j.g.reiter,b.allen,t.antal,k.chatterjee,p.shah,y.s.moon,a.yaqubie,n.kelly,d.t.le,e.j.lipson,p.b.chapman,l.a.diaz,jr.,b.vogelstein和m.a.nowak(2013)."癌症响应靶向联合疗法的进化动力学(evolutionarydynamicsofcancerinresponsetotargetedcombinationtherapy.)"elife2:e00747.
brana,i.和l.l.siu(2012)."临床开发磷脂酰肌醇3-激酶抑制剂用于癌症治疗(clinicaldevelopmentofphosphatidylinositol3-kinaseinhibitorsforcancertreatment.)"bmcmed10:161.
cancergenomeatlas,n.(2012)."人结和直肠癌的全面分子鉴定(comprehensivemolecularcharacterizationofhumancolonandrectalcancer.)"nature487(7407):330-337.
chandarlapaty,s.(2012)."对靶向癌症疗法的负反馈和适应性耐药(negativefeedbackandadaptiveresistancetothetargetedtherapyofcancer.)"cancerdiscov2(4):311-319.
chapman,p.b.,d.b.solit和n.rosen(2014)."raf和mek抑制的组合用于治疗braf突变黑素瘤:不促进反馈(combinationofrafandmekinhibitionforthetreatmentofbraf-mutatedmelanoma:feedbackisnotencouraged.)"cancercell26(5):603-604.
chatterjee,m.s.,j.e.purvis,l.f.brass和s.l.diamond(2010)."成对激动剂扫描预测对组合刺激的细胞信号转导反应(pairwiseagonistscanningpredictscellularsignalingresponsestocombinatorialstimuli.)"natbiotechnol28(7):727-732.
chou,t.c.和p.talalay(1981)."用于分析米氏抑制和高阶动力学系统的广义方程,用2种或更多互相排斥和非排他性抑制剂(generalizedequationsfortheanalysisofinhibitionsofmichaelis-mentenandhigher-orderkineticsystemswithtwoormoremutuallyexclusiveandnonexclusiveinhibitors.)"eurjbiochem115(1):207-216.
devita,v.t.,jr.(1975)."单一药剂相比联合化疗(singleagentversuscombinationchemotherapy.)"cacancerjclin25(3):152-158.
doebele,r.c.,a.b.pilling,d.l.aisner,t.g.kutateladze,a.t.le,a.j.weickhardt,k.l.kondo,d.j.linderman,l.e.heasley,w.a.franklin,m.varella-garcia和d.r.camidge(2012)."alk基因重排非小细胞肺癌患者中耐受克唑替尼的机制(mechanismsofresistancetocrizotinibinpatientswithalkgenerearrangednon-smallcelllungcancer.)"clincancerres18(5):1472-1482.
druker,b.j.(2008)."费城染色体转化成cml疗法(translationofthephiladelphiachromosomeintotherapyforcml.)"blood112(13):4808-4817.
duncan,j.s.,m.c.whittle,k.nakamura,a.n.abell,a.a.midland,j.s.zawistowski,n.l.johnson,d.a.granger,n.v.jordan,d.b.darr,j.usary,p.f.kuan,d.m.smalley,b.major,x.he,k.a.hoadley,b.zhou,n.e.sharpless,c.m.perou,w.y.kim,s.m.gomez,x.chen,j.jin,s.v.frye,h.s.earp,l.m.graves和g.l.johnson(2012)."三阴性乳腺癌中蛋白激酶组响应靶向mek抑制的动态重编程(dynamicreprogrammingofthekinomeinresponsetotargetedmekinhibitionintriple-negativebreastcancer.)"cell149(2):307-321.
faber,a.c.,e.m.coffee,c.costa,a.dastur,h.ebi,a.n.hata,a.t.yeo,e.j.edelman,y.song,a.t.tam,j.l.boisvert,r.j.milano,j.roper,d.p.kodack,r.k.jain,r.b.corcoran,m.n.rivera,s.ramaswamy,k.e.hung,c.h.benes和j.a.engelman(2014)."mtor抑制通过阻遏mcl-1特异性地使带kras或braf突变的结直肠癌对bcl-2/bcl-xl抑制敏感(mtorinhibitionspecificallysensitizescolorectalcancerswithkrasorbrafmutationstobcl-2/bcl-xlinhibitionbysuppressingmcl-1.)"cancerdiscov4(1):42-52.
fearon,e.r.(2011)."结直肠癌的分子遗传学(moleculargeneticsofcolorectalcancer.)"annurevpathol6:479-507.
horn,t.,t.sandmann,b.fischer,e.axelsson,w.huber和m.boutros(2011)."通过rnai的合成遗传相互作用分析对信号转导网络绘图(mappingofsignalingnetworksthroughsyntheticgeneticinteractionanalysisbyrnai.)"natmethods8(4):341-346.
ji,z.,c.n.njauw,m.taylor,v.neel,k.t.flaherty和h.tsao(2012)."通过hdm2拮抗作用的p53挽救可阻遏黑素瘤生长和加强mek抑制(p53rescuethroughhdm2antagonismsuppressesmelanomagrowthandpotentiatesmekinhibition.)"jinvestdermatol132(2):356-364.
katayama,r.,a.t.shaw,t.m.khan,m.mino-kenudson,b.j.solomon,b.halmos,n.a.jessop,j.c.wain,a.t.yeo,c.benes,l.drew,j.c.saeh,k.crosby,l.v.sequist,a.j.iafrate和j.a.engelman(2012)."alk重排肺癌中获得性克唑替尼耐药的机制(mechanismsofacquiredcrizotinibresistanceinalk-rearrangedlungcancers.)"scitranslmed4(120):120ra117.
lehar,j.,a.krueger,g.zimmermann和a.borisy(2008)."高阶组合效应和生物顽强性(high-ordercombinationeffectsandbiologicalrobustness.)"molsystbiol4:215.
lito,p.,n.rosen和d.b.solit(2013)."肿瘤适应和抗raf抑制剂(tumoradaptationandresistancetorafinhibitors.)"natmed19(11):1401-1409.
pau,g.,f.fuchs,o.sklyar,m.boutros和w.huber(2010)."ebimage--r包用于应用于细胞表型的图像处理(ebimage--anrpackageforimageprocessingwithapplicationstocellularphenotypes.)"bioinformatics26(7):979-981.
porter,k.,a.babiker,k.bhaskaran,j.darbyshire,p.pezzotti,k.porter,a.s.walker和c.collaboration(2003)."引入haart后跟随hiv-1血清转化的生存决定因素(determinantsofsurvivalfollowinghiv-1seroconversionaftertheintroductionofhaart.)"lancet362(9392):1267-1274.
robert,c.,b.karaszewska,j.schachter,p.rutkowski,a.mackiewicz,d.stroiakovski,m.lichinitser,r.dummer,f.grange,l.mortier,v.chiarion-sileni,k.drucis,i.krajsova,a.hauschild,p.lorigan,p.wolter,g.v.long,k.flaherty,p.nathan,a.ribas,a.m.martin,p.sun,w.crist,j.legos,s.d.rubin,s.m.little和d.schadendorf(2015)."联用达拉菲尼和曲美替尼改善黑素瘤的总体生存(improvedoverallsurvivalinmelanomawithcombineddabrafenibandtrametinib.)"nengljmed372(1):30-39.
safaeeardekani,g.,s.m.jafarnejad,l.tan,a.saeedi和g.li(2012)."braf突变在结直肠癌和黑素瘤中的预后价值:系统性回顾和meta分析(theprognosticvalueofbrafmutationincolorectalcancerandmelanoma:asystematicreviewandmeta-analysis.)"plosone7(10):e47054.
shoemaker,r.h.(2006)."nci60人肿瘤细胞系抗癌药物筛选(thenci60humantumourcelllineanticancerdrugscreen.)"natrevcancer6(10):813-823.
solit,d.b.和n.rosen(2014)."针对raf抑制剂抗性的统一模型(towardsaunifiedmodelofrafinhibitorresistance.)"cancerdiscov4(1):27-30.
sullivan,r.j.和k.t.flaherty(2013)."黑素瘤中对braf靶向疗法的抗性(resistancetobraf-targetedtherapyinmelanoma.)"eurjcancer49(6):1297-1304.
turner,n.c.,j.ro,f.andre,s.loi,s.verma,h.iwata,n.harbeck,s.loibl,c.huangbartlett,k.zhang,c.giorgetti,s.randolph,m.koehler和m.cristofanilli(2015)."激素受体阳性晚期乳腺癌中的帕布昔利布(palbociclibinhormone-receptor-positiveadvancedbreastcancer.)"nengljmed.
- 还没有人留言评论。精彩留言会获得点赞!
- 一锅法合成托匹司他的方法
- 草本水杨梅在制备黄嘌呤氧化酶抑制剂中的应用
- 5-芳基吡唑并嘧啶衍生物的合成方法
- 5-(4-氟苯基)-N–吗啉基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(4-氯苯基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 2-(5-(4-氟苯基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺)戊二酸二乙酯的合成方法
- 一种黄嘌呤脱氢酶截断体及其应用
- 5-(4-氟苯基)-N–(3-(二甲基氨基)丙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 5-(4-氟苯基)-N–(4-氯苯乙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 一种5-芳基吡唑并[1,5-a]嘧啶衍生物的合成方法