一种利用发夹引物进行多重PCR的方法与流程
本发明涉及核酸检测领域,特别是涉及一种利用发夹引物进行多重PCR并对多个样本目标区段DNA序列进行富集,并减少引物二聚体和非特异扩增的多重PCR技术。
背景技术:
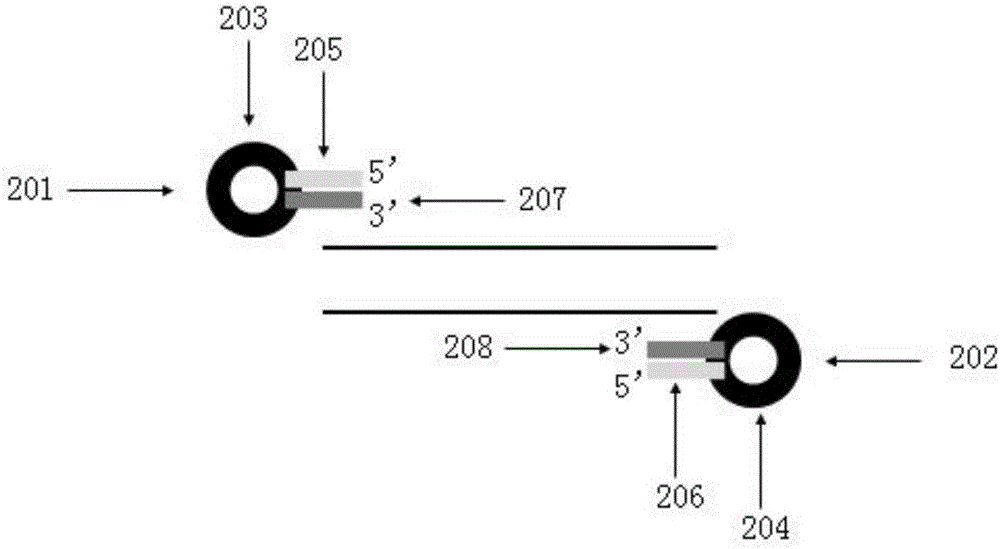
:由于低廉的价格和简便的操作,多重PCR是目标区段富集技术的重要手段。面对大量复杂基因组样本的目标区段捕获,多重PCR技术由于特异性强、价格低、重复性好等优点,已成为首选技术。与此同时,商业化的多重PCR试剂盒和非商业化的多重PCR技术不断涌现。根据文献“Target-enrichmentstrategiesfornext-generationsequencing”(Naturemethods,2010,2010,7(2):111-118)和“Genotyping‐in‐Thousandsbysequencing(GT‐seq):AcosteffectiveSNPgenotypingmethodbasedoncustomampliconsequencing”(Molecularecologyresources,2015,15(4):855-867)报道,ThermoFisherScientific和illumina等供应商提供针对肿瘤检测的商业化多重PCR试剂盒;非商业化技术也有长片段PCR、微液滴PCR、补丁PCR等。与其他目标区段富集技术相比,多重PCR的特异性强、价格低廉、不需要昂贵的硬件设施。但是传统多重PCR的通量受到引物对数的限制。在传统多重PCR反应中,随着引物对数的增加,引物错配和非特异扩增产物会急剧增加。多对引物间的相互作用,不但引入了大量非特异扩增产物,还导致不同目标片段的拷贝数差异巨大;所以传统多重PCR一般被限制为10-20对引物扩增,超过该限制则无法得到均一的产物。如何优化多重PCR反应条件,减少引物二聚体、引物错配和非特异扩增是增加多重PCR通量的关键因素,近些年有一些多重PCR优化策略被报道,但仍有待进一步提高。技术实现要素:所要解决的技术问题本发明所要解决的技术问题是提供一种利用发夹引物进行多重PCR的方法,提升多重PCR通量,同时克服传统多重PCR技术中因多对引物产生错配、非特异性扩增导致目标产物不均一的缺陷。技术方案本发明的第一个方面是提供一种利用发夹引物进行多重PCR的方法,其中,与目标区段序列特异性配对的引物是发夹引物。优选地,所说发夹引物的3’端与目标区段序列特异性配对,发夹引物的5’端与3’端序列配对。更加优选地,所说的发夹引物3’末端的碱基与5’末端的碱基互补配对,以使所说的发夹引物5’末端和3’末端封闭。如本发明所述,“发夹引物”指具备“发夹结构”或称“茎环结构”的引物。作为一种优选的实施方式,发夹引物的3’端有约20bp的目标区段特异序列,5’端序列与3’端序列互补配对,形成茎部。其余部分为环部。除非特别指明,发夹引物指上游和下游均为发夹结构的一对引物。优选地,在同一PCR反应中所述的上下游发夹引物数为两对或者两对以上。作为本发明的一种实施方式的同时,上述方法的一个优选方式为,所说发夹引物的解链温度不低于80℃,且低于多重PCR的变性温度。优选地,所说发夹引物的解链温度为81℃-86℃。在常温或者低于解链温度下,发夹引物会自身形成茎环结构,在PCR反应中的变性温度下,茎环结构的5’端序列与3’端序列配对杂交的发夹结构打开。发明人出乎意料地发现,当发夹引物3’端特异序列与目标区段完全配对时进行多重扩增反应,可以极大地减少非特异扩增和引物二聚体的产生。上述方法的另一个优选方式为,所说发夹引物除5’端与3’端配对序列之外的环部包含通用序列。所述的通用序列,作为本发明的一种具体实施方式,例如,当目标序列为人类基因的时候,为非人类基因序列。本发明的第二个方面是提供一种基于上述方法的多重PCR试剂盒,包括多对所说的发夹引物。本发明的第三个方面是提供一种利用发夹引物构建二代测序平台文库的方法,包括如下步骤:(1)在针对目标区段设计的特异引物序列的上下游5’端分别添加上游通用序列和下游通用序列,并在通用序列5’端各添加与目标区段3’端部分序列完全互补配对的序列,使解链温度不低于80℃,获得末端封闭的上下游发夹引物;(2)第一轮PCR反应,利用两对或多对上下游发夹引物对目标区段进行10-30个循环,优选15-25个循环的扩增;(3)设计接头引物,包括三部分序列,从5’端开始依次为二代测序接头引物、条形码(即barcode)序列、和通用序列;其中,每一对接头引物的条形码序列不同,以区分不同模板;(4)利用第一轮PCR扩增产物的原液或者稀释液为模板,添加上下游接头引物进行第二轮PCR反应,将不同样本所获得的第二轮扩增产物混合,即可构成二代测序平台文库。本发明的第四个方面是提供一种基于上述方法的构建二代测序平台文库的试剂盒,包括多对所说的发夹引物和接头引物。进一步地,上述技术方案所采用的具体方式,举例而言,可以包括以下步骤:发夹引物的设计:针对需要测序的目标区段序列设计引物,根据NCBI数据库中的序列,使用Primer3在线软件(http://frodo.wi.mit.edu/primer3/)进行设计,并人工在设计的引物上下游5’端分别添加与目标区段3’末端部分序列完全互补配对序列,构成末端完全封闭发夹引物,同时保证其发夹引物解链温度不低于80℃。应注意,针对本发明的第三方面:利用发夹引物构建二代测序平台文库时,可在目标区段5’端添加通用序列后,再在通用序列5’端添加与目标区段3’末端部分序列完全互补配对序列,获得末端完全封闭的特异发夹引物。应理解,是否在目标区段3’端添加通用序列不构成本发明的限制。利用发夹引物进行多重PCR反应:利用多对特异发夹引物进行反应,进行有限的循环数对目标区段进行富集。例如,PCR扩增体系具体为:25ngDNA加入到30μLTaq酶体系中,其中Taq酶体系包含0.01μM每对特异引物,200μMdNTPs,0.2mMMgCl2,1XPCRBuffer,1.2UTaq酶,并使用矿物油覆盖;同时设阴性对照,反应程序为:95℃变性3min;94℃变性30s,60℃复性4min,28个循环。上述体系和反应程序可以依照通常的多重PCR反应的常规进行适当调整,例如变性、复性、延伸的温度和时间,以及循环数,使之能够达到对数线性扩增、或者均一性等考量的要求。发明人通过多次试验,测试第一轮多重PCR所能够允许的引物对数,亦即多重PCR反应的“重”数,发现,在20重(即20对引物同时在一个反应体系中扩增)以下、50-60重,以及最多测试了89重的情形下,都能够获得均一的适合二代测序的平台文库,获得所预期的技术效果。尽管未进行更多重PCR的试验,但是,发明人相信利用本发明的方法,100重、甚至150重,都是可能成功实现本发明构思的。更多重的反应或许需要对多重PCR的反应体系或者反应条件做出优选,但是,应当仍然是在本发明的技术方案之内的优选方式。同时,对于多重PCR的重数,并非影响最终平台文库的构建及其均一性的决定或唯一因素,所以,不应当构成对本发明构思的限制,实施本发明过程中,可以根据实际需要选择重数。在试验中,发明人发现,若是省略本发明的发夹引物设计,直接进行传统的两轮PCR扩增,那么,由于反应体系中有超过20对引物,不可避免产生大量引物二聚体或者错配,导致无明显目的条带出现,产物无法作为二代测序文库。所以在本领域,多重PCR一般被限制为10-20对引物扩增,超过该限制则无法得到目的产物。有益效果本发明提供了现有技术文献中所缺乏的利用发夹引物结合多重PCR对多个目标区段进行富集的方案。在PCR扩增时,利用发夹引物进行多重PCR,减少多对引物之间引物二聚体和非特异扩增的形成,增加了不同扩增子的均一性;同时扩增的引物对数目远超普通多重PCR,提高了多重PCR的效率和通量。本发明的多重PCR方案实现了针对上数十个样本进行的平行捕获的能力,极大地节约了成本,有效地提高了捕获效率。下面结合附图和实施例对本发明及其效果进一步说明。附图说明图1是本发明的引物示意图。图中,101.上游接头引物,102.下游接头引物,103.上游特异发夹引物,104.下游特异发夹引物。图2是本发明特异发夹引物示意图。图中,201.上游特异发夹引物,202.下游特异发夹引物,203.上游通用序列,204.下游通用序列,205.与上游特异引物目标区段3’末端部分序列完全互补配对序列,206.与下游特异引物目标区段3’末端部分序列完全互补配对序列,207.上游特异引物序列,208.下游特异引物序列。图3是本发明接头引物示意图。图中,301.上游接头引物,302.下游接头引物,303.二代测序上游接头引物,304.二代测序下游接头引物,305.上游接头引物条形码序列,306.下游接头引物条形码序列,307.通用引物上游序列,308.通用引物下游序列,309.特异发夹引物扩增产物。图4是本发明利用IontorrentPGM平台检测单核苷酸多态性时有效扩增子的分布图。图中,401.扩增子数目,402.扩增子测序深度。具体实施方式下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,如分子克隆操作手册,或按照试剂制造厂商所建议的条件。所有无机化学试剂和有机溶剂购自上海化学试剂有限公司,TaqDNA聚合酶、dNTPs均购自美国Promega公司,引物由上海翰宇生物科技有限公司合成,大鼠DNA抽提采用生工生物工程(上海)股份有限公司动物基因组快速抽提试剂盒,琼脂糖凝胶回收测序文库使用天根生化科技有限公司的回收试剂盒,测序试剂盒IonPGMTMSequencing200Kitv2购自ThermoFisherScientific公司。实施例1:利用IontorrentPGM平台检测单核苷酸多态性(singlenucleotidepolymorphism,SNP)。序列查找和引物设计:根据NCBI的SNP数据库,查找出大鼠基因组中共计89段携带SNP位点的目标区段序列,使用Primer3在线软件按照一般规则进行引物设计,并人工在设计的引物上下游5’端分别添加上下游通用序列,并在通用序列的5’端添加与目标区段3’端大约13bp完全互补配对的序列,构成末端完全封闭发夹引物,同时保证其发夹引物解链温度不低于80℃,然后进行引物合成(表1)。表1.SNP检测特异发夹引物DNA抽提:44组大鼠DNA采用动物基因组快速抽提试剂盒,按照说明书进行抽提得到DNA,以0.8%琼脂糖凝胶电泳确定DNA质量和浓度。44组DNA与接头引物对应如表2所示。44个样本的接头引物不同,以区分不同模板的文库(表3)。表2.样本编号和接头引物对应表表3.接头引物序列表对44组样本DNA分别进行两轮PCR扩增。第一轮PCR选取表1中所有89对上下游特异引物,混合在同一反应体系中作为扩增引物,对单个样本进行89重PCR反应。PCR扩增体系具体为:每个样本的25ngDNA分别加入到各自的30μLTaq酶体系中,其中Taq酶体系包含0.01μM每对特异引物,200μMdNTPs,2mMMgCl2,1XPCRBuffer,1.2UTaq酶,并使用矿物油覆盖;同时设阴性对照,反应程序为:95℃变性3min;94℃变性30s,60℃复性4min,22个循环。第二轮PCR选取第一轮PCR产物作为模板,PCR扩增体系具体为:1μL第一轮PCR产物分别加入到各自的第二轮10μLTaq酶体系中,其中Taq酶体系包含1μM上下游接头引物,200μMdNTPs,2mMMgCl2,1XPCRBuffer,0.4UTaq酶,并使用矿物油覆盖;反应程序为:95℃变性3min;94℃变性30s,65℃复性2min,72℃延伸30s,10个循环。扩增结束后,将44个样本的扩增产物等体积均匀混合在一起,作为构建完成的测序文库,对44个样本同时进行IontorrentPGM平台测序分型。测序文库按照琼脂糖凝胶回收试剂盒说明书进行纯化。纯化后的文库按照PGM商业化操作流程进行后续乳液PCR和富集。富集后的产物利用318芯片和IonPGMTMSequencing200Kitv2进行二代测序,步骤严格按照供应商要求使用。测序结果如图4所示。通过检测不同扩增子的测定序列数(reads)来判断多重PCR的均一性。44个样本的所有扩增子在PGM上进行测序的结果表明,99.6%的扩增子至少有1个测定序列数(reads),而87.8%的扩增子有大于等于20和小于5000个测定序列数。本实施例检测到的扩增子中,有效的扩增子(≥20并且<5000reads)差异在1-2个数量级。图4结果显示:72.8%、90.1%、97.6%、99.2%和99.7%的有效扩增子差异分别在25倍、50倍、100倍、150倍和200倍以内。对比参考文献“Methodsforgenomicpartitioning”(AnnualReviewofGenomics&HumanGenetics,2009,10:263-284)和“Ahigh-plexPCRapproachformassivelyparallelsequencing”(Biotechniques,2013,55(2):69-74)报道的数据,利用发夹引物进行多重PCR的均一性与
背景技术:
中一些已公布的多重PCR方法相比毫不逊色(表4)。表2的结果显示,不仅针对每一个扩增子的均一性差异被缩小,针对每一个样本的均一性差异也同样减小。44个样本中,100%的样本差异在27倍以内,保证了每一个样本都有足够的测序通量。表4.不同扩增方案均一性比较扩增方法均一性实施例1的发夹引物多重PCR99.7%的扩增子差异在200倍以内High-plexPCR98.33%在100倍以内分子内探针58%在10倍以内,88%在100倍以内补丁PCR75%在50倍以内液体杂交80%在25倍以内当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1